
Abstract
In barbiturate-anesthetized rats, we induced 3 hours of permanent middle cerebral artery occlusion (MCAO) by an intraluminal thread (n = 6), or 1 hour MCAO followed by 2 hours of reperfusion (n = 6). Through a closed cranial window over the parietal cortex, the production of reactive oxygen species (ROS) was measured in the infarct border using online in vivo chemiluminescence (CL) while monitoring the appearance of peri-infarct depolarizations (PID). The borderzone localization of the ROS and direct current (DC) potential measurements was confirmed in additional experiments using laser-Doppler scanning, mapping regional CBF changes through the cranial window after permanent (n = 5) or reversible (n = 5) MCAO. CL measurements revealed a short period (10 to 30 minutes) of reduced ROS formation after vessel occlusion, followed by a significant increase (to 162 ± 51%; baseline = 100%; P < .05) from 100 minutes of permanent MCAO onward. Reperfusion after a 1-hour period of MCAO led to a burst-like pattern of ROS production (peak: 489 ± 330%; P < .05). When the experiments were terminated 3 hours after induction of MCAO, CL was still significantly increased above baseline after permanent and reversible MCAO (to 190 ± 67% and 211 ± 64%, respectively; P < .05). Simultaneous DC potential recordings detected 6.4 ± 2.7 PID in the first, 4.7 ± 2.3 in the second, and 2.8 ± 2.0 in the third hour after permanent MCAO. In animals with reversible MCAO, PID were abolished from 15-minutes recirculation onward. There was no temporal relationship between ROS production and peri-infarct DC potential shifts. In conclusion, using a high temporal resolution ROS detection technique (CL), we found that permanent MCAO (after an initial decrease) was accompanied by a steady increase of ROS production during the 3-hour observation period, while reperfusion after 1 hour of MCAO produced a burst in ROS formation. Both patterns of ROS production were not related to the occurrence of PID.
Oxygen-derived free radicals have been implicated in the pathogenesis of cerebral infarction and vasogenic edema formation after cerebral ischemia (Traystman et al., 1991; Ikeda and Long 1990; Braughler and Hall, 1989). In transient global ischemia, free radicals produced during reperfusion may contribute to a disturbed membrane function, resulting in a critical intracellular calcium accumulation (Siesjö et al., 1995). In focal cerebral ischemia, free radicals may play a key role in the pathophysiologic cascade leading to ischemic tissue damage (Matsuo et al., 1995). The production of reactive oxygen species (ROS) may occur especially in ischemic brain regions where the local oxygen tension is partially preserved by residual collateral flow, i.e., in the ischemic penumbra (Back et al., 1994b). In the infarct border, other important predisposing factors for the generation of ROS are also present, such as tissue acidosis (Back et al., 1994a; Hossmann et al., 1985), accumulation of ADP (Kogure et al., 1980), postischemic leukocyte activation (Kochanek and Hallenbeck, 1992), and excessive production of nitric oxide (Iadecola et al., 1995).
Because it is technically difficult to measure free radicals directly, most of the previous studies provided indirect evidence based on the effects of ROS scavenging drugs (Liu et al., 1989; Watanabe et al., 1994; Oh and Betz, 1991; Abe et al., 1988). It was shown that focal cerebral ischemia in transgenic mice expressing three-fold higher than normal levels of copper-zinc (CuZn)-SOD have smaller volumes of infarction and a lesser degree of brain edema compared with nontransgenic littermates (Kinouchi et al., 1991). In a recent study, hydroxyl radical trapping by salicylate microdialysis was used in reversible focal ischemia (Morimoto et al., 1996). In the infarct borderzone, significant increases of 2,3- and 2,5-dihydroxybenzoic acid (DHBA) as indicators for hydroxyl radicals were measured both during ischemia and even to a higher degree during reperfusion. The inverse correlation between 2,3-DHBA levels and CBF during recirculation suggested that less complete degrees of postischemic reperfusion may favor the production of hydroxyl radicals in the penumbra (Morimoto et al., 1996; Ginsberg et al., 1994). Based on the simultaneous observation of increased excitotoxic neurotransmitter concentrations, a link between excessive activation of glutamate receptors (excitotoxic injury) and ROS-mediated brain damage has been suggested (Morimoto et al., 1996; Ginsberg et al., 1994; Pellegrini-Giampietro et al., 1990).
At present, little is known about the interrelationship between ROS production and other important mechanisms of brain damage, such as peri-infarct depolarizations (PID). Transient depolarizations are known to occur at irregular intervals in the borderzone of evolving brain infarcts (Back et al., 1994b; Nedergaard and Astrup, 1986) and are triggered by local increases in extracellular potassium and/or glutamate concentrations. Alternatively, they may be also be evoked by fluctuations of local CBF (Nedergaard and Hansen, 1993). The significant enlargement of ischemic damage in middle cerebral artery (MCA) territory infarcts after the elicitation of those depolarizations indicates PID as a key pathophysiologic event for the dynamic evolution of morphologic damage in focal ischemia (Back et al., 1996). The purpose of the present study was to monitor the formation of oxygen-free radicals in the acute phase of focal cerebral ischemia, induced by MCA occlusion (MCAO), and to compare ROS production in permanent MCAO with that occurring after reperfusion. Recently, we have described a novel approach for the online monitoring of oxygen-free radicals in vivo with high temporal resolution (seconds) using lucigenin-enhanced chemiluminescence (CL) (Dirnagl et al., 1995). This technique enables the detection of superoxide radicals derived mainly from the intracellular compartment of cortical cells (Dirnagl et al., 1995). Using this technique we were for the first time able to address the question of whether there is a correlation between the occurrence of PID and ROS production.
MATERIALS AND METHODS
General preparation
Male Wistar rats (n = 28; body weight 270 to 310 g) were anesthetized with 100 mg/kg body weight thiopental sodium (Trapanal, BYK Pharmaceuticals, Konstanz, Germany), tracheotomized, and artificially ventilated (Small rodent respirator, Effenberger, Pfaffing/Attel, Germany). The femoral artery and vein were cannulated and a continuous intravenous saline infusion (1 mL/h) was started. Body temperature was measured by a rectal probe and maintained at 36.5°C to 37.5°C with a feedback-controlled heating pad. Systemic arterial pressure (RFT Biomonitor, Zwönitz, Germany) and end-expiratory pCO2 (CO2 Monitor EGM I; Heyer, Bad Ems, Germany) were monitored continuously. Arterial Pao2, Pa
Middle cerebral artery occlusion
A ventral midline incision was made in the neck. The right common carotid artery (CCA) was mobilized and proximally ligated (5-0 suture) with careful conservation of the vagal nerve. The distal part of the CCA was temporarily occluded by a miniature clip to prevent retrograde bleeding, then the CCA was incised. A 4-0 monofilament thread (0.15 mm in diameter, length 15 cm, the tip rounded and coated, tip diameter: 0.3 mm) was introduced into the proximal part of the CCA guided by a plastic catheter (inner diameter 0.38 mm, outer diameter 1.9 mm, length 12 cm, polyethylene tubing) which was fixed by two 5-0 sutures. At the distal end of the catheter, an additional 7-0 suture was loosely fixed around the CCA, the miniature clip removed, and the occluding thread was advanced through the internal carotid artery until it reached the base of the skull (Nagasawa and Kogure, 1989; Longa et al., 1989). In the sham-operated controls, the thread remained in this position. To induce MCAO, the thread was advanced by 5 to 6 mm until a resistance could be felt and EEG amplitude declined indicating successful vessel occlusion. Reperfusion was achieved by pulling back the thread to the initial position.
Cranial window preparation, electrophysiology, and laser-Doppler scanning
A craniotomy was performed over the right parietal cortex (covering an area 0.5- to 7.5-mm posterior to the bregma and 1.5- to 6.5-mm lateral to the midline) using a saline-cooled drill. The dura mater was removed and a closed cranial window was implanted (Lindauer et al., 1993). The cortical surface underlying the window was continuously superfused with artificial CSF (aCSF) at a superfusion rate of 2 to 3 mL/h. The composition of the aCSF was: Na+ 152 mmol/L, K+ 3 mmol/L, Ca2+ 1.5 mmol/L, Mg2+ 1.2 mmol/L, HCO3– 24.5 mmol/L, Cl− 135 mmol/L, glucose 3.7 mmol/L, and urea 6.7 mmol/L. The aCSF was equilibrated with a gas mixture containing 6.6% O2, 5.9% CO2, and 87.5% N2. The actual gas concentrations in the aCSF amounted to a pO2 of 92 ± 13 mm Hg, pCO2 of 44 ± 3 mm Hg, and pH of 7.357 ± 0.029 (mean ± SD; n = 24, for technical reasons only 24 experiments could be included). The electrical activity of the cortical surface was monitored inside the closed cranial window using an Ag-AgCl wire inserted into the space between the cortex and the coverslip. The signal was introduced into an electrometer (WPI, Sarasota, FL) and two amplifiers (J. Meyer, Munich, Germany) to record the direct current (DC) potential and the EEG. Both variables were displayed on a chart recorder and recorded using a PC running the Asyst data acquisition program (Macmillan, New York, NY).
The spatial and temporal profiles of the blood flow changes beneath the cranial window were studied by laser-Doppler scanning. For those experiments, a laser-Doppler flow probe (Vasamedics BPM2, Troy, MI) with a spatial resolution of approximately 1 mm3 and a temporal resolution of 0.1 second was used (Dirnagl et al., 1989). Controlled by a PC, the laser-Doppler flow probe was repeatedly positioned by a x-y-z-micromanipulator (Luigs & Neumann, SMI, Ratingen, Germany) at 20 predefined locations on the cranial window before MCAO, after MCAO and MCA reperfusion. The 20 individual positions were equally distributed over the cranial window surface by 1 mm (medial-to-lateral, four positions) and 1.5-mm (anterior-to-posterior, five positions) interdistance. Flow values were averaged over a 15-second period at each position and expressed as percent of baseline.
Lucigenin-enhanced CL
As previously described (Dirnagl et al., 1995), CL was measured using a cooled photomultiplier (Hamamatsu R943-02) with a dark count of 3 cps at −30°C. The counts of the photomultiplier were amplified and counted by a Hewlett-Packard Universal counter (HP 5316 B) connected to a PC for data acquisition and storage. Photon counts were summed over intervals of 6 seconds. To exclude photons from other sources than the brain, the animal in the stereotaxic frame was wrapped in aluminum foil and placed in two dark boxes, the outer one equipped with metal shielding (Faraday cage). Only the cranial window was left unshielded and positioned under a reflector collecting the photons from the exposed brain onto the photon-sensitive area of the photo cathode. Inside the dark box, only materials with zero or very low phosphorescence were used. To record the formation of ROS from the surface of the brain and the outer layers of the brain cortex, topical superfusion with lucigenin (N,N′-dimethyl-diacridinium, Sigma Chemicals), a CL enhancer, was used. Lucigenin-induced CL is particularly sensitive to intracellularly produced superoxide radicals (Paky et al., 1993; Archer et al., 1989). After the cranial window preparation, the animals were transferred to the dark box and superfusion of 0.33 mmol/L lucigenin in aCSF was started at a rate of 3 mL/h for the initial 60 minutes and, thereafter, continued at a rate of 2 mL/h. Because of the diffusion of lucigenin into the extracellular space and into the cells of the cortex, about 60 minutes were required to obtain a stable photon count. A CL baseline was recorded for at least 15 minutes before the MCA was occluded inside the dark box. Without changing the position of the animal, the intraluminal thread was advanced (occlusion) or pulled back (reperfusion).
Experimental groups and protocol
Five groups of animals were studied as summarized in Figure 1. In group I (n = 6), sham-operated controls were positioned in the dark box and lucigenin-enhanced CL, DC potential, and EEG were measured for 3 hours. In group II (n = 6), the same variables were measured before and throughout a 3-hour period of MCAO. In group III (n = 6), a 1-hour period of occlusion was followed by reperfusion for 2 hours. Measured variables were identical with groups I and II. To confirm that the cranial window includes the ischemic MCA territory, the spatial and temporal profiles of changes in CBF were studied in two further experimental groups. The protocol of flow mapping experiments of group IV (n = 5) corresponded to group II experiments (permanent MCAO for 3 hours), the protocol of group V experiments (n = 5) to group III (1-hour MCAO followed by 2-hour reperfusion). In these experiments, aCSF contained lucigenin in the same concentration as used in the CL-experiments. In group IV, laser Doppler flowmetry (LDF) mapping was performed before vascular occlusion and at 5, 60, and 180 minutes postocclusion. In experiments of group V, LDF scanning was performed before occlusion, 5 minutes postocclusion, and 5 and 120 minutes postreperfusion. Sensitivity of the CL measurement to hyperoxia (inhalation of 100% oxygen for 1 minute) was tested in all experiments of groups I, II, and III.
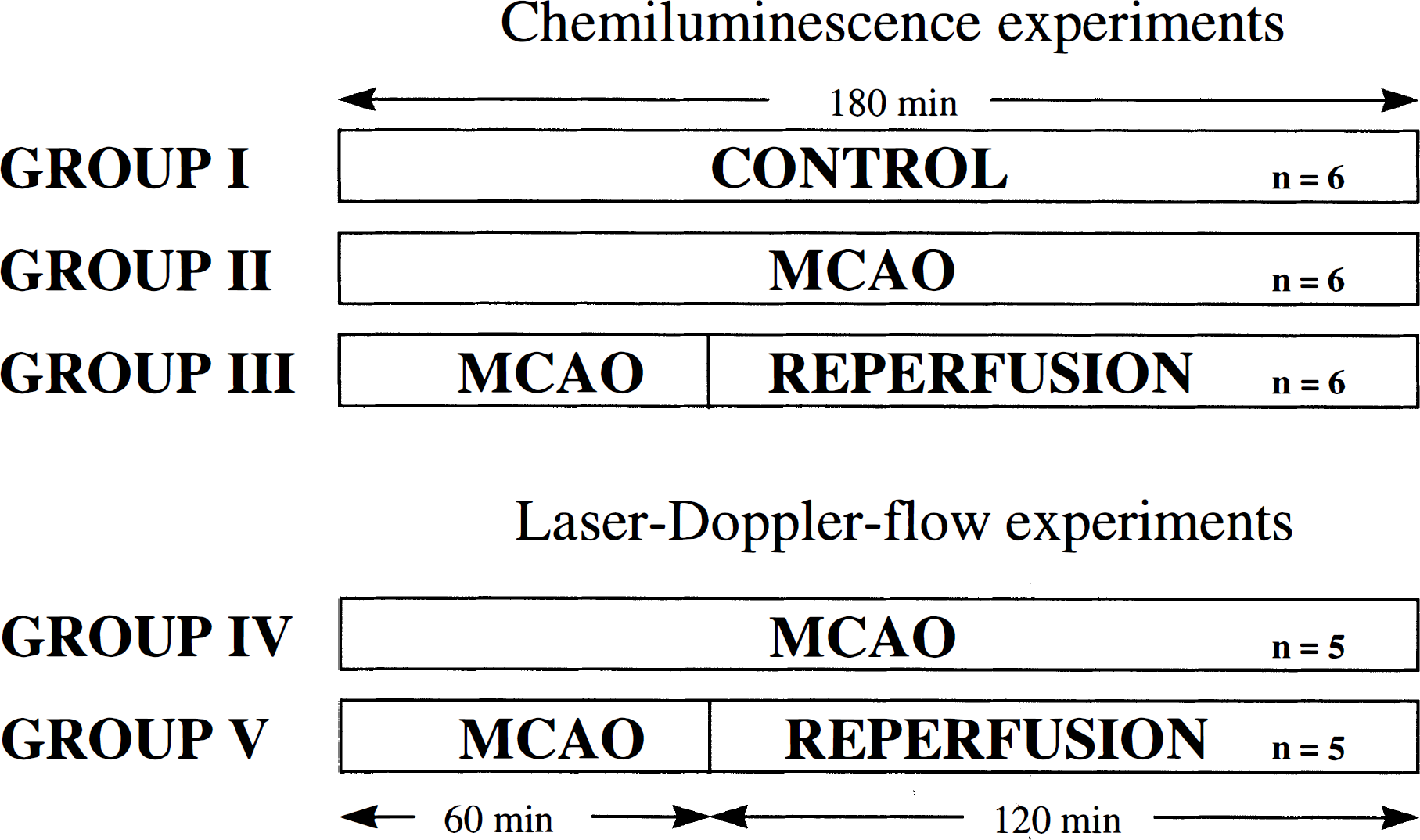
The paradigm of experimental groups I to V is shown. EEG and DC potential were measured in all experiments.
Experiments were terminated by intravenous application of KCl (n = 25). In three animals of group II, thiopental anesthesia was extended for 6 hours after insult to obtain histopathology. Under 4% halothane anesthesia, the rats were decapitated. Brains were removed within 2 minutes and frozen in liquid nitrogen. Coronal sections (40 μm) with 500-μm interdistance were cut on a cryostat and stained with vanadium acid fuchsin (VAF, 100 mg of acid fuchsin dissolved in 75 mL of distilled water, 25 mL of 0.5% ammonium metavanadate solution and 1 mL of 100 % acetic acid added) and counterstained with toluidine blue (TB; 25 mg TB dissolved in 100 mL of acetate buffer). VAF staining is capable of revealing irreversibly damaged brain tissue comparable to conventional hematoxylin and eosin staining (Victorov and Barskow, 1993). The infarct distribution was evaluated in relation to the position of the cranial window.
Statistical analysis
All data are given as means ± SD. CL data and physiologic variables were analyzed for statistical differences between experimental groups using analysis of variance followed by the nonparametric Mann-Whitney U test. Repeated LDF measurements of mapping experiments were tested for significant differences by repeated measures analysis of variance followed by a paired samples Student's t-test. P values less than .05 were accepted as statistically significant. DC potential data of groups II + IV and III + V were merged because the experimental protocol and design were comparable. The LDF measurements of groups IV and V were also merged for the timepoint of 5 minutes postocclusion because of an identical experimental protocol at that stage.
RESULTS
Physiologic variables
The physiologic variables are summarized in Table 1. Except for a 1-minute period of induced hyperoxia in groups I, II, and III (test of sensitivity of the CL measurement, data not shown), all the physiologic variables were within normal limits. No significant differences were detected between experimental groups.
Physiologic variables
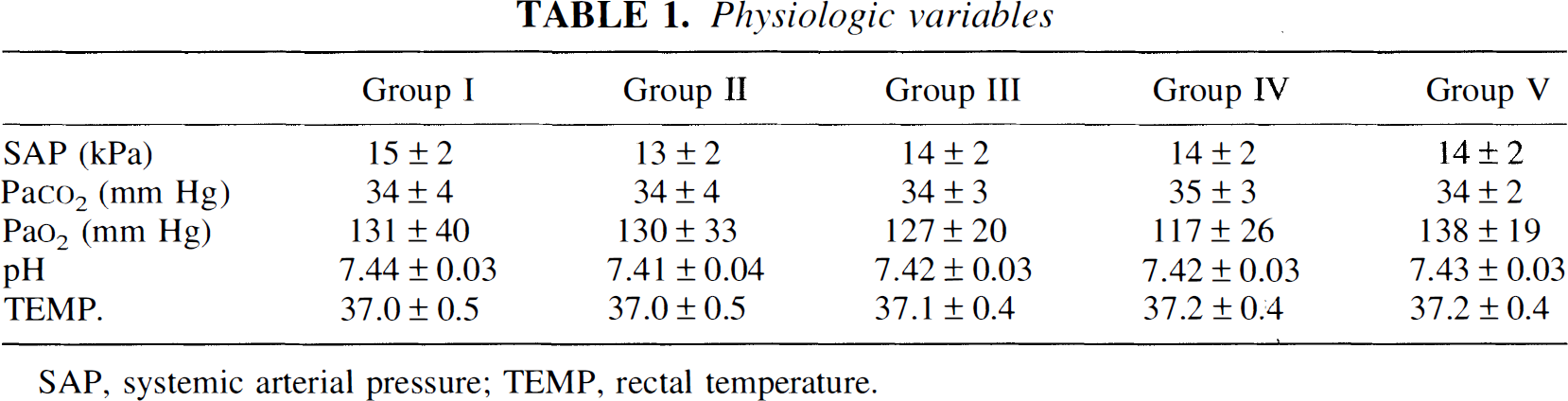
SAP, systemic arterial pressure; TEMP, rectal temperature.
EEG and DC potential measurements
Typical examples of EEG and DC potential recordings are shown in Figure 2. Immediately after MCAO (groups II–V) the EEG amplitude decreased and, after a latency of a few minutes, the first negative DC deflection appeared (Fig. 2A and B). This postocclusion pattern of EEG and DC potential changes indicated successful vessel occlusion; animals not showing these specific changes were excluded from the study. In groups III and V, reperfusion induced a normalization of the EEG amplitude (Fig. 2B).
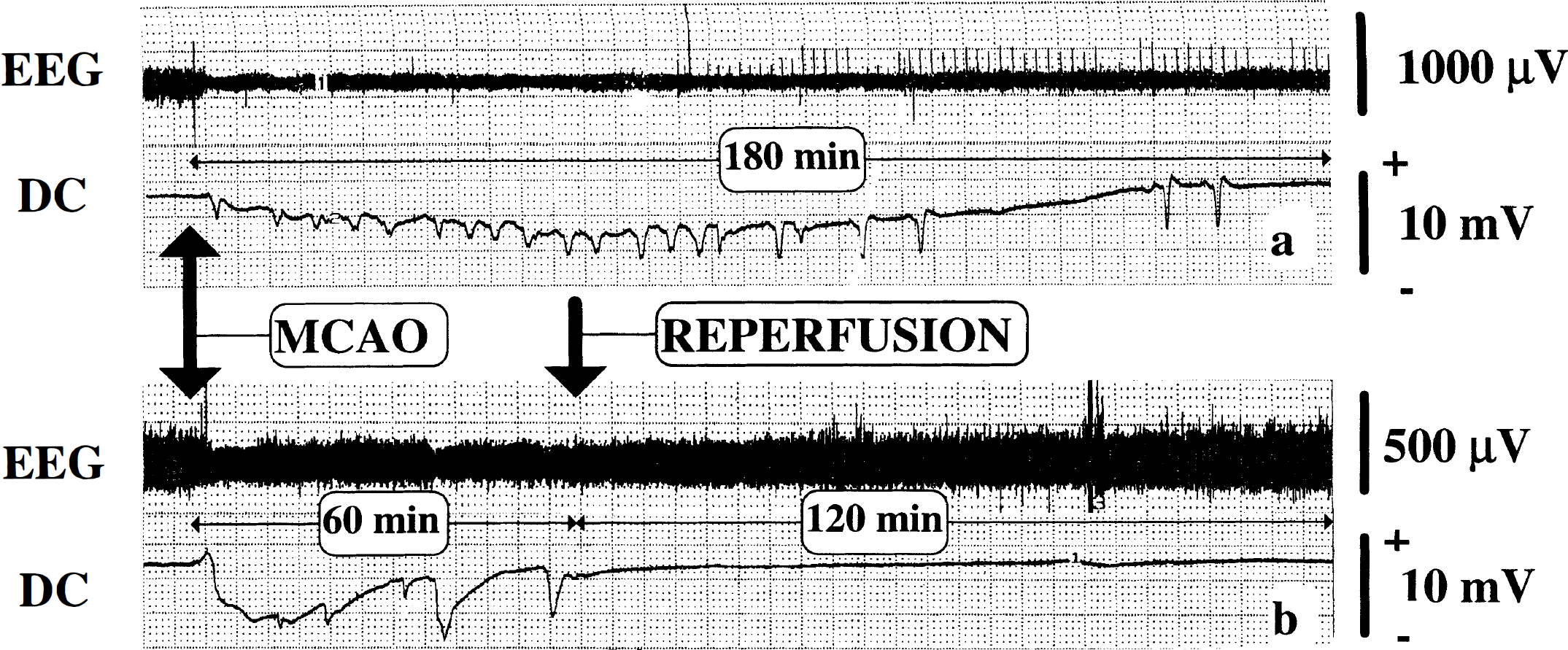
Typical recordings of simultaneous EEG and DC potential recordings are shown in an example of permanent MCAO of 3-hour duration
Figure 3 shows the frequency distribution of PID over time. In summary, PID tended to occur less frequently in the third hour of MCAO, whereas reperfusion nearly abolished the occurrence of PID (only in 4 of 11 animals PID appeared during the initial 15 minutes after the initiation of recirculation). In animals with permanent MCAO (groups II and IV), the amplitude and duration of the DC shifts observed amounted to 3.5 ± 1.8 mV and 3.5 ± 2.2 minutes, respectively (mean ± SD). In groups III and V (reversible MCAO), PID were measured with an amplitude of 2.9 ± 2.0 mV and a duration of 3.0 ± 1.9 minutes. They did not differ from the DC potential changes seen in groups II and IV.
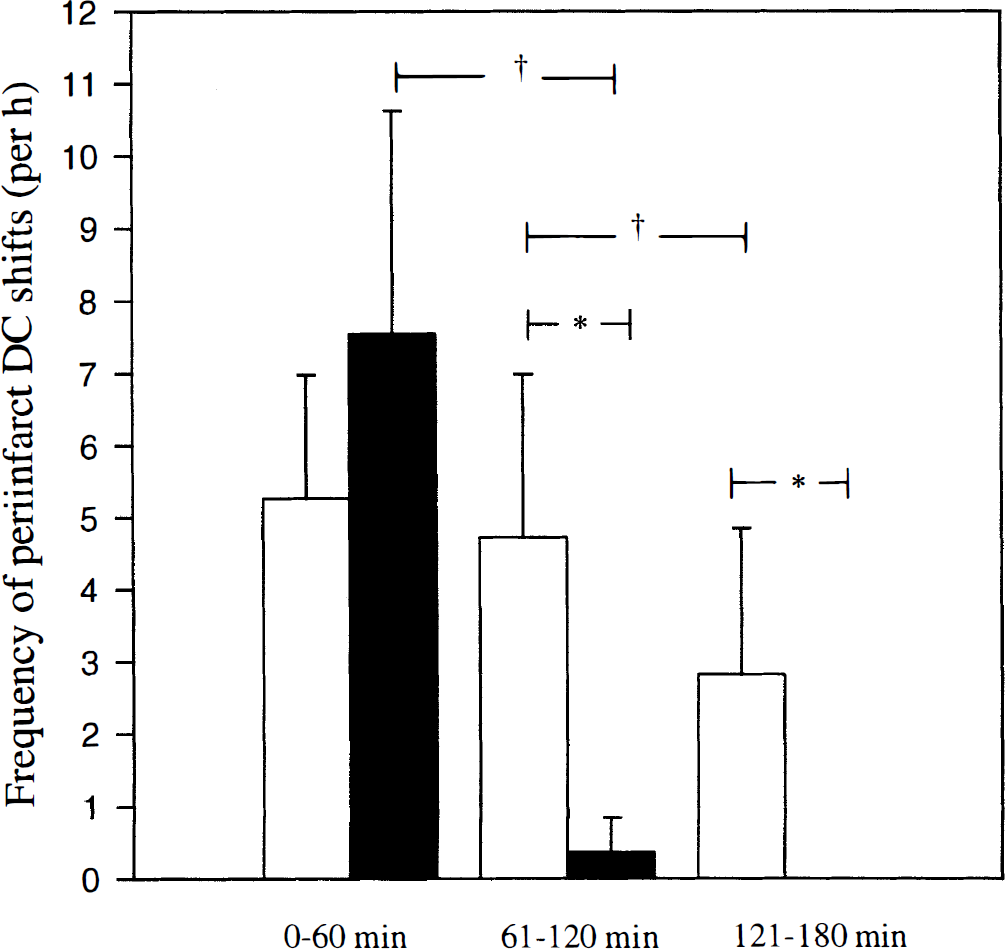
Frequency distribution of peri-infarct DC potential shifts resembling PID during the initial, second, and third hour of observation. Open bars denote merged data of groups II and IV (permanent 3-hour MCAO), solid bars show merged data of groups III and V (1-hour MCAO followed by 2-hour reperfusion). The DC shifts measured in reperfusion experiments all occurred within the initial 15 minutes after recirculation of the MCA territory. 'Group II + IV (n = 11) MCAO without reperfusion. Group III + V (n = 11) reperfusion after 1 hour MCAO. † Marks significant differences within groups, * denotes a significant difference between groups II and IV compared to groups III and V.
Lucigenin-enhanced CL
Superfusion of aCSF containing 0.33 mmol/L lucigenin led to an increase of the photons emitted from the cortical surface. A stable photon count was achieved within 1 hour. Sensitivity of the system was tested by inducing hyperoxia (increase in photon counts by inhalation of 100% oxygen for 1 minute). Baseline measurements were taken for at least 15 minutes.
In group I (control), CL remained stable throughout the observation period of 3 hours. In group II, the photon count decreased to 78 ± 8% (P < .05) within 10 minutes postocclusion and remained below baseline for 10 minutes until the count started to increase (Fig. 4). In group III, a similar pattern was observed: the lowest level of CL, 81 ± 11% of baseline (P < .05), was measured 20 minutes after occlusion, while the 10- and 30-minute postocclusion values were also significantly reduced. Within the initial 60 minutes, the photon count increased to 116 ± 49% (group II) and 109 ± 30% of control (group III), respectively. In group II (permanent MCAO), CL was significantly enhanced from 100 minutes postocclusion onward and peaked to 193 ± 51% of control at 150 minutes after MCAO. In group III (reversible MCAO), reperfusion led to a sharp increase of CL with a maximum of 489 ± 330% of baseline (P < .05); thereafter, the signal decreased rapidly. In both experimental groups, the photon count did not return to baseline and remained increased at approximately two-fold levels throughout the observation period (group II: 190 ± 67% and group III: 211 ± 64%, respectively).
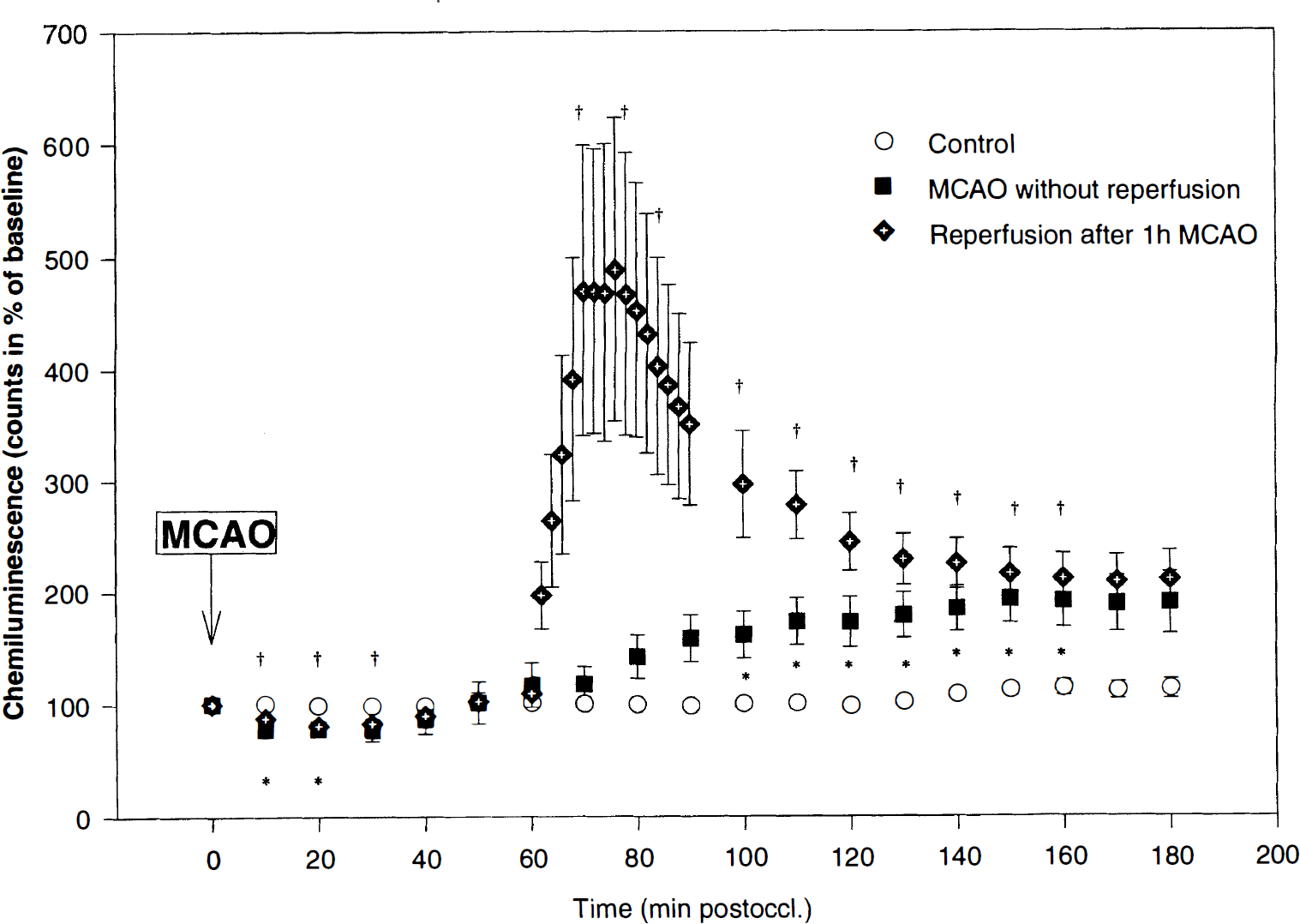
Lucigenin-enhanced CL for monitoring reactive oxygen species during control condition (open circle), permanent MCAO (solid square), and reperfusion for 2 hours after 1 hour MCAO (solid diamond). N = 6 for each experimental group. The data points are presented as means ± SEM. * Significant difference of measurements during permanent MCAO (group II data) compared to control; †significant difference of group III data (reperfusion after 1-hour MCAO) compared to control (P < .05). During the early reperfusion phase (60 to 90 minutes after recirculation) only data-points with 10-minute interval were used for statistical testing to meet the identical timepoints of the experiments of groups I and II.
Relationship between free radical production and peri-infarct DC deflections
In Figure 5, a typical example of the simultaneous CL and DC potential recordings is shown in an animal with permanent MCAO. PID spread at irregular intervals across the parietal cortex, whereas the photon count continuously increased except for the initial decrease of CL after vessel occlusion. Using the high temporal resolution of CL, depolarization-induced concurrent changes in CL could not be detected (Fig. 5B). Analysis of all DC deflections measured simultaneously with lucigenin-enhanced CL, did not reveal a significant interrelationship between PID and ROS production. At the end of the experiment, intravenous KCl led to a sudden decrease of the CL signal and a large DC negativation (terminal depolarization).
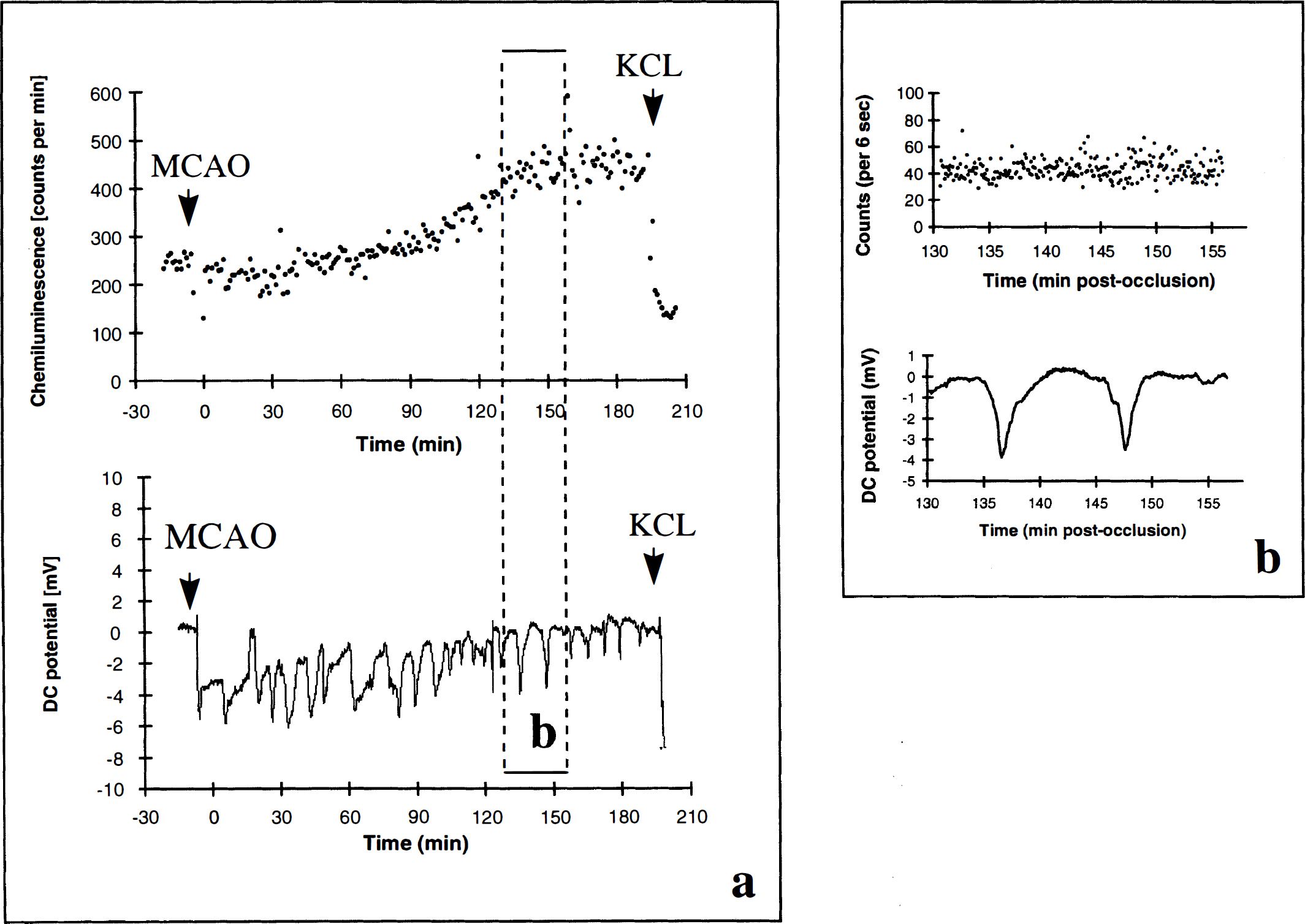
Time-matched comparison of CL measurements (in counts per minute [
LDF mapping
In accordance with the topography of the MCA territory, 5 minutes after MCAO, significant reductions of CBF to 50 ± 12 % of control were measured in 14 of 20 positions, all of them located in the lateral part of the cranial window (merged LDF data of groups IV and V, P < .05; Fig. 6). In some experiments, the points of measurement facing the saggital suture (anterior cerebral artery territory) showed hyperperfusion immediately after MCAO. As shown in Fig. 6, LDF measurements revealed significant reductions in 10 of 20 positions 60 minutes and 180 minutes after vessel occlusion (group IV LDF data; mean flow reduction to 51 ± 10% and 58 ± 10% of control, respectively, P < .05). In group V, reperfusion was established promptly within 5 minutes after reopening of the MCA. This was true in particular for the previously ischemic cortex as present under the lateral part of the cranial window. At 13 window positions, mean flow values were greater than 150% at this timepoint (because of high SD, flow was significantly different from control only at five positions). Two hours after reperfusion, no significant differences compared with baseline were detected (Fig. 6).
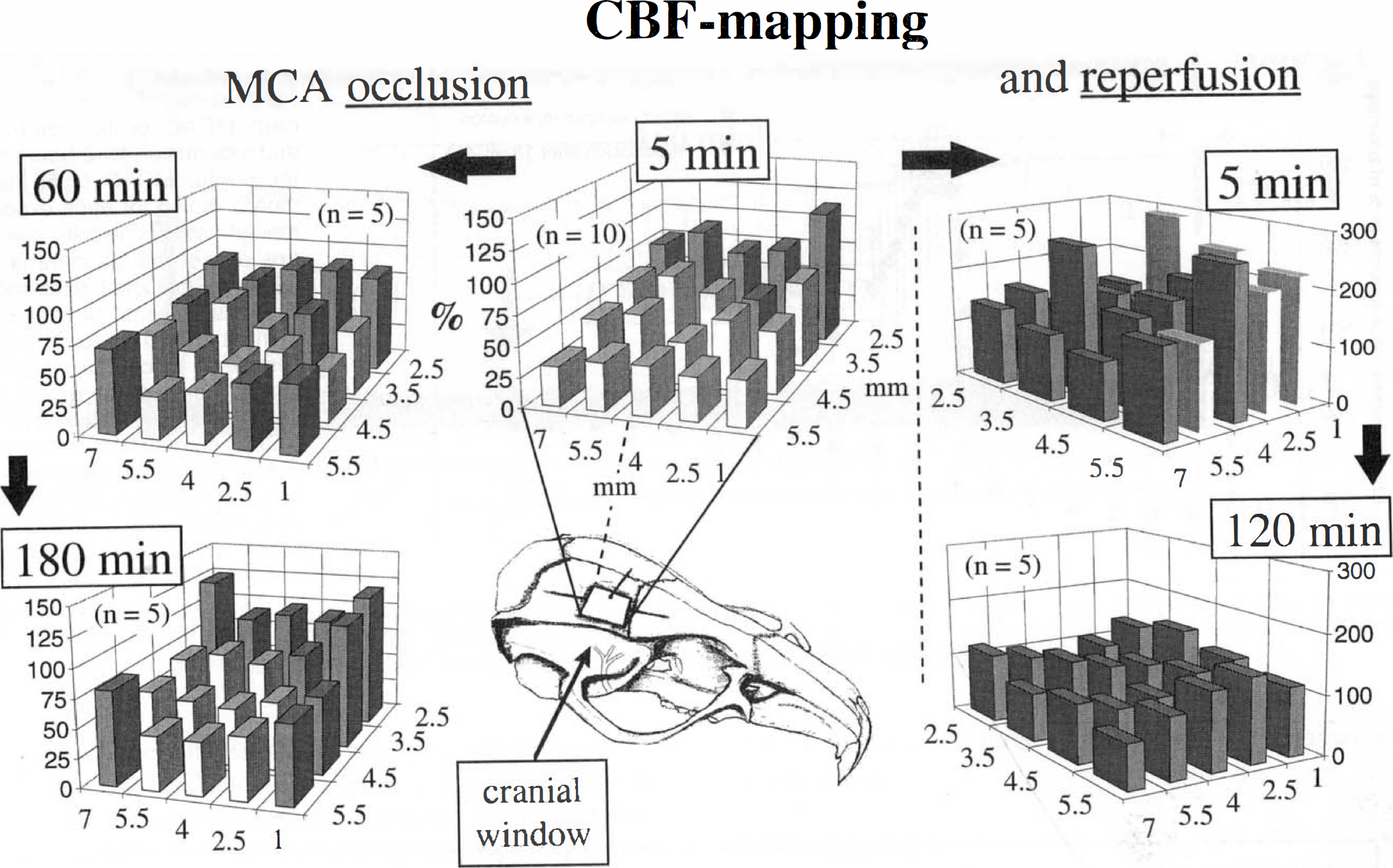
LDF mapping in permanent (left column) and reversible MCAO experiments (right column). Twenty points of CBF measurement were equally distributed over the surface of the cranial window positioned in the parietal part of the skull as shown in the figure. The points of LDF measurement are described in millimeters of distance from the sutura sagittalis (z-axis) or bregma (x-axis), and are shown as bars (flow in % of control). The white bars denote significant differences in flow compared to the control condition preocclusion. Note the prompt recirculation at 5 minutes postreperfusion and the homogenous flow distribution after 120 minutes of reperfusion.
Histology
Coronal sections, stained with VAF and counterstained by TB, confirmed ischemic lesions in the striatum and in the frontoparietal cortex at the level of the cranial trepanation (not shown). Lesions were in good spatial correspondence with the position of the cranial window.
DISCUSSION
In the present study, we applied the newly developed technique of lucigenin-enhanced CL to a model of focal cerebral ischemia to monitor the production of ROS online. CL has been shown to measure predominantly the formation of superoxide radicals derived from the intracellular compartment of cortical cells (Dirnagl et al., 1995). For the first time, we showed an early sustained increase of superoxide radicals in the infarct border after both permanent and reversible MCAO. The comparison between both experiments revealed that the initiation of recirculation was accompanied by a steep short-lasting increase of ROS production by nearly five-fold levels, while permanent vessel occlusion led to a more gradual increase by about two-fold levels at 3-hour postocclusion. This finding is in good agreement with the effect of 20-minute global cerebral ischemia on CL measurements: after reperfusion an increase of CL to 225% of control was observed that did not return to normal over the subsequent 2 hours of observation (Dirnagl et al., 1995). The condition of focal ischemia that is characterized by a graded flow reduction in the infarct border (still allowing oxygen transport to a limited degree) is a better stimulus for the production of ROS compared with global ischemia where oxygen transport is greatly exhausted.
In a rat model of reversible MCAO, Morimoto et al. (1996) showed by microdialysis sampling of salicylate hydroxylation products that the pattern of increased concentrations of hydroxyl radicals after ischemia/reperfusion was similar, however less pronounced, compared with our observations. During the 2-hour MCAO period, the levels of 2,5 and 2,3-DHBA (as indicators of hydroxyl radicals) were increased gradually to 2- to 2.5-fold values above baseline, and further enhanced to nearly four-fold values at 30 minutes postreperfusion without a clear peak after recirculation (Morimoto et al., 1996). In contrast, reperfusion in our experiments prompted a steep maximum increase of ROS concentrations within only 10 minutes, which may be explained by the biochemical pathway of oxygen radicals. Superoxide radicals are an immediate radical product of mitochondrial electron flow that has been shown to constantly produce oxygen radicals by 2% to 5% of the corresponding electrons (Boveris and Chance, 1973). Hydroxyl radicals, however, are shorter lived than superoxide radicals and secondarily formed in the presence of trace metal iron and H2O2 or, alternatively, in the presence of NO radicals outside the mitochondria (Chan, 1996).
Our study provides direct evidence for the role of ROS, especially superoxide radicals, in the pathophysiologic cascade leading to ischemic injury. In models of focal ischemia, the participation of free radicals in the production of ischemia/reperfusion injury has mainly been suggested by the effectiveness of radical-scavenging drugs (Watanabe et al., 1994; Oh and Betz, 1991; Abe et al., 1988) and SOD (Imaizumi et al., 1990; Kinouchi et al., 1991; He et al., 1993). Mice overexpressing human CuZn-SOD are highly resistant to reperfusion injury after focal cerebral ischemia (Yang et al., 1994). Our measurements emphasize that the deleterious effect of superoxide radicals may be enhanced by reperfusion compared to permanent MCAO. However, only future studies using various timepoints of reperfusion will be able to clarify to which degree the early restoration of flow is able to minimize infarct development despite the generation of additional oxidant-related stress to the ischemic tissue.
Peri-infarct depolarization occurring at irregular intervals in the infarct border have been identified as a major determinant of infarct volume in focal cerebral ischemia (Back et al., 1996; Mies et al., 1993). The appearance of PID is known to produce marked metabolism/flow uncoupling (Back et al., 1995), to induce episodes of tissue hypoxia due to the absence of an appropriate flow response (Back et al., 1994b), to stimulate anaerobic glycolysis leading to lactacidosis (Gyngell et al., 1994), and to compromise energy metabolism (Takeda et al., 1993). Therefore, important predisposing conditions for the generation of oxygen-free radicals, such as reduced blood flow, acidosis, accumulation of ADP, and activation of microglia (Korematsu et al., 1994) are present in the infarct borderzone. On the other hand, it is known that cortical spreading depression when applied 3 days before subsequent focal ischemia reduced the infarct volume by nearly 50% (Matsushima et al., 1996). This neuroprotective effect was attributed to the increased expression of both CuZn and Mn isoforms of SOD as recently shown by preliminary data (Caggiano and Kraig, 1997). Therefore, it seemed debatable whether the spreading depression-like depolarizations in the borderzone of focal ischemia would alter the levels of ROS. Using lucigenin-enhanced CL with a temporal resolution of 6 seconds and the online measurement of the DC potential via an Ag/AgCl electrode positioned under the closed cranial window, we were unable to detect any relationship between superoxide formation and DC shifts resembling PID.
It may be inferred that the CL measurements do not allow differentiation between different sources of photon counts under the cranial window and may not be specific for the detection of intracellularly generated superoxide radicals. In a brain slice preparation, however, it could be shown that free SOD, which is restricted to the extracellular space, did not attenuate the CL response to reoxygenation after a 15-minute period of hypoxia. In contrast, liposome-entrapped SOD, which is able to enter cells (Yusa et al., 1984), strongly impaired the hyperoxygenation CL burst (Dirnagl et al., 1995). It appears that lucigenin is taken up by the cells and reduced intracellularly. Considering that lucigenin-enhanced CL in vitro can be almost completely abolished by SOD treatment but not by catalase (Archer et al., 1989), a relative specifity for superoxide radicals may be assumed. Despite the low spatial resolution and the lack of quantitative data on ROS, there are good reasons to believe that this technique is sensitive enough to uncover signal changes due to PID. Consider that a wave of PID travels through the cranial window from lateral (infarct core) to medial, that the speed amounts to 4 mm/min, and with duration of at least 1 minute, the depolarization would cover 4 of 5 mm in the medial-to-lateral direction and the entire anterior-to-posterior span of the window until the repolarization phase started at the lateral rim. Even assuming less ideal spatial patterns, the depolarizations should cover approximately 50% of the window surface when travelling across and, therefore, 50% of the area contributing to the CL signal would undergo a transient change from the repolarized to the depolarized state. The fact that we almost never observed PID during reperfusion when ROS generation peaked supports the view that the phenomenon of PID is a pathophysiologic factor independent of the formation of ROS. It may be speculated that the enhancement of superoxide radicals after the reopening of the MCA contributes to the type of brain damage known as reperfusion injury (Dietrich, 1994). Reperfusion injury is accompanied by an early dysfunction of the blood-brain barrier, subsequent vasogenic edema formation and irreversible neuronal damage and has been documented in a model of reversible thrombotic MCAO (Dietrich et al., 1989). In contrast, the spread of depolarizations to the non-ischemic cortex of the affected hemisphere may trigger genomic responses (e.g., expression of heat shock proteins, CuZn and Mn SOD) which, to a later timepoint, may act neuroprotective (Nowak and Jacewicz, 1994; Matsushima et al., 1996; Caggiano and Kraig, 1997).
In addition to the CL experiments, we studied the pattern of blood flow reduction as present under the cranial window to better understand the relationship between the generation of ROS and the degree of ischemia. The LDF mapping data clearly show that the position of the cranial window was located in the vicinity of the ischemic penumbra exhibiting a mean flow reduction to approximately 50% of control in the majority of points measured. These flow values are distinctly too high to produce consistent infarction in rats (Tyson et al., 1984). Therefore, it may be assumed that the cortical tissue exposed to the CL measurement did not suffer from critical ischemia and contained viable cells. In agreement with this notion, the histologic sections obtained 6 hours after induction of MCAO proved that the infarct rim approached the lateral part of the cranial window, but did not reach into it. At this site, the initial flow reduction amounted to 20% to 30% of control. The parallel CL measurements revealed an initial significant decrease of photon counts which may be explained by a concurrent decrease of oxygen transport due to the pronounced reduction of flow. With increasing ischemia time, the regional flow values tended to slightly increase probably due to an improved collateral circulation. On the other hand, reperfusion of the MCA led to a prompt recirculation of the parietal cortex as early as 5 minutes after pulling back the occluding thread. The restoration of flow preceded the generation of ROS which peaked at about 10 minutes postreperfusion. Despite a homogenous normalization of flow 2 hours after recirculation, the photon counts still revealed a significant two-fold increase in superoxide radicals. Because this level of increased ROS formation was exactly comparable to the CL value measured after 3-hour permanent MCAO, we conclude that the effect of reperfusion was to augment at early times the basal steady increase of postischemic free radical production. It is tempting to argue that the source of excess ROS production is located in the (dysfunctional) mitochondria of cells which have not been exposed to critical ischemia as shown by the two-dimensional flow profile of the LDF mapping. However, it appears to be likely that other potential sources such as blood-brain barrier breakdown, microglial activation, or perturbation of radical scavenging systems (SOD, glutathione peroxidase, catalase) are also involved. Although the lucigenin-enhanced CL measured in vivo does not provide quantitative data, we are convinced that the pronounced changes in ROS levels seen in this study are of pathophysiologic significance.
In contrast to previously reported data (Back et al., 1996; Back et al., 1994b; Mies et al., 1993), this investigation revealed a higher frequency of peri-infarct DC potential shifts (5 to 7.5 DC shifts/h) probably due to the lack of halothane anesthesia. Halothane has been used in most of the previous studies and is known to decrease the DC shift frequency in a dose-dependent manner (Back et al., 1997). The amplitude and duration of PID were in good agreement with earlier findings (Back et al., 1994b; Kohno et al., 1995b). For the first time it could be shown that complete and rapid recirculation was able to inhibit the occurrence of further depolarizations from 15 minutes postreperfusion onward (PID appeared during the initial phase of reperfusion in only 4 of 10 animals). This observation supports the previous finding that partial reperfusion in a model of thrombotic MCAO coupled with permanent ipsilateral and reversible contralateral CCA occlusions reduced significantly the frequency of PID from 4 to 0.8 DC shifts per hour (Back et al., 1995). It is conceivable that the restoration of flow is able to reverse the accumulation of glutamate and potassium in the extracellular space which trigger the depolarizations in the infarct border (Hossmann, 1997).
CONCLUSION
The present study characterized the dynamic pattern of free radical production after permanent and reversible MCAO using the lucigenin-enhanced CL technique which mainly detects superoxide radicals generated in the mitochondria during ischemic stress. We showed a steady increase in ROS production in permanent occlusion and a burst-like pattern of enhanced ROS production after early reperfusion—both unrelated to the occurrence of PID. We conclude that the mechanism of superoxide radical-related brain injury is independent of the PID-related increase in scattered neuronal injury after focal cerebral ischemia. It is possible that superoxide radicals play a major role in the mechanism of reperfusion injury which, however, needs to be shown in future investigations.