
Abstract
Casein zymographic assays were performed to identify changes in μ-calpain and m-calpain activity in naive, sham-injured, and injured rat cortex at 15 minutes, 3 hours, 6 hours, and 24 hours after unilateral cortical impact brain injury. Cortical samples ipsilateral and contralateral to the site of injury were separated into cytosolic and total membrane fractions. Marked increases in μ-calpain activity in cytosolic fractions in the ipsilateral cortex occurred as early as 15 minutes, became maximal at 6 hours, and decreased at 24 hours to levels observed at 15 minutes after injury. A similar temporal profile of cytosolic μ-calpain activity in the contralateral cortex was observed, although the increases in the contralateral cortex were substantially lower than those in the ipsilateral cortex. Differences were also noted between cytosolic and total membrane fractions. The detection of a shift in μ-calpain activity to the total membrane fraction first occurred at 3 hours after traumatic brain injury and became maximal at 24 hours after traumatic brain injury. This shift in μ-calpain activity between the two fractions could be due to the redistribution of μ-calpain from the cytosol to the membrane. m-Calpain activity was detected only in cytosolic fractions. m-Calpain activity in cytosolic fractions did not differ significantly between ipsilateral and contralateral cortices, and increased in both cortices from 15 minutes to 6 hours after injury. Relative magnitudes of m-calpain versus μ-calpain activity in cytosolic fractions differed at different time points after injury. These studies suggest that traumatic brain injury can activate both calpain isoforms and that calpain activity is not restricted to sites of focal contusion and cell death at the site of impact injury but may represent a more global response to injury.
Traumatic brain injury (TBI) can result in excitotoxic consequences that have largely been attributed to pathological increases in intracellular calcium (Fineman et al., 1993; Nilsson et al., 1993; Shapira et al., 1989; Nadler et al., 1995, for reviews see Hayes et al., 1992, McIntosh, 1996). Loss of calcium homeostasis can result in activation of calcium-dependent proteases, calpains, that may be one of the principle causes of pathology after TBI (for reviews see Wang and Yuen, 1994; Yuen and Wang, 1996; Kampfl et al., 1997). TBI produces significant degradation of all three major classes of cytoskeletal proteins, including microtubule-associated proteins (MAP2; Taft et al., 1992; Hicks et al., 1995; Posmantur et al., 1995), intermediate filament proteins (i.e., low and high molecular weight neurofilament proteins; Posmantur et al., 1994; 1996a,b) and microfilaments (i.e., spectrin; Kampfl et al., 1996; Posmantur et al., 1996a,b; Saatmann et al., 1996a). These cytoskeletal proteins are all substrates for calpain proteolysis. Some studies of cytoskeletal proteolysis after TBI have reported the accumulation of calpain-specific breakdown products (Posmantur et al., 1994; Saatmann et al., 1996b; Kampfl et al., 1996; Newcomb et al., 1997). Further, inhibitors of calpain (and other proteases) have been shown to attenuate cytoskeletal protein loss (Posmantur et al., 1997) and provide behavioral protection (Saatmann et al., 1996b; Hayes et al., unpublished data) after experimental brain injury in vivo. Finally, subunit autolysis of μ-calpain, an event thought to accompany calpain activation, has been reported between 15 minutes and 24 hours after TBI (Kampfl et al., 1996).
Despite a number of congruent indirect lines of evidence for the role of calpain in pathological responses to TBI, no studies to date have provided direct evidence of increased calpain activity after injury. Moreover, no studies of central nervous system injury have systematically compared changes in activity of the major calpain isoforms. The absence of such data may be attributable to difficulties in examining calpain activity in vivo partly due to the presence of the endogenous calpain inhibitor, calpastatin. Recently, a zymographic assay for calpains using nondenaturing casein-containing polyacrylamide gels has been developed that circumvents this limitation (Raser et al., 1995). In addition, the technique allows for the differential and concurrent measurement of the two major isoforms of calpain, μ-calpain and m-calpain. The technique also provides the opportunity for analyzing protease activity in cytosolic and total membrane fractions, an important consideration because translocation of calpain may be a determinant of its attack on membrane-bound cytoskeletal protein targets (Saido et al., 1994). The present study represents the first application of this technique to in vivo studies of central nervous system injury. We report that TBI results in increases in μ-calpain and m-calpain activity that occur as early as 15 minutes after injury and persist as long as 24 hours after injury. Increased cytosolic μ-calpain activity is associated with translocation of this isoform to the membranes, while increased m-calpain activity remained in the cytosol.
MATERIALS AND METHODS
Chemicals
Dithiothreitol (DTT), ethylenediaminetetraacetic acid (EDTA), ethylenebis (oxyethylenenitrilo) (EGTA), phenylmethylsulfonyl fluoride (PMSF), 4-(2-aminoethyl)-benzenesulfonylfluoride (AEBSF), leupeptin, pepstatin and casein were obtained from Sigma Chemical Company (St. Louis, MO). Tris base, glycine, sodium dodecyl sulfate, N,N,N′,N′-tetramethyletheylenediamine and Commassie blue were obtained from Bio-Rad Laboratories (Hercules, CA, U.S.A.).
Rat model of traumatic brain injury
A controlled cortical impact device was used to induce TBI as previously described (Dixon et al., 1991). Briefly, adult male Sprague Dawley rats (250 to 300 g) were anesthetized with 2% halothane in a 2:1 mixture of N2O/O2. After craniotomy adjacent to the central suture, midway between lambda and bregma, injury was induced by impacting the right cortex (2.5-mm deformation) with a 6-mm diameter tip at a rate of 6 m/sec. The injury produces focal contusion and necrosis in the ipsilateral cortex at the site of impact (Kampfl et al., 1996; Posmantur et al., 1996a,b). Sham-injured animals underwent identical surgery, but did not receive impact injury. Naive rats were not exposed to any surgical procedures.
Assessment of calpain activity
Casein zymogram.
As has been previously described (Raser et al., 1995), casein [0.2%, weight/volume (w/v), sodium salt] was copolymerized in 12% (w/v) acrylamide separating gel (375 mmol/L Tris-HCI, pH 8.8). A 4% (w/v) acrylamide gel (330 mmol/L Tris-HCI, pH 6.8) was used as the stacking gel. The casein gels were pre-run at 150 V for 2 hours at 4°C in a running buffer (25 mmol/L Tris base, 192 mmol/L glycine, 1 mmol/L EGTA, and 1 mmol/L DTT, pH 8.3). One hundred fifty micrograms of each sample was mixed with one fifth volume of sample buffer [150 mmol/L Tris-HCI (pH 6.8), 20% glycerol, 2 mmol/L 2-mercaptoethanol, 0.004% (w/v) bromphenol blue], loaded in each well, and given electrophoresis at 80 V, 4°C for 16 to 18 hours. The gel was rinsed in incubation buffer [20 mmol/L Tris-HCI (pH 7.6 for μ-calpain; pH 7.3 for m-calpain), 10 mmol/L DTT, 3 mmol/L CaCl2] twice, then incubated in the same incubation buffer at 32 ± 2°C for 24 hours. The gel was stained in 0.2% Commassie blue for 2 hours and incubated in a destaining solution (5% methanol, 8% acetic acid in distilled water) overnight. Computer image analysis software vi. 56 was used to analyze the pictures on a Macintosh computer. Results were normalized as the optical density (arbitrary densitometric units) of the lysed region (cleared band) divided by the protein amounts for each sample run. A Polaroid film negative was used for quantification.
Assessment of m-calpain protein levels
Data analysis
Statistical analysis of the data was performed using an analysis of variance with the Waller-Duncan K ratio to determine group differences or Student's t-test. Significance was taken as P ≤ .05. Variance was expressed as SEM.
RESULTS
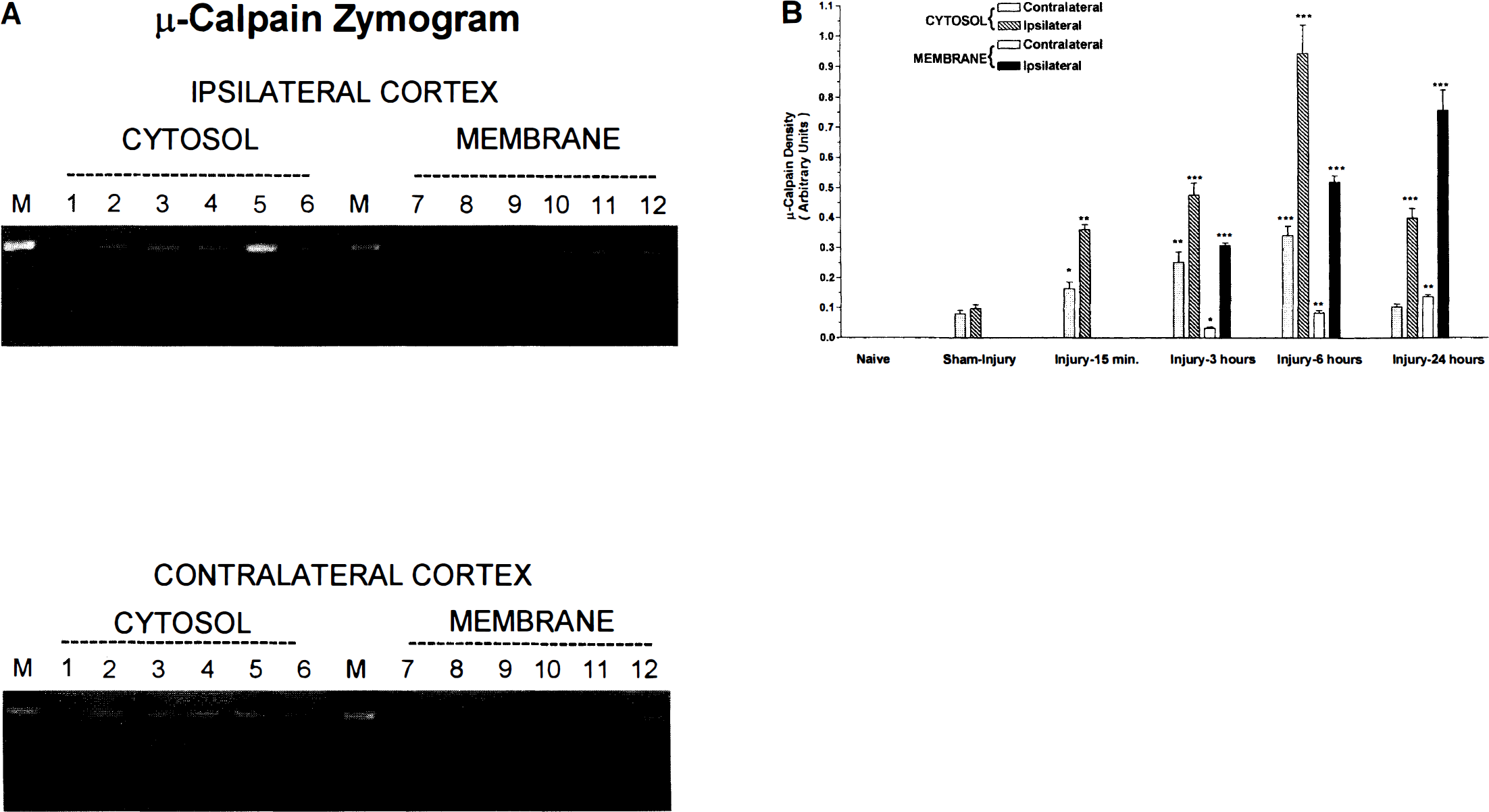
μ-Calpain analysis
Densitometric analyses detected little μ-calpain activity in naive rats and low levels of activity in sham-injured rats (Fig. 1). Cortical impact injury resulted in significant increases in μ-calpain activity in cytosolic fractions from the ipsilateral cortex that occurred as early as 15 minutes after injury, peaked at 6 hours after injury, and remained above sham-injury levels at 24 hours after injury (P ≤ .05, compared to sham injury). Increases in μ-calpain activity in total membrane fractions from the ipsilateral cortex were not detected until 3 hours and were maximal at 24 hours after injury. In the contralateral cortex, μ-calpain activity was significantly elevated in cytosolic fractions at 3 hours after injury, peaked at 6 hours after injury (P ≤ .01 compared to sham injury) and did not differ from sham-injury values by 24 hours after injury (P ≤ .05). Evidence of μ-calpain activity was not detectable in membrane fractions from the contralateral cortex until 3 hours after injury and was maximal at 24 hours after injury. Levels of μ-calpain activity in the contralateral cortex (total membrane and cytosolic fractions) were markedly lower than values observed in the cortex ipsilateral to injury (P ≤ .01).
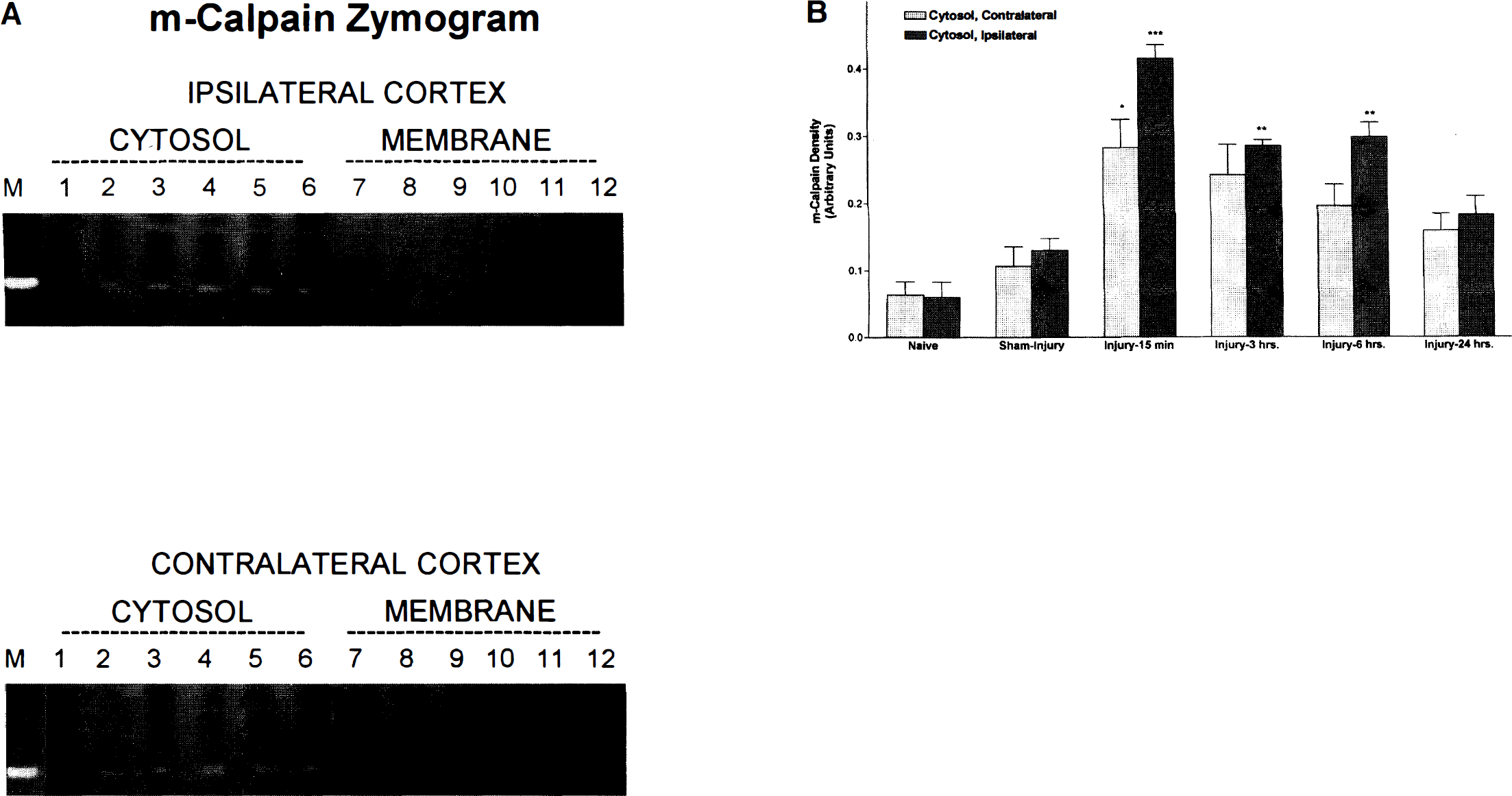
m-Calpain analysis
Densitometric analysis detected no significant differences in m-calpain activity between naive and sham-injured rats (Fig. 2). Cortical impact injury resulted in significant increases in m-calpain activity in cytosolic fractions from 15 minutes to 6 hours after injury in the ipsilateral cortex (P ≤ .01 compared to sham injury). By 24 hours after injury, m-calpain activity in cytosolic fractions from the ipsilateral cortex was not significantly different from sham-injury values. In the contralateral cortex, m-calpain activity was significantly increased in cytosolic fractions only at 15 minutes after injury (P ≤ .05 compared to sham injury). There was no detectable m-calpain activity in total membrane fractions. Levels of m-calpain activity in cytosolic fractions from the contralateral cortex were not significantly different from values observed in the cortex ipsilateral to injury.
Western blotting analysis detected prominent 80-kd immunoreactive bands in the absence of lower molecular weight bands, suggesting no autolysis or further processing of the parent protein (Fig. 3). There was no apparent change in m-calpain immunoreactivity in cytosolic or total membrane fractions from samples taken at various times after TBI. Immunoreactivity to m-calpain was more apparent in cytosolic than in total membrane fractions and did not differ in samples taken from cortices ipsilateral and contralateral to the site of injury.
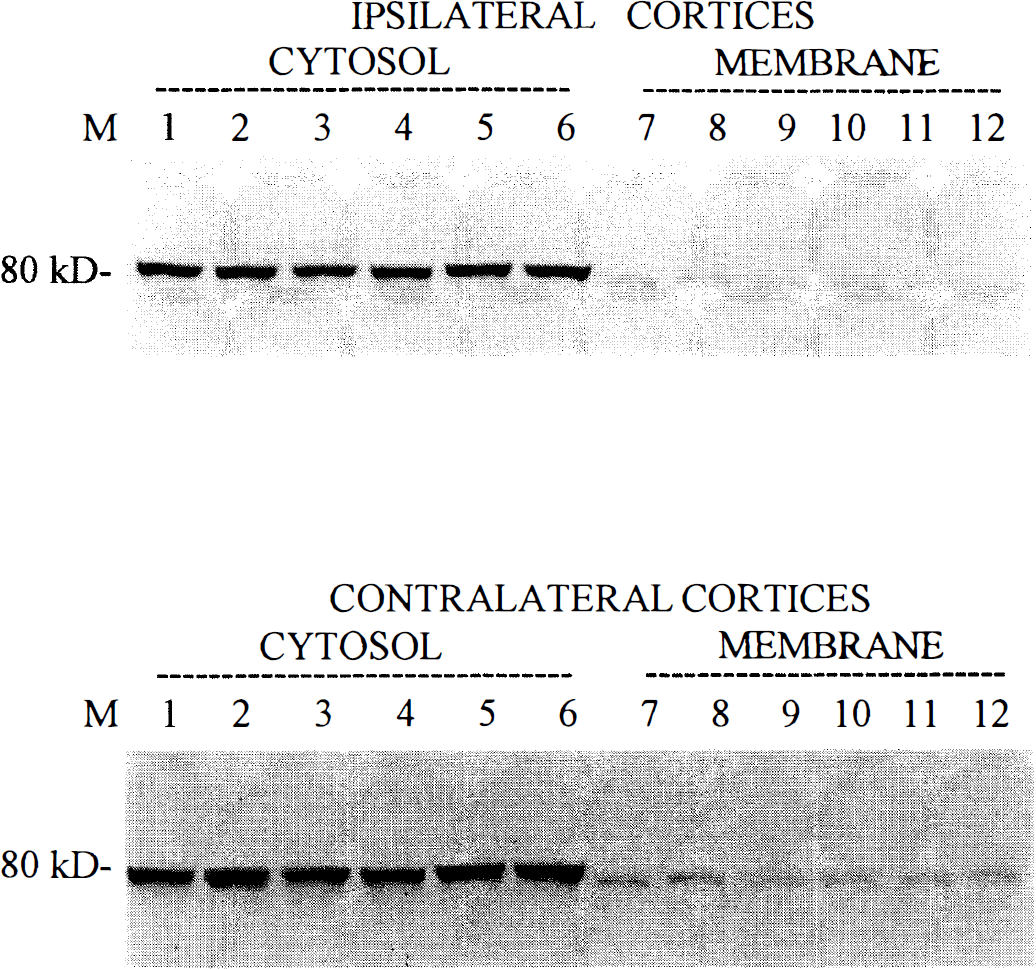
Western blots of m-calpain protein in cytosolic (supernatant) and total membrane (pellet) fractions in ipsilateral and contralateral cortices after experimental traumatic brain injury. Analyses detected prominent 80-kd immunoreactive bands to m-calpain that were more evident in cytosolic than total membrane fractions. There was no apparent change in m-calpain immunoreactivity from naive (lanes 1, 7), sham injured (lanes 2, 8), and injured cortices at 15 minutes (lanes 3, 9), 3 hours (lanes 4, 10), 6 hours (lanes 5, 11), and 24 hours (lanes 6, 12) after TBI.
Comparison of μ-calpain m-calpain activity
Because incubation buffers for calpain isoforms require different pH values for optimal detection of calpain activity (pH 7.5 for μ-calpain; pH 7.3 for m-calpain), experiments routinely used separate gels to assess m-calpain and μ-calpain activity. The use of separate gels prevents direct comparisons of activity of μ-calpain and m-calpain. However, Table 1 summarizes the ratios of differences in optical densities for cytosolic fractions of samples assaying μ-calpain and m-calpain activity after TBI. These ratios represent an indirect assessment of the relative magnitude of m-calpain versus μ-calpain activity in cytosolic fractions at different time points after injury.
DISCUSSION
The present study represents the first application of casein zymography to study calpain activity after experimental brain injury in vivo. These observations also provide the first systematic comparisons of changes in the two major calpain isoforms after injury to the central nervous system. We observed significant increases in cortical μ-calpain activity as early as 15 minutes after cortical impact injury that persisted for up to 24 hours. Separate examinations of cytosolic and total membrane fractions provided evidence for translocation of calpain activity from the cytosol to membrane by 3 hours after injury. In contrast, TBI produced relatively less activation of m-calpain detectable only in cytosolic fractions and which persisted no longer than 6 hours.
Our data on the temporal profile of μ-calpain activation using casein zymography show both similarities and differences to previously published observations on μ-calpain activation after TBI. Kampfl et al. (1996), using concurrent assessments of μ-calpain autolysis and accumulation of calpain-specific breakdown products to α-spectrin, concluded that μ-calpain activation after cortical impact injury occurred within 15 minutes, but returned to controlled levels within 24 to 48 hours after injury. The present data are in general agreement with an early onset of μ-calpain activation at the site of injury. Although Saatman et al. (1996), did not detect calpain-specific breakdown products to α-spectrin at 30 minutes after injury, those studies used a different model (lateral fluid percussion) of TBI. The data presented here also indicate that μ-calpain activation may persist beyond the 24-hour duration suggested by studies of μ-calpain autolysis (Kampfl et al., 19%). This difference may be attributable to differences in mechanical injury magnitude in different experiments and/or differences in the sensitivities of casein zymography and μ-calpain autolysis to detect changes in calpain activity. Some reports have suggested that μ-calpain does not require autolysis for activation (Cong et al., 1989; Edmonds et al., 1991; Zhang et al., 1996; Guttmann et al., 1997).
This study provides the first evidence of m-calpain activation after TBI. As observed for μ-calpain, the data suggest rapid activation of m-calpain. Moreover, m-calpain activation persisted for 6 hours after injury, a substantially briefer period than μ-calpain activity which persisted for 24 hours. Although μ-calpain has micromolar sensitivity to calcium actuation while m-calpain has a millimolar sensitivity to calpain activation (Homokubo et al., 1986), the relative differences in m-calpain activity and μ-calpain activity are not an artifact because the buffer used in these studies had sufficient Ca++ (3.0 mmol/L) to fully activate both m-calpain and μ-calpain. The preferential activation of μ-calpain over m-calpain is also surprising in view of reports that the total amount of m-calpain exceeds by one to two orders of magnitude the amount of μ-calpain (Kawashima et al., 1988), and similar profiles have been published for the content of the two isoforms in rat brain membranes (Siman et al., 1983).
The present study provides evidence that calpain activation was substantially higher at the site of cortical impact injury, a region associated with focal contusion and necrosis. The rapid onset of both μ-calpain and m-calpain activation, especially at the site of impact-induced focal contusion and necrosis, suggests that calpain activation may play an important role in cortical neuronal degeneration and even precede the full expression of evolutionary histopathologic changes characteristic of cortical impact injury (see Kampfl et al., 1996). Our study also provides evidence of μ-calpain and m-calpain activation in the contralateral cortex, a region not associated with focal contusions in this model system. Calpain-specific breakdown products to α-spectrin have also been detected in the contralateral cortex after cortical impact injury (Posmantur et al., 1997), and recent work in our laboratory has shown evidence of diffuse histopathology and cell death in the contralateral cortex after injury (Newcomb et ·al., 1997), However, Kampfl et al. did not detect evidence of μ-calpain autolysis or accumulation of calpain-specific breakdown products to α-spectrin in the contralateral cortex after cortical impact injury, an observation possibly attributable to use of a lower injury level than used in the present study. In any case, our data suggest the possibility that calpain activation can extend into brain regions not associated with focal contusions. This interpretation is consistent with reports of degradation of cytoskeletal proteins in regions distal from the site of cortical impact or lateral fluid percussion injury (Posmantur et al., 1996a, 1997; Saatman et al., 1996; Newcomb et al., 1997).
Ratios of m-calpain versus μ-calpain activation after injury
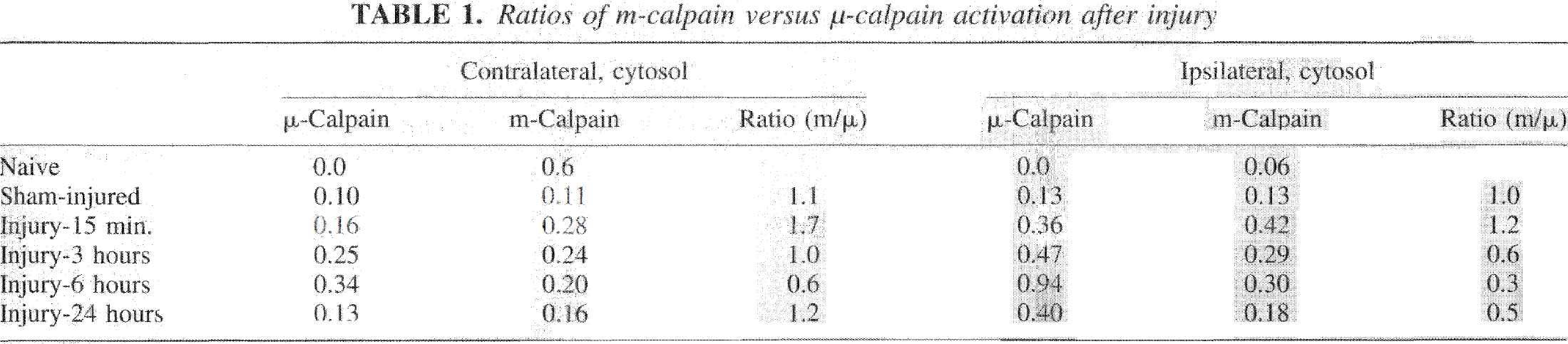
Casein zymography allows concurrent analyses of both cytosolic and total membrane fractions. μ-Calpain activation in the cytosol preceded later activation at the cell membrane. Saido et al., have argued that the pre-autolysis form of μ-calpain migrates from the cytosol to membranes where it becomes activated by autolysis (e.g., Saido et al., 1993). Kampfl et al. (1996) reported that autolysis of μ-calpain after cortical impact injury was most readily detectable in pellet fractions. Although our data suggest that substantial activation of μ-calpain can occur in the cytosolic fraction, there was evidence of later translocation to the membrane associated with more prolonged calpain activation. The detection of m-calpain activity in cytosolic rather than total membrane fractions is consistent with previous reports of cytosolic localization of m-calpain, although m-calpain has also been detected in purified myelin (Chakrabarti et al. 1996). While Western blots in this study detected m-calpain in total membrane fractions, m-calpain remained preferentially in cytosolic fractions after TBI.
Our studies did not directly examine mechanisms mediating calpain activation after TBI. However, it is unlikely that observed increases in μ-calpain activity are due to increased amounts of μ-calpain in the protein samples because previous studies by Kampfl et al. (1996) did not detect increased levels of μ-calpain at similar time points after cortical impact injury. Similarly, increases in m-calpain activity are not related to an increased amount of protein because Western blotting did not detect any changes in m-calpain immunoreactivity. It is also unlikely that changes in calpain-calpastatin interactions influenced detection of calpain in the casein zymogram because the method eliminates interactions between calpain and its endogenous inhibitor, calpastatin (Raser et al., 1995). Moreover, increases in calpain activity measured by casein zymography in this current study are not likely due to calpain autolysis because unautolyzed calpain would also be activated in the development procedure. Although a clear autolyzed μ-calpain form in casein zymography has been detected only from cultured cells under maximal pathological conditions (Raser et al., 1995), past and current in vivo studies in our laboratory for unknown reasons have yet to detect the shift in electrophoretic mobility associated with the autolyzed μ-calpain form (unpublished observations, 1995). Our in vivo data sampled calpain activity up to 24 hours after injury, More prolonged activation of calpain in vivo could ultimately reduce activity of the enzyme. For example, in vitro studies have suggested that lysates containing calpain pools which have undergone sustained activation would be predicted to contain less casein hydrolysis because the autolyzed form would be unstable and undergo further degradation (Raser et al., 1995). A probable cause of the increased casein hydrolysis in samples obtained after TBI may be due to post-translational changes in calpain including interaction with phospholipids or activator proteins that could enhance calpain activity (Saido et al., 1992: Chakrabarti et al., 1996). Alternatively, nonactivated calpains (μ- and m-) may be compartmentalized in resting neurons and become accessible to calcium-dependent autolytic activation only in injured neurons or protein-separating techniques. Our Western blotting also did not detect autolytic breakdown products to m-calpain (Fig. 3). Although autolysis of m-calpain occurs both in in vitro preparation and in cultured cells when m-calpain is activated, little is known about m-calpain autolysis in vivo, and no studies have addressed m-calpain autolysis after neuronal injury (Nishimura and Goll, 1991; Wang et al., 1996; Nath et al., 1996).
In conclusion, the present study has provided strong evidence that cortical impact injury results in rapid and sustained increases in calpain activity. TBI also produces relatively greater and more sustained increases in μ-calpain activity than in m-calpain activity. Future studies need to examine mechanisms mediating calpain activity after TBI. Because calpain activation has been implicated in other brain regions such as the hippocampus and thalamus (Posmantur et al., 1996; Saatman et al., 1996), additional brain regions should be examined.