
Abstract
Glial-neuronal interchange of amino acids was studied by 13C nuclear magnetic resonance spectroscopy of brain extracts from fluoroacetate-treated mice that received [1,2-13C]acetate and [1-13C]glucose simultaneously. [13C]Acetate was found to be a specific marker for glial metabolism even with the large doses necessary for nuclear magnetic resonance spectroscopy. Fluoroacetate, 100 mg/kg, blocked the glial, but not the neuronal tricarboxylic acid cycles as seen from the 13C labeling of glutamine, glutamate, and γ-aminobutyric acid. Glutamine, but not citrate, was the only glial metabolite that could account for the transfer of 13C from glia to neurons. Massive glial uptake of transmitter glutamate was indicated by the labeling of glutamine from [1-13C]glucose in fluoroacetate-treated mice. The C-3/C-4 enrichment ratio, which indicates the degree of cycling of label, was higher in glutamine than in glutamate in the presence of fluoroacetate, suggesting that transmitter glutamate (which was converted to glutamine after release) is associated with a tricarboxylic acid cycle that turns more rapidly than the overall cerebral tricarboxylic acid cycle.
According to the commonly accepted idea of the glutamate-glutamine cycle, glutamine is formed in glia and transferred to neurons as a precursor for transmitter glutamate and γ-aminobutyric acid (GABA). After release during neurotransmission, glutamate and GABA are taken up by glial cells and converted to glutamine (Balázs et al., 1970; Van den Berg, 1973). Much of the evidence in favor of such a cycle has been obtained from studies in vitro. Evidence for a glutamine cycle in the brain includes the localization of glutamine synthetase in glial cells only (Martinez-Hernandez et al., 1977) and its presence in glial processes in the vicinity of glutamatergic synapses (Derouiche and Frotcher, 1991). The extracellular level of glutamine is high in brain, ∼0.25 mmol/L (Hagberg et al., 1985), and during inhibition of the glial tricarboxylic acid (TCA) cycle the level decreases (Paulsen and Fonnum, 1989) in agreement with extensive glial export of glutamine under physiologic conditions.
The role of glutamine as a precursor for transmitter amino acids has been more difficult to ascertain in vivo, e.g., with radiolabeled glutamine which only labeled GABA after prolonged intracerebral infusion (Thanki et al., 1983). Instead, several authors have used labeled acetate, which to a large extent is rapidly converted to glutamine by the intact brain (O'Neal and Koeppe, 1966; Van den Berg et al., 1969). The labeling of GABA from acetate has been interpreted to reflect transfer of labeled glutamine from glia to neurons (Van den Berg, 1973; Cerdan et al., 1990; Hassel et al., 1995). There remains a need, however, to determine in vivo whether acetate is a substrate for glia only, or whether it may also be metabolized by neurons. If acetate is a substrate for neurons, labeling of GABA from [14C]acetate or [13C]acetate cannot serve as an indicator of glial–neuronal transfer of glutamine.
Uptake of glutamate and GABA by glia in vivo is suggested by the presence of transport proteins for GABA and glutamate in the glial plasma membrane (Radian et al., 1990; Pines et al., 1992; Storck et al., 1992; Rothstein et al., 1994), by the elevated extracellular level of glutamate in brains of rats lacking glial glutamate transporters (Rothstein et al., 1996), and by the rapid labeling of glutamine when radiolabeled glutamate and GABA are injected into the brain (Berl et al., 1961; Berl and Purpura, 1966; Hassel et al., 1992). It should be noted, however, that injection of radiolabeled glutamate and GABA does not reproduce normal neurotransmission, because to a large extent the injected amino acids may reach nonsynaptic brain tissue. As neuronal glutamate and GABA are rapidly labeled from glucose (Cremer, 1964; O'Neal and Koeppe, 1966; Van den Berg et al., 1969; Fitzpatrick et al., 1990), labeling of glutamine from glucose could indicate transfer of glutamate and GABA from neurons to glia. However, labeling of glutamine from [13C]glucose or [14C]glucose may also reflect glial metabolism of glucose. To distinguish between the two routes for labeling of glutamine from glucose, the use of fluoroacetate, an inhibitor of aconitase of the glial TCA cycle (Clarke et al., 1970; Cheng et al., 1972; Muir et al., 1986), seems attractive, as in the presence of this toxin, the labeling of glutamine from glucose should mainly reflect transfer of glutamate from neurons to glia.
The purpose of the present study was to determine to what degree glial uptake of amino acid transmitters takes place in vivo, and to what extent glutamine is a likely glial precursor of neuronal glutamate and GABA, using 13C-labeled glucose and acetate with 13C nuclear magnetic resonance (NMR) spectroscopy. Therefore, it was also necessary to determine whether [13C]acetate may be used as a specific marker for glial metabolism even in the large doses needed for 13C NMR spectroscopy. These are important issues considering the possible use of relabeled substrates in conjunction with 13C NMR spectroscopy in the study of human cerebral metabolism (Gruetter et al., 1994; Mason et al. 1995).
In the current study [1,2-13C]acetate was administered intravenously together with [1-13C]glucose to awake mice, and brain extracts were analyzed by 13C NMR spectroscopy and by gas chromatography/mass spectrometry (GC/MS). Coadministration of [1,2-13C]acetate and [1-13C]glucose has been used successfully in vitro to assess glial–neuronal interactions (Taylor et al., 1996). Incorporation of the coupled 13C atoms of [1,2-13C]acetate into amino acids gives rise to isotopomer patterns in the NMR spectra that are different from those obtained by incorporation of the single 13C atom of [1-13C]glucose (Taylor et al., 1996; Badar-Goffer and Bachelard, 1991). The contribution of [1,2-13C]acetate to the labeling of metabolites may therefore be distinguished from the labeling obtained from [1-13C]glucose. If acetate is metabolized exclusively by glia, this allows determination of the glial versus the neuronal contribution to the labeling of amino acids. Fluoroacetate was used to selectively inhibit glial TCA cycle activity to allow further differentiation between the glial and neuronal formation of amino acids.
MATERIALS AND METHODS
Materials
Female NMRI mice (Bomholt, Ry, Denmark), 30 g body weight, were caged in groups of 10 with free access to food and tap water, with a 12-hour light/dark cycle, a relative humidity of 50%, and a room temperature of 25°C. Before experiments the animals were fasted for 24 hours with free access to tap water. Sodium [1,2-13C]acetate and [1-13C]glucose (both 99% 13C enrichment) were from Sigma (St. Louis, MO, U.S.A.). A solution containing either sodium [1,2-13C]acetate + [1-13C]glucose, both 0.5 mol/L (pH 7.4) or sodium [1,2-13C]acetate + unlabeled glucose was prepared immediately before the experiments. Sodium fluoroacetate (Sigma) was dissolved in 30 mmol/L NaCl to a concentration of 12 mg/mL (120 mmol/L), and pH was adjusted to 7.4.
Experimental protocols
Four experimental groups of mice were used. The animals were unanesthetized during the study. One group of mice (n = 8) was dosed with 0.3 mL of the solution containing the 13C-labeled substrates. The injection was done in a tail vein and was completed in 5 seconds. Another group of animals (n = 7) received 0.25 mL of the fluoroacetate solution, corresponding to 100 mg fluoroacetate/kg body weight, intraperitoneally. This dose has previously been shown not to interfere with cerebral levels of ATP (Goldberg et al., 1966). At 15 minutes these animals received 0.3 mL of the solution containing the 13C-labeled substrates intravenously. To examine whether fluoroacetate blocks the uptake of [1,2-13C]acetate into glial cells, the 13C labeling of citrate from [1,2-13C]acetate was determined in two groups of mice (both n = 5) that received 0.3 mL sodium [1,2-13C]acetate and unlabeled glucose, both 0.5 mol/L, intravenously. One group was pretreated with fluoroacetate 100 mg/kg body weight intraperitoneally 15 minutes before the [1,2-13C]acetate and unlabeled glucose. If citrate labeling did not occur in the presence of fluoroacetate, it would indicate inhibition of the uptake of [1,2-13C]acetate rather than inhibition of aconitase (Peters, 1957; Brand et al., 1973). Because the enrichment of citrate could not be reliably quantified by NMR spectroscopy, these brain extracts were analyzed by GC/MS gas chromatography/mass spectrometry.
All animals were killed by cervical dislocation and decapitation 15 minutes after injection of the 13C-labeled substrates. The heads were immediately immersed in liquid N2, and blood (∼500 μL) was collected from the severed vessels. The brains were removed in the frozen state, and were homogenized in 2 mL ice-cold perchloric acid, 7% (vol/vol). The homogenizer was rinsed with 1 mL double-distilled water that was added to the homogenate. Protein was removed by centrifugation (20,000g for 20 minutes), and the supernatant was neutralized with 9 mol/L KOH. The precipitate, KClO4, was removed by centrifugation (20,000g for 20 minutes), and 300 μL (∼10%) of the supernatant was used for determination of total amounts of amino acids, lactate, and glucose; the rest was lyophilized to dryness. Serum was obtained by centrifugation of blood samples at 5,000g for 5 minutes, and 10 μL serum was used to determine serum glucose. The rest was mixed with 1 mL 3.5% perchloric acid and frozen at −70°C. After centrifugation, neutralization with KOH, and another centrifugation to remove KClO4, 1.2 mL of the supernatant was lyophilized to dryness. The animals were handled in strict accordance with the institutional and national guidelines for animal research.
Amino acids were measured by high-pressure liquid chromatography (Varian Model 5000) and fluorescence detection (CMA/280, CMA Microdialysis, Sweden) after derivatization with o-phthaldialdehyde (Sigma), using an LC-DB 18 column, 25 cm length, 4 mm inner diameter, 5 μm pore size (Pharmacia Biotech, Uppsala, Sweden). The mobile phase was 75% 50 mmol/L phosphate buffer, pH 5.25, and 25% methanol, changing gradually to 25% phosphate buffer and 75% methanol during 20 minutes. α-Amino adipic acid (0.5 mmol/L) was added 1:1 to the samples as internal standard. A mixture of the amino acids of interest in equimolar concentration was run as an external standard. Citrate was measured spectrophotometrically (UV 1201, Shimadzu, Kyoto, Japan) using an enzymatic kit from Boehringer (Mannheim, Germany) (Cat. No. 139 076). The content of glucose and lactate in extracts of brain and serum as well as the glucose concentration in serum was measured by reflectance spectrophotometry using an Ektachem DT60 (Kodak, Rochester, NY, U.S.A.).
NMR spectroscopy
Proton-decoupled 125.7 MHz 13C NMR spectra were obtained on a Bruker Avance DRX-500 spectrometer (Bruker, Zurich, Switzerland). Lyophilized samples were redissolved in D2O containing 0.1% (vol/vol) dioxane as an internal standard. Spectra were accumulated using a 35° pulse angle and 31-KHz spectral width. The acquisition time was 1.049 seconds and an additional relaxation delay of 5 seconds was used. The number of scans was typically 4,000. Some spectra were also broadband decoupled only during acquisition to avoid nuclear Overhauser effects. From the two sets of spectra, factors for the nuclear Overhauser effects of different carbon atoms were obtained and applied to all spectra. Inversion recovery experiments were performed to obtain relaxation (T1) values. The chemical shift of dioxane was set to 67.4 ppm. The total amount of 13C in the resonance of a particular metabolite was calculated from peak integrals using dioxane as an internal standard. The percent enrichment was calculated after subtraction of naturally abundant 13C (Badar-Goffer et al., 1990). The naturally abundant 13C (1.1% of total carbon) was calculated from the total amounts of amino acids, lactate, and glucose as determined biochemically. Analysis of isotopomers with two or more consecutive 13C atoms does not require correction for naturally abundant 13C, because the resulting multiplets generally do not overlap with the singlets. Incorporation of 13C-13C from [1,2-13C]acetate into amino acids and lactate was calculated by dividing the total amount of 13C as determined from the doublets representing [4,5-13C]glutamate, [4,5-13C] glutamine, [1,2-13C]GABA, [1,2-13C]aspartate, [3,4-13C] aspartate, and [1,2-13C]lactate by the total amount of the metabolite. [1-13C]Glucose could also yield [1,2-13C]aspartate and [3,4-13C]aspartate by cycling of the label in the TCA cycle, but this was not considered to be quantitatively important in the current study (see Discussion). The 13C enrichment of the C-3 positions of glutamate, glutamine, and GABA was calculated in fluoroacetate-treated animals only. In controls the labeling of these positions from [1,2-13C]acetate and [1-13C]glucose could not be distinguished.
Gas chromatography/mass spectrometry (GC/MS)
Samples of neutralized perchloric acid extracts or plasma were adjusted to pH ∼2.5, dried under N2, and derivatized with N-methyl-N-t-butyldimethylsilyl trifluoroacetamide (Sigma) at 70°C (Mawhinney et al., 1986) for all metabolites except glucose. We found that this compound was unsuitable for glucose, but achieved success with TriSil reagent (Pierce & Warriner [UK] Ltd., Chester, U.K.) at 80°C. This product contains hexamethyldisilazane as reagent with trimethylchlorosilane as catalyst, and yields only one fragment containing the C-1 of glucose (mass 191, TMSO-1CH-OTMS), where TMS is trimethylsilyl, —S(CH3)3, molecular weight 73 (see Kamerling and Vliegenhart, 1989). Each derivatized sample (1 to 2 μL) was analyzed in a Hewlett Packard 5890 Series II gas chromatograph linked to a Hewlett Packard 5970 mass spectrometer (Hewlett-Packard Ltd., Wokingham, Berkshire, U.K.). 13C Atom percent excess of the various metabolites was determined after allowing for naturally abundant 13C and silicon (from silyl groups) as described by Biemann (1962). This yielded the percent singly labeled (M + 1) and doubly labeled (M + 2) fragments of the intermediates.
Statistics
Values are given as means ± SD. Differences between groups were analyzed by the two-tailed Student's t-test. Differences in percent enrichments between carbon positions within a molecule or between molecules within the same animal were analyzed by the two-tailed paired Student's t-test.
RESULTS
Behavioral changes
Intravenous injection of acetate and glucose into control mice did not cause behavioral changes. Five minutes after injection of fluoroacetate the animals showed intermittent “freezing” in a posture for 5 to 20 second, and they tended to lie down more than their untreated litter-mates. At 10 minutes the animals would lie down, but could readily be awakened to walk about the cage for short distances. At 15 minutes, when the animals received the intravenous injection, they appeared very somnolent and were not readily aroused. They would, however, resist being rolled over on the side. At this stage two of the seven animals had general convulsions that lasted for ∼5 s. The somnolent state, which commenced before the intravenous injection, lasted for approximately 5 minutes. Thereafter the animals became more alert, with short episodes of spontaneous walking. Respiration appeared unaffected, with a respiratory frequency of 140 to 200 breaths/min. Throughout the time of exposure to the 13C-labeled substrates, the animals were clearly affected by the fluoroacetate.
Effects of fluoroacetate on total levels of brain metabolites and on serum metabolites
Flouroacetate caused a 200% increase in the cerebral level of citrate and a 78% increase in the level of glucose (Table 1). The level of aspartate decreased by 32% and that of alanine increased by 120%, indicating shifts in the equilibria of aspartate and alanine aminotransferases. The other measured metabolites remained constant.
Total amounts of cerebral amino acids, lactate and glucose (nmol/mg protein) in controls and mice treated with fluoroacetate
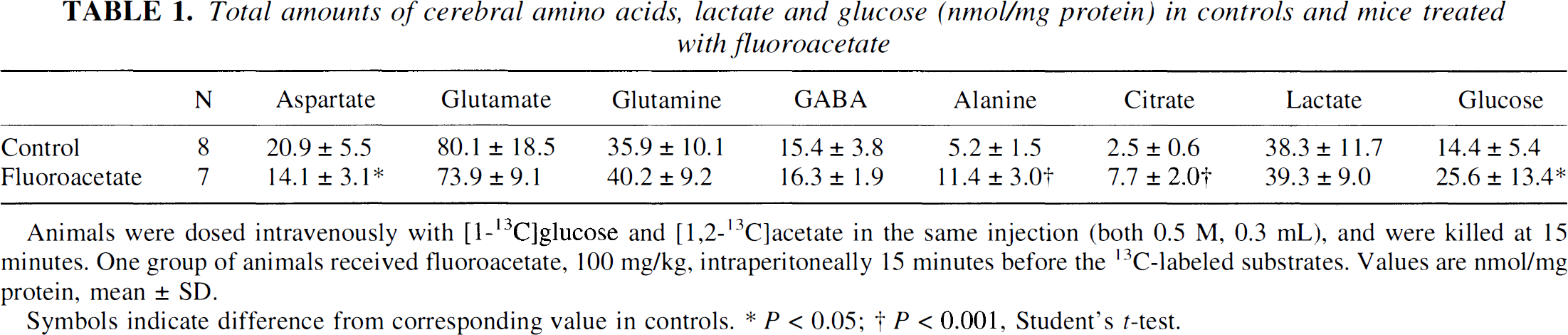
Animals were dosed intravenously with [1-13C]glucose and [1,2-13C]acetate in the same injection (both 0.5 M, 0.3 mL), and were killed at 15 minutes. One group of animals received fluoroacetate, 100 mg/kg, intraperitoneally 15 minutes before the 13C-labeled substrates. Values are nmol/mg protein, mean ± SD.
Symbols indicate difference from corresponding value in controls.
P < 0.05
P < 0.001, Student's t-test.
Fifteen minutes after injection of the 13C-labeled substrates, serum glucose was 13.1 ± 2.9 mmol/L in the control group and 14.7 ± 3.3 mmol/L in the fluoroacetate group (means ± SD, n = 8 and 7, respectively). The percent enrichment of glucose C-1 was 40.9% ± 3.3% in controls and 38.9% ± 6.3% in fluoroacetate-treated animals as detected by GC/MS. 13C Nuclear magnetic resonance spectra of serum extracts showed single peaks of glucose C-1 and lactate C-2 and C-3. No peaks of other isotopomers of [13C]glucose were seen, which shows that hepatic gluconeogenesis was negligible; nor were there any detectable peaks of citrate (C-2 + C-4) in the sera of fluoroacetate-treated animals or in controls. [1,2-13C]Acetate was not seen in the sera of any animals.
Labeling of cerebral metabolites from [1-13C]glucose and [1,2-13C]acetate
[1-13C]Glucose gave a higher 13C enrichment in glutamate C-4 and GABA C-2 than in glutamine C-4, ∼12% in glutamate and GABA, and ∼6% in glutamine(Table 2). [1,2-13C]Acetate, on the other hand, gave a higher 13C enrichment in glutamine C-4 (∼15%) than in glutamate C-4 and GABA C-2 (4% to 5%) (Table 3). The glutamine C-4/glutamate C-4 enrichment ratio obtained with [1-13C]glucose was thus 0.54 ± 0.04. The ratio obtained with [1,2-13C]acetate was 3.4 ± 1.6 (n = 8), confirming predominantly neuronal metabolism of [1-13C]glucose and glial metabolism of [1,2-13C]acetate (see Discussion).
Percent enrichment of cerebral amino acids labeled from [1-13C]glucose in fluoroacetate-treated mice and controls
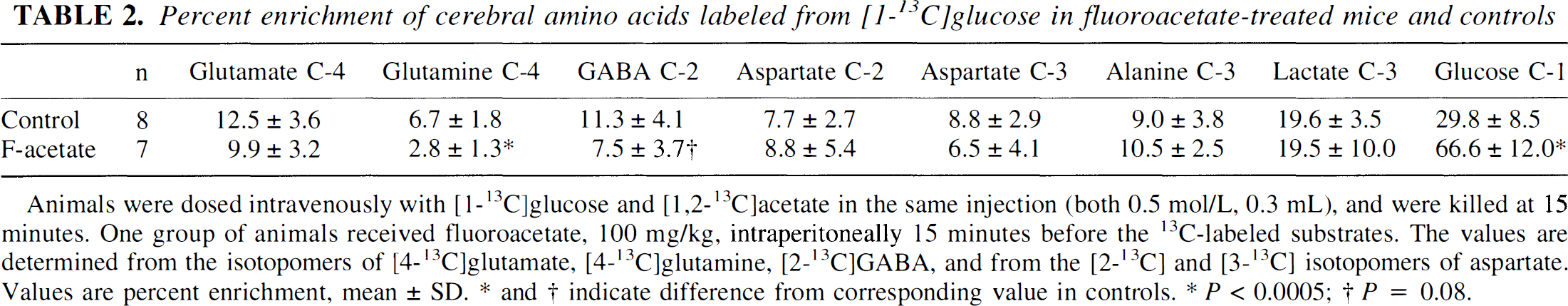
Animals were dosed intravenously with [1-13C]glucose and [1,2-13C]acetate in the same injection (both 0.5 mol/L, 0.3 mL), and were killed at 15 minutes. One group of animals received fluoroacetate, 100 mg/kg, intraperitoneally 15 minutes before the 13C-labeled substrates. The values are determined from the isotopomers of [4-13C]glutamate, [4-13C]glutamine, [2-13C]GABA, and from the [2-13C] and [3-13C] isotopomers of aspartate. Values are percent enrichment, mean ± SD. * and † indicate difference from corresponding value in controls.
P < 0.0005
P = 0.08.
Percent enrichment of cerebral amino acids from [1,2-13C]acetate in fluoroacetate-treated mice and controls

Animals were dosed intravenously with [1-13C]glucose and [1,2-13C]acetate in the same injection (both 0.5 mol/L, 0.3 mL), and were killed at 15 minutes. One group of animals received fluoroacetate, 100 mg/kg, intraperitoneally 15 minutes before the 13C-labeled substrates. The values are determined from [4,5-13C] glutamate, [4,5-13C]glutamine, [1,2-13C]GABA, and from the [1,2-13C] and [3,4-13C] isotopomers of aspartate. Values are percent enrichment, mean ± SD. ND, not detectable; NS, not significantly different from zero
Difference from control, P < 0.05.
[1-13C]Glucose gave an enrichment of ∼8% in aspartate C-2 and C-3 (Table 2), whereas [1,2-13C]acetate gave an enrichment of ∼2% in both carbon positions (Table 3). The percent enrichment of aspartate C-2 was not significantly different from the C-3 position with either of the two 13C-labeled precursors.
Glucose C-1, which had a 13C enrichment of approximately 30% (Table 2), was the only carbon position of glucose that was visible in the spectra. [1,2-13C]Acetate was not present in the spectra.
Effects of fluoroacetate on the labeling of cerebral metabolites
In fluoroacetate-treated animals labeling of glutamine from [1,2-13C]acetate was hardly detectable by NMR, as seen by the reduction of peaks representing [4,5-13C]glutamine (Fig. 1; Table 3). Likewise, [1,2-13C]GABA, [1,2-13C]aspartate, and [3,4-13C]aspartate were not detected in the brain extracts of fluoroacetate-treated animals, whereas [4,5-13C]glutamate was reduced by 68% (Fig. 1; Table 3), indicating preserved labeling of glial glutamate (Hassel et al., 1992).
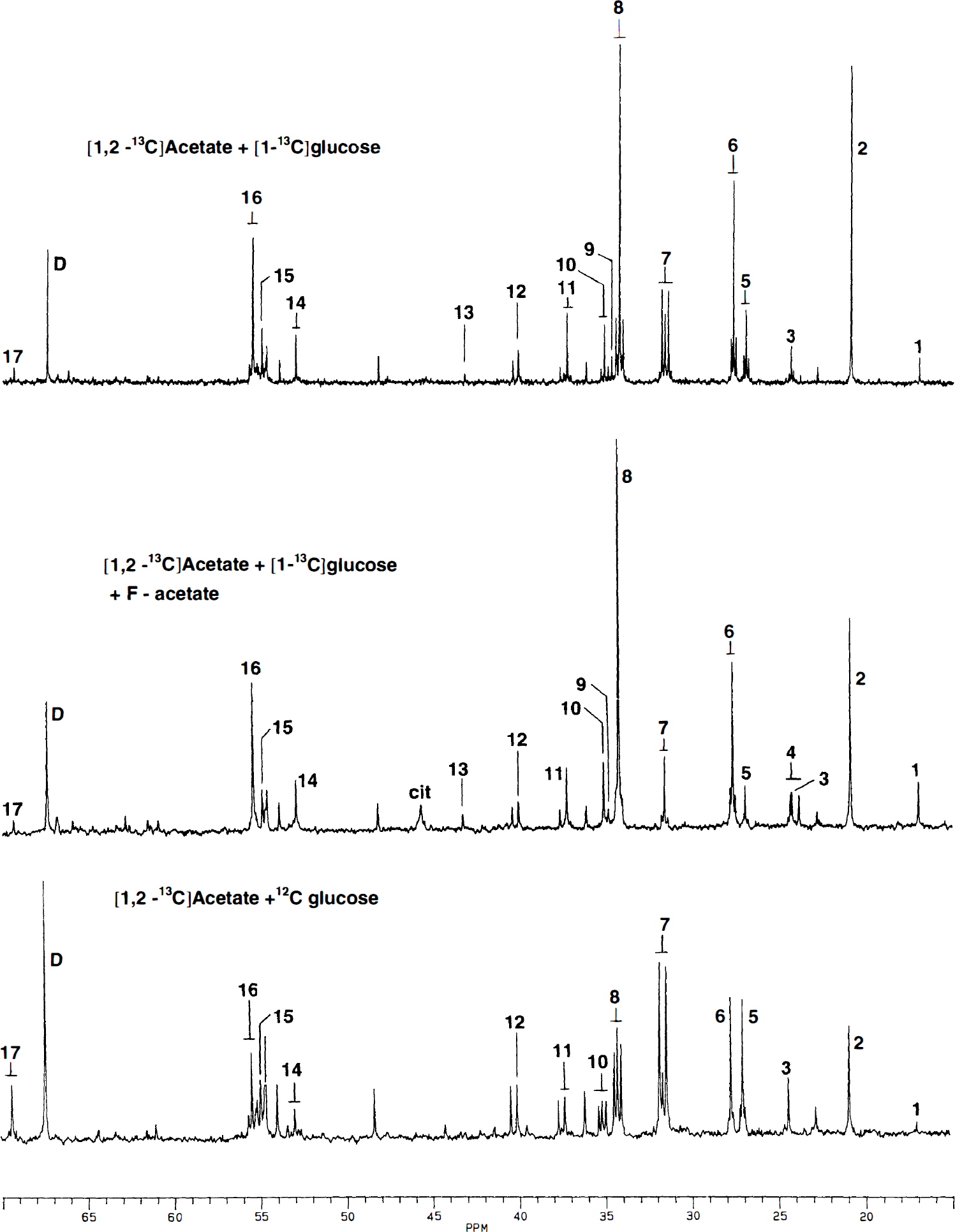
Typical spectra of mouse brain extracts. Upper panel: [1,2-13C]Acetate + [1-13C]glucose were given in the same intravenous bolus. Middle panel: Effect of pretreatment with fluoroacetate (100 mg/kg, intraperitoneally) 15 minutes before [1,2-13C]acetate + [1-13C]glucose. Lower panel: [1,2-13C]Acetate + [12C]glucose were given intravenously. In all experimiments the intravenous bolus was 0.3 mL and both substrates were 0.5 mol/L. Survival time after injection of 13C-labeled substrates was 15 minutes. Peaks identified by numbers: 1, alanine C-3; 2, lactate C-3; 3, GABA C-3; 4, acetate C-2; 5, glutamine C-3; 6, glutamate C-3; 7, glutamine C-4; 8, glutamate C-4; 9, succinate C-2 + C-3; 10, GABA C-2; 11, aspartate C-3; 12, GABA C-4; 13, malate C-3; 14, aspartate C-2; 15, glutamine C-2; 16, glutamate C-2; and 17, lactate C-2. Cit, citrate C-2 + C-4; D, dioxan (internal standard).
In contrast to the lack of labeling of glutamine C-4 from [1,2-13C]acetate, the labeling of glutamine from [1-13C]glucose remained at 42% of control (Table 2). Fluoroacetate did not, however, cause a significant reduction in the labeling of glutamate C-4 from [1-13C]glucose, but it reduced the glutamine C-4/glutamate C-4 enrichment ratio obtained with [1-13C]glucose from 0.54 ± 0.04 to 0.29 ± 0.12 (P < 0.001). The enrichment of GABA C-2 from [1-13C]glucose was reduced by 33% with marginal significance (P = 0.08) in the presence of fluoroacetate. The labeling of aspartate C-2 and C-3 from [1-13C]glucose was not significantly reduced by fluoroacetate (Tables 2 through 4). However, in fluoroacetate-treated animals, aspartate C-2 was more highly enriched than the C-3 (P = 0.01, paired Student's t-test).
The C-3/C-4 enrichment ratios in glutamate and glutamine are indices of the number of times that the 13C has passed through a TCA cycle (Hassel et al., 1995; Hassel and Sonnewald 1995a). During fluoroacetate treatment the C-3/C-4 enrichment ratios in glutamate and glutamine were 0.27 ± 0.05 and 0.55 ± 0.15, respectively. These values were significantly different (P = 0.008, paired Student's t-test), indicating that the 13C in glutamine had cycled more in a TCA cycle than the 13C in glutamate. The C-3/C-2 enrichment ratio in GABA (0.51 ± 0.33) was not significantly different from the corresponding C-3/C-4 enrichment ratios in either glutamine or glutamate.
The percent enrichment of glucose C-1 increased 123%, from approximately 30% to approximately 67% (Table 2), in the presence of fluoroacetate, which, in view of the observation that the enrichment of serum glucose was the same in the two groups of animals, indicates cerebral accumulation of [1-13C]glucose. In contrast, the percent enrichments of alanine C-3 and lactate C-3 were similar to controls, indicating that glycolysis was not inhibited by fluoroacetate treatment. In the fluoroacetate-treated brains [1,2-13C]acetate was seen as a small doublet, with one peak on each side of the GABA C-3 peak (Fig. 1). A peak corresponding to citrate C-2 + C-4 was also clearly visible in these brains. However, no peak corresponding to citrate C-3 was seen, indicating that the label in the accumulated citrate had not passed a full turn through the TCA cycle. The two animals that convulsed during fluoroacetate exposure did not stand out with respect to percent enrichment of any metabolite.
Labeling of glutamine, citrate, and lactate from [1,2-13C]acetate alone. Effects of fluoroacetate
To determine whether fluoroacetate blocked the uptake of [1,2-13C]acetate, a separate experiment was performed in which [1,2-13C]acetate and unlabeled glucose was given intravenously to fluoroacetate-treated animals and controls. Analysis by GC/MS showed that double labeling of cerebral citrate was unaffected by fluoroacetate, whereas the double labeling of glutamine was reduced by ∼90% (Table 4). This confirms that [1,2-13C]acetate was taken up and converted to citrate, and that the metabolic block caused by fluoroacetate was distal to the formation of citrate, but before the formation of glutamine. The identity of the double-labeled glutamine in control animals as predominantly representing [4,5-13C]glutamine was verified by 13C NMR spectroscopy (Fig. 1). The percent enrichment of glutamine C-4 was 9.5% ± 1.8% as determined from the [4,5-13C]isotopomer of glutamine.
The double labeling of citrate and glutamine from [1,2-13C]acetate in fluoroacetate-treated animals and controls
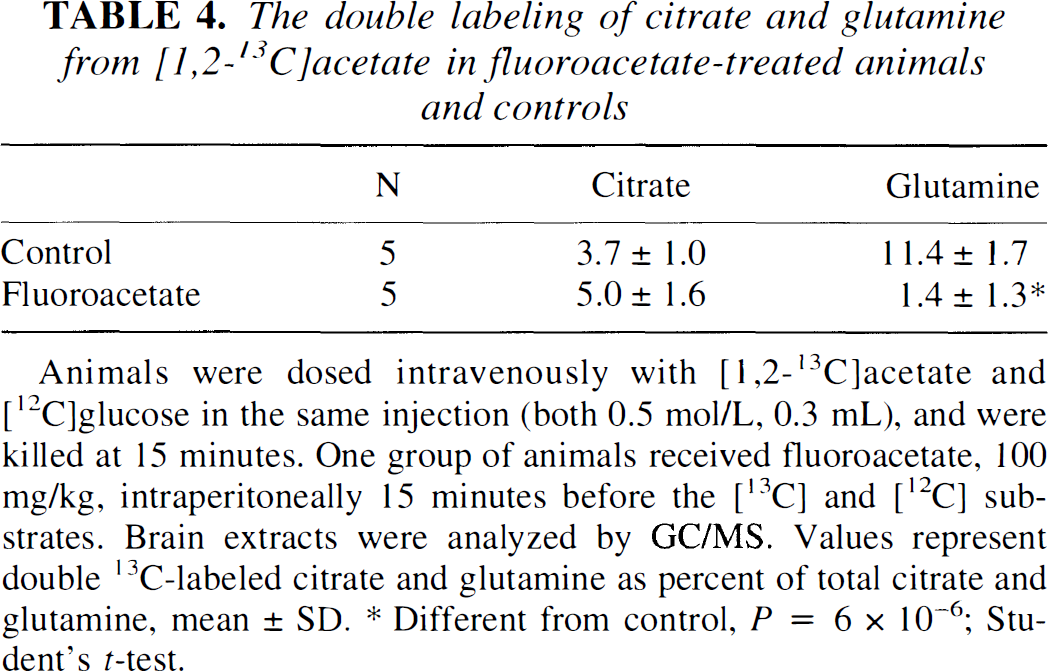
Animals were dosed intravenously with [1,2-13C]acetate and [12C]glucose in the same injection (both 0.5 mol/L, 0.3 mL), and were killed at 15 minutes. One group of animals received fluoroacetate, 100 mg/kg, intraperitoneally 15 minutes before the [13C] and [12C] substrates. Brain extracts were analyzed by GC/MS. Values represent double 13C-labeled citrate and glutamine as percent of total citrate and glutamine, mean ± SD.
Different from control, P = 6 × 10−6; Student's t-test.
Lactate C-2 was present as a small doublet (Fig. 1) representing an enrichment of 0.3% ± 0.07% as determined by 13C NMR spectroscopy. This was confirmed by GC/MS, which showed 0.4% ± 0.2% doubly labeled lactate. No labeling of lactate was seen in the presence of fluoroacetate.
DISCUSSION
In the present study glial–neuronal interactions were identified by simultaneously introducing 13C into the glial and neuronal TCA cycles with [1,2-13C]acetate and [1-13C]glucose, respectively. The glial metabolism of [1,2-13C]acetate and the predominantly neuronal metabolism of [1-13C]glucose was confirmed by the resulting glutamine/glutamate enrichment ratios being greater and less than one, respectively (O'Neal and Koeppe, 1966; Van den Berg et al., 1969; Hassel et al., 1992), which reflects the strictly glial localization of glutamine synthetase (Martinez-Hernandez et al., 1977). These indices of metabolic compartmentation, originally defined by amino acid labeling by trace amounts of radiolabel, were thus maintained even with the large amounts of 13C-containing substrates used in the current study. The glial metabolism of acetate was further demonstrated by the effect of fluoroacetate, which blocked the formation of [4,5-13C]glutamine from [1,2-13C]acetate almost completely, but had little effect on the formation of [4-13C]glutamate and [2-13C]GABA from [1-13C]glucose. The latter observation confirms that fluoroacetate did not interfere with the neuronal TCA cycles. During fluoroacetate treatment the formation of [1,2-13C]GABA was abolished even though the neuronal TCA cycles were not inhibited by fluoroacetate. This shows that acetate is not a substrate for neurons, and that [13C]acetate may be used as a highly specific marker for glial metabolism in the high doses needed for 13C NMR spectroscopy.
A notable finding in the present study was the labeling of glutamine from [1-13C]glucose during fluoroacetate treatment. This was intriguing since the glial TCA cycle was inhibited, and the labeling of glutamine from [1,2-13C]acetate was blocked. The neuronal TCA cycles were not inhibited by fluoroacetate, however, and the [4-13C]glutamine was probably formed from glutamate that was labeled from [1-13C]glucose in neurons, released during neurotransmission, and subsequently taken up by glia. This indicates considerable glial uptake and inactivation of neuronal, i.e., transmitter, glutamate. This also shows that glucose, and not only glutamine, may label endogenous transmitter glutamate.
From the foregoing it appears that two pools of glutamate serve as quantitatively important precursors for glutamine, the glial pool and the neuronal transmitter pool. The latter is quantitatively important, because in the present study approximately 40% of the [4-13C]glutamine formed from [1-13C]glucose was derived from glutamate released from neurons. Distinguishing between the various pools of glutamate serving as precursors for glutamine may become important with the clinical use of 13C NMR spectroscopy. For instance, in brain areas of neurodegeneration and gliosis, the astrocytic pool of glutamate will probably be of greater importance for glutamine synthesis than the transmitter pool. Another interesting finding was the higher C-3/ C-4 enrichment ratio in glutamine than in glutamate during fluoroacetate treatment. The C-3/C-4 enrichment ratio is an index of the average number of times that the label has cycled in a TCA cycle (Hassel et al., 1995; Hassel and Sonnewald 1995a). In the presence of fluoroacetate, because the labeling of glutamine from [1-13C]glucose can only be explained by glial uptake of neuronally released glutamate, this glutamate must have had a higher C-3/C-4 enrichment ratio than average, indicating that it was associated with a TCA cycle that turned more rapidly than the overall cerebral TCA cycle. This agrees with previous findings that the transmitter pool of glutamate turns over more rapidly than the nontransmitter, or metabolic, pool of glutamate (Cotman and Hamberger, 1978; Engelsen et al., 1986). The fluoroacetate-induced blockade of the glial TCA cycle prevented cycling of label in glia, which could otherwise have explained the difference in the C-3/C-4 enrichment ratio. It should be emphasized that [2-13C]GABA, being a four-carbon molecule, must pass through the glial TCA cycle before its label can be transferred to glutamine. This would lead to labeling of glutamine C-2 and C-3, but not of the C-4. In the presence of fluoroacetate, however, this probably did not occur because of the inhibition of the glial TCA cycle.
The conclusion that acetate is not a substrate for GABAergic neurons implies that [1,2-13C]acetate labeled GABA by some glial metabolite that was able to label GABA in the C-1 and C-2 positions. Such labeling requires that the glial precursor of GABA is a five- or a six-carbon molecule. This is because GABA cannot be formed directly from a four-carbon molecule by reversal of succinic semialdehyde dehydrogenase and GABA transaminase (Möhler et al., 1974). Therefore, four-carbon molecules like malate and succinate, which were suggested to be transferred from glia to neurons (Shank and Campbell., 1984; Westergaard et al., 1994), could not explain the formation of [1,2-13C]GABA. Citrate, 2-oxoglutarate, and glutamine are the five- and six-carbon metabolites that are released by astrocytes (Sonnewald et al., 1991; Westergaard et al., 1994; Hassel et al., 1994). The level of citrate in the cerebrospinal fluid is 0.3 to 0.6 mmol/L, similar to the level of glutamine (Petroff et al., 1986), but its physiologic role remains unknown. Citrate has been suggested as a substrate for neurotransmitter synthesis (Sonnewald et al., 1991). In the present study fluoroacetate blocked the labeling of glutamate and GABA from [1,2-13C]acetate, whereas the labeling of glial citrate was preserved. This shows that endogenous glial citrate is not a substrate for neurons in vivo. Previously, radiolabeled citrate and 2-oxoglutarate have been shown to be taken up by glial cells and not by neurons after intracerebral injection in vivo (Hassel et al., 1992). The only glial metabolite that could explain the labeling of GABA seen in the present study is, therefore, glutamine.
Glucose accumulated in the brains of fluoroacetate-treated animals as seen from the increase in the cerebral pool size and percent enrichment of glucose in fluoroacetate-treated animals, although serum glucose was maintained at control level. This accumulation could reflect the inhibitory effect of citrate on phosphofructokinase (Garland et al., 1963), which would inhibit glial glycolysis. The overall neuronal glycolytic rate was not reduced, however, because the percent enrichment of lactate and alanine from [1-13C]glucose was the same in fluoroacetate-treated animals and controls. The block in the glial metabolism of [1-13C]glucose to [4-13C] glutamine could fully account for the reduction in the labeling of GABA C-2.
[1,2-13C]Acetate labeled lactate in controls, but not during fluoroacetate treatment. This confirms a recent observation that glial cells may convert TCA cycle intermediates into pyruvate and lactate in vivo (Hassel and Sonnewald, 1995b).
Treatment with fluoroacetate led to an increase in the level of citrate in agreement with conversion of fluoroacetate into fluorocitrate, which is an inhibitor of aconitase (Peters, 1957). The increase in citrate compared well with the reduction in aspartate, which suggests that glial aspartate was converted to oxaloacetate and consumed in the synthesis of citrate. The reduction in aspartate implies stoichiometric formation of glutamate by the aspartate aminotransferase reaction. However, the level of glutamate was decreased by fluoroacetate, whereas the level of alanine increased. This indicates catabolism of glutamate in the alanine aminotransferase reaction. The observation that the decrease in aspartate was nearstoichiometric with the increase in alanine suggests that the two aminotransferases operate in a concerted manner to regulate the level of TCA cycle intermediates and related amino acids. As mentioned in Methods, [1,2-13C]aspartate and [3,4-13C]aspartate were probably labeled from [1,2-13C]acetate, and not by cycling of the label from [1-13C]glucose, as [1,2-13C]aspartate and [3,4-13C]aspartate were not present in fluoroacetate-treated animals, whereas the levels of [2-13C]aspartate and [3-13C]aspartate were similar in fluoroacetate-treated animals and controls. In controls aspartate C-2 and C-3 were equally well enriched from [1-13C]glucose. Interestingly, with fluoroacetate the aspartate C-2 became more highly enriched than the C-3. This could reflect incomplete scrambling of label in the neuronal TCA cycle. It may be that glial pyruvate carboxylation, which leads to preferential labeling of aspartate C-3 from [1-13C]glucose masks this phenomenon in the control situation.
The lack of effect of fluoroacetate on the labeling of lactate, alanine, glutamate, GABA, and aspartate from [1-13C]glucose shows that neuronal glycolysis and TCA cycle activity remained unaffected by the toxin. This should, however, not be taken to indicate that neuronal electrical activity was not affected by the fluoroacetate. As has been shown previously, fluorocitrate, the toxic product of fluoroacetate, affects neuronal activity by interfering with astrocytic maintenance of extracellular potassium homeostasis and pH (Largo et al., 1996). In the current study, such indirect effects of fluoroacetate on neuronal activity were illustrated by the behavioral changes in the treated animals.
The high percent enrichment of glutamine from [1,2-13C]acetate, which was higher than that of glutamate from [1-13C]glucose, suggests that 13C-labeled acetate would be as suitable a substrate as [1-13C]glucose (Gruetter et al., 1994; Mason et al., 1995) for in vivo studies in humans. [13C]Acetate has the advantage of yielding high enrichments of cerebral amino acids in spite of high serum levels of glucose (Hassel and Sonnewald, 1995b; this study). Therefore, glucose clamping with insulin is not necessary during infusion of [13C]acetate. The use of [13C]acetate should be thought of as complementary to the use of [13C]glucose, because [13C]acetate elucidates glial metabolism and glial-neuronal interactions which do not become apparent with [13C]glucose alone.
In conclusion, the present study gives in vivo evidence that [13C]acetate given in large amounts is metabolized by glia and not by neurons, and that [13C]acetate labels GABA through glutamine. Furthermore, endogenous neuronal glutamate is to a large extent taken up by glial cells and converted to glutamine. This demonstrates the validity of the concept of a glutamine-glutamate cycle in the brain in vivo.