
Abstract
Previously, it had been observed that nitric oxide (NO) contributes to hypoxia-induced pial artery dilation in the newborn pig. Additionally, it was also noted that activation of ATP-sensitive K+ channels (KATP) contribute to cGMP-mediated as well as to hypoxia-induced pial dilation. Although somewhat controversial, adenosine is also thought to contribute to hypoxic cerebrovasodilation. The present study was designed to investigate the role of NO, cyclic nucleotides, and activation of KATP channels in the elicitation of adenosine's vascular response and relate these mechanisms to the contribution of adenosine to hypoxia-induced pial artery dilation. The closed cranial window technique was used to measure pial diameter in newborn pigs. Hypoxia-induced artery dilation was attenuated during moderate (PaO2 ≈ 35 mm Hg) and severe hypoxia (PaO2 ≈ 25 mm Hg) by the adenosine receptor antagonist 8-phenyltheophylline (8-PT) (10–5 M) (26 ± 2 vs. 19 ± 2 and 34 ± 2 vs. 22 ± 2% for moderate and severe hypoxia in the absence vs. presence of 8-PT, respectively). This concentration of 8-PT blocked pial dilation in response to adenosine (8 ± 2, 16 ± 2, and 23 ± 2 vs. 2 ± 2, 4 ± 2, and 6 ± 2% for 10–8, 10–6, and 10–4 M adenosine before and after 8-PT, respectively). Similar data were also obtained using adenosine deaminase as a probe for the role of adenosine in hypoxic pial dilation. Adenosine-induced dilation was associated with increased CSF cGMP concentration (390 ± 11 and 811 ± 119 fmol/ml for control and 10–4 M adenosine, respectively). The NO synthase inhibitor, L-NNA, and the cGMP antagonist, Rp 8-bromo cGMPs, blunted adenosine-induced pial dilation (8 ± 1, 14 ± 1, and 20 ± 3 vs. 3 ± 1, 5 ± 1, and 8 ± 3% for 10–8, 10–6, and 10–4 M adenosine before and after L-NNA, respectively). Adenosine dilation was also blunted by glibenclamide, a KATP antagonist (9 ± 2, 14 ± 3, 21 ± 4 vs. 4 ± 1, 8 ± 2, and 11 ± 2% for 10–8, 10–6, and 10–4 M adenosine before and after glibenclamide, respectively). Finally, it was also observed that adenosine-induced dilation was associated with increased CSF cAMP concentration and the cAMP antagonist, Rp 8-bromo cAMPs, blunted adenosine pial dilation. These data show that adenosine contributes to hypoxic pial dilation. These data also show that NO, cGMP, cAMP, and activation of KATP channels all contribute to adenosine induced pial dilation. Finally, these data suggest that adenosine contributes to hypoxia-induced pial artery dilation via cAMP and activation of KATP channels by NO and cGMP.
Keywords
Although somewhat controversial, adenosine is generally thought to contribute to hypoxic cerebrovasodilation. For example, adenosine concentration has been observed to increase during hypoxia in brain tissue and interstitial fluid of the adult rat (Van Wylen et al., 1986; Winn et al., 1981), while the adenosine receptor antagonist, theophylline, attenuated hypoxic hyperemia in the adult dog (Emerson and Raymond, 1981) and pial artery dilation in the adult rat (Morii et al., 1987). On the other hand, 8-phenyltheophylline and adenosine deaminase had no effect on the cerebral hyperemic response to hypoxia in the adult cat or rat (Reid et al., 1995; Dora et al., 1984). The role of adenosine in hypoxic cerebrovasodilation has been less well characterized in the newborn, but the observations have been equally inconsistent. For example, brain interstitial fluid adenosine concentration is increased during hypoxia in the newborn pig (Park et al., 1987) and 8-phenyltheophylline attenuates hypoxic hyperemia while adenosine deaminase blunted hypoxic dilation in the newborn pig (Laudignon et al., 1990; Wagerele and Mishra, 1986). Alternatively, theophylline was also observed to have no effect on hypoxic hyperemia in the newborn pig (McPhee and Maxwell, 1987).
Several other mechanisms have also been suggested to be involved in hypoxic cerebrovasodilation. For example, nitric oxide (NO) synthase inhibitors have been reported to attenuate the increase in cerebral blood flow and pial artery dilation elicited by hypoxia in the adult (Iwamoto et al., 1992) and newborn pig (Armstead, 1995a), while μ and δ opioid receptor antagonists attenuate hypoxic pial dilation in the piglet (Armstead, 1995a, b ). Inhibitors of the ATP-sensitive K+ channel (KATP) have similarly been observed to attenuate hypoxic hyperemia and pial artery dilation in the adult and newborn (Shankar and Armstead, 1995; Reid et al., 1993; Taguchi et al., 1994).
Recently, it has been observed that activation of KATP channels contributes to cGMP-mediated vasodilation of pial arteries (Armstead, 1996). It has also been observed that NO synthase inhibition attenuates the cerebral hyperemic response to adenosine in the adult rat (Dirnagl et al., 1994) while inhibition of KATP channels attenuates the dilation of porcine coronary arteries by adenosine (Kuo and Chancellor, 1995). On the other hand, inhibition of KATP channels has also been observed to only attenuate the coronary vasoactivity of adenosine analogues and not native adenosine (Niiya et al., 1994). Since significant development of cerebral adenosine receptors, particularly in terms of density and coupling to second messengers may occur postnatally (Geiger et al., 1984; Marangos et al., 1982), the contribution of cyclic nucleotides and KATP channel activation to adenosine dilation in the newborn cerebral circulation is uncertain.
Therefore, the present study was designed to investigate the role of NO, cyclic nucleotides, and activation of KATP channels in the elicitation of adenosine's vascular response and relate these mechanisms to the contribution of adenosine to hypoxia induced pial artery dilation in the newborn pig.
MATERIALS AND METHODS
Newborn pigs (1–5 days old) of either sex were used in these experiments. They were initially anesthetized with ketamine hydrochloride (33 mg/kg i.m.) and acepromazine (3.3 mg/kg i.m.). Anesthesia was maintained with α-chloralose (30–50 mg/kg initially, supplemented with 5 mg/kg/h i.v.). A catheter was placed into the right femoral artery to record blood pressure and to obtain blood samples for determination of blood gases and pH. Another catheter was inserted into the right femoral vein for the injection of drugs and fluids. The trachea was cannulated, and the animals were ventilated with room air. Core body temperature was monitored with a rectal probe and maintained at 37 ± 2°C with an overhead radiant heater.
For the insertion of the cranial window, the scalp was removed and an opening was made in the skull over the parietal cortex. The dura was cut and retracted over the cut bone edge. The cranial window was placed in the hole and filled with artificial CSF of the following composition (in milligrams per liter): 220 KCl, 132 MgCl2, 221 CaCl2, 7,710 NaCl, 402 urea, 665 dextrose, and 2,066 NaHCO3; with pH at 7.33, P
Pial arteries were observed with a microscope, a television camera mounted on the microscope, and a video monitor. Vessel diameter was measured with a video microscaler (model VPA 550, For-A-Corp, Los Angeles, CA, U.S.A.).
Experimental protocol
Diameters of pial vessels (small artery 120–160 μm, arteriole 50–70 μm) were measured every minute for a 10-min exposure period after injection under the window of artificial CSF containing no drug and after injection of CSF containing a drug. Two milliliters of CSF was flushed through a window over 30 s. Needles incorporated into the side of the window allowed infusion of CSF under the window and runoff of excess CSF. We measured the peak response. We waited at least 20 min after flushing the highest concentration of any drug before topically applying another agent. Hypoxia was produced by decreasing inspired oxygen sufficiently to reduce and maintain arterial P
Influence of moderate and severe hypoxia on pial artery diameter
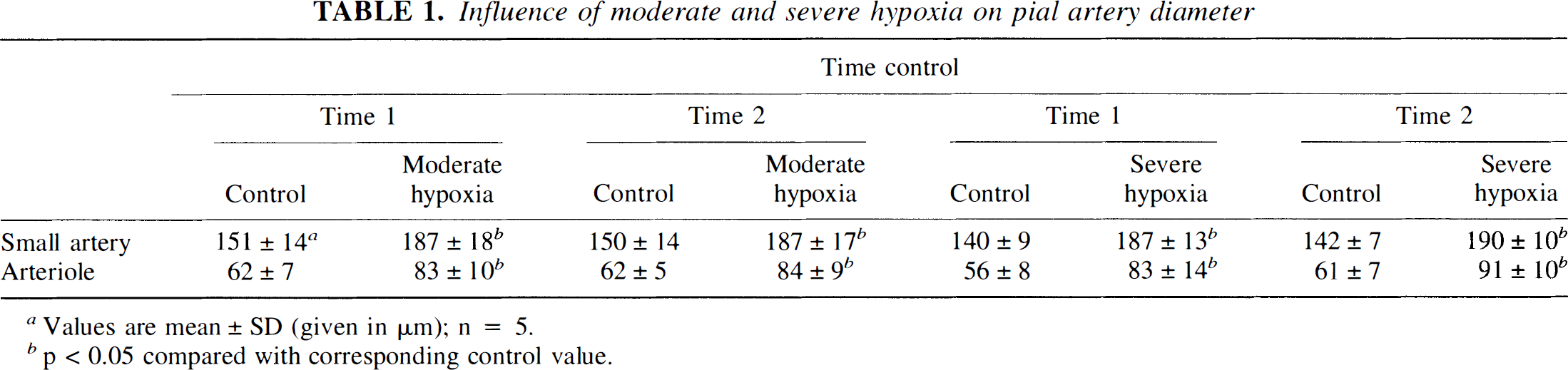
Values are mean ± SD (given in μm); n = 5.
p < 0.05 compared with corresponding control value.
Influence of adenosine on pial artery diameter
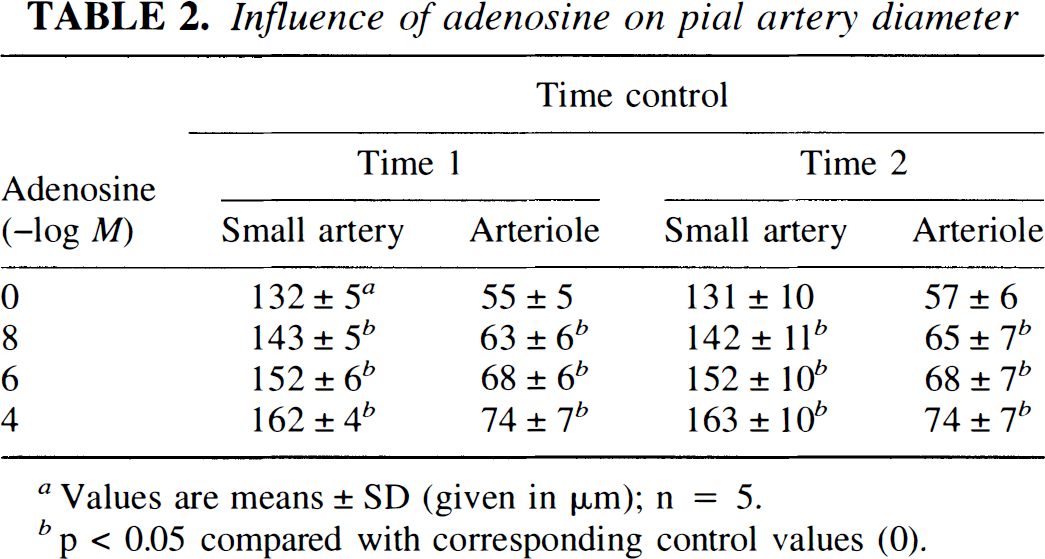
Values are means ± SD (given in μm); n = 5.
p < 0.05 compared with corresponding control values (0).
The selectivity of the adenosine antagonists was assessed by obtaining responses to adenosine and sodium nitroprusside (SNP) (10–8, 10–6, 10–4 M) (Sigma) before and after 8-phenyltheophylline or adenosine deaminase. Additionally, responses to adenosine were obtained in the absence and presence of L-NNA, glibenclamide, or the cAMP antagonist, Rp 8-bromo cAMPs (10–5 M) (Biolog Life Science Institute, LaJolla, CA, U.S.A.), and a CSF sample for cyclic nucleotide or opioid analysis was collected at the end of the 10-min exposure period. CSF (300 μl) was collected by slowly infusing CSF into one side of the window and allowing the CSF under the window to drip freely into a collection tube on the opposite side.
The stock glibenclamide solution (10–3 M) was made by initially dissolving this agent in a small amount of dimethyl sulfoxide (200 μl) and the balance in ethanol. This vehicle was then diluted 1:1000 in CSF to make the working solution. This CSF–dimethyl sulfoxide–ethanol vehicle had no effect on pial artery diameter (157 ± 6 vs. 158 ± 8 μm, n = 5). Appropriate aliquots of the vehicle for all other agents (0.9% saline) were added to the CSF infused under the window. This CSF vehicle had no effect on pial artery diameter, as reported previously (Armstead, 1995a, b ).
cGMP and cAMP analysis
CSF samples collected after a 10-min exposure to a drug were analyzed for cGMP or cAMP using scintillation proximity assay methods. Commercially available kits for cGMP and cAMP (Amersham, Arlington Heights, IL, U.S.A.) were used. Briefly, this assay determines cyclic nucleotide concentration for binding to an antiserum, which has a high specificity for either cGMP or cAMP. The antibody-bound cyclic nucleotide is then reacted with an anti-rabbit second antibody bound to fluoromicrospheres. Labeled cyclic nucleotide bound to the primary rabbit antibody can then be measured by determining the amount of light emitted by the fluoromicrospheres using a beta-scintillation counter. All unknowns were assayed at two dilutions. The concentration of unlabeled cyclic nucleotides is calculated from the standard curve via linear regression analysis.
Opioid analysis
CSF samples were immediately acidified with 1 N acetic acid to prevent peptide degradation, rapidly frozen, and stored at −20°C. Radioimmunoassay kits for methionine enkaphalin and leucine enkephalin are commercially available (Incstar, Stillwater, MN, U.S.A.; Peninsula, Belmont, CA, U.S.A.). The radioimmunoassay uses simultaneous addition of sample, rabbit anti-opioid antibody, and the 125I derivative of the opioid. After an overnight incubation at 4°C, free opioid was separated from opioid bound to antibody by the addition of saturated ammonium sulfate in the presence of rabbit carrier γ-globulin. Following centrifugation at 760 g for 10 min, the supernate was decanted and the pellet counted using a γ-scintillation counter. All samples and standards were assayed in duplicate. Data are calculated as % B/B0 versus concentration, where % B/B0 = (avg. cpm of sample – avg. cpm of nonspecific binding tube)/B0 × 100 and B0 = avg. cpm of total binding tube – avg. cpm of nonspecific binding tube. We have used these methods previously to quantify opioid concentration in periarachnoid cortical CSF (Armstead et al., 1991).
Statistical analysis
Pial arteriolar diameter, systemic arterial pressure, cyclic nucleotide, and opioid levels were evaluated using analysis of variance for repeated measures. If the value was significant, the Fisher test was performed. An α level of p < 0.05 was considered significant in all statistical tests. Values are presented as means ± SD of absolute values or as percentages of change from control values. Data presented as percent change were compared by nonparametric means using the Wilcoxon signed rank test.
RESULTS
Influence of adenosine antagonists on hypoxia-induced pial artery dilation
Moderate hypoxia (Pa
Hypoxia-induced vasodilation of the small pial arteries and arterioles was attenuated by topical 8-phenyltheophylline (10–5 M) or adenosine deaminase (20 U/ml) (Figs. 1 and 2; n = 5). On a percentage basis, 8-phenyltheophylline attentuated the moderate hypoxia-induced vasodilation of small arteries and arterioles by 27 ± 10 and 29 ± 9%, respectively, while adenosine deaminase attenuated dilation of these vessels by 24 ± 9 and 28 ± 7%, respectively. Vasodilation due to severe hypoxia was decreased in small arteries and arterioles in the presence of 8 phenyltheophulline by 37 ± 9 and 47 ± 8% and by 41 ± 12 and 45 ± 6% in the presence of adenosine deaminase, respectively.
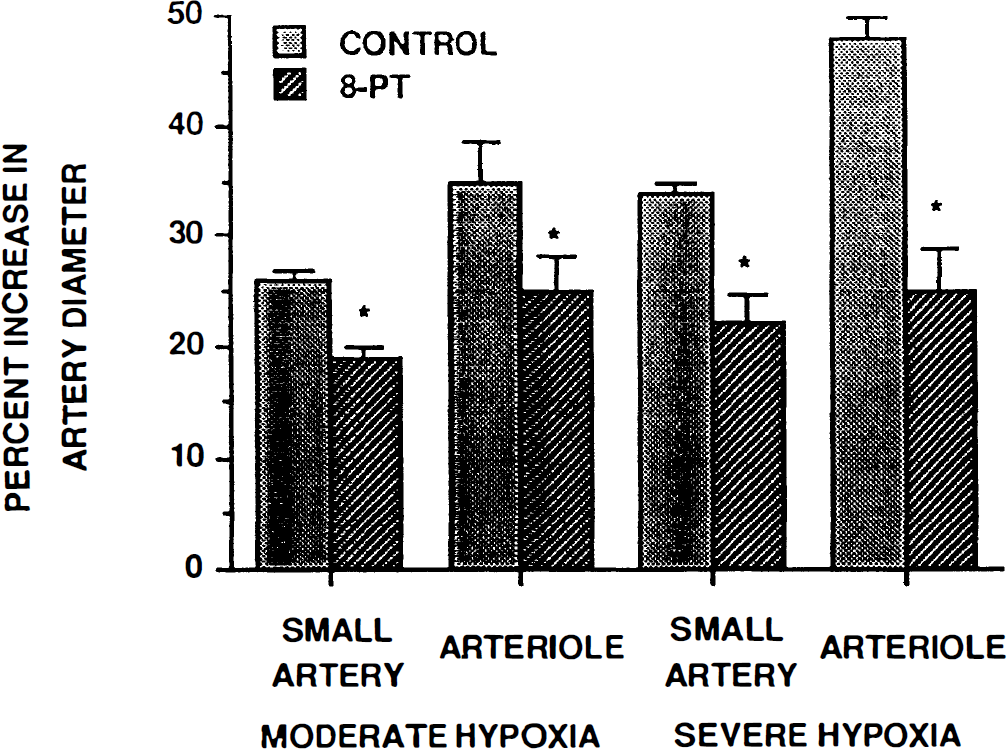
Influence of moderate [arterial P
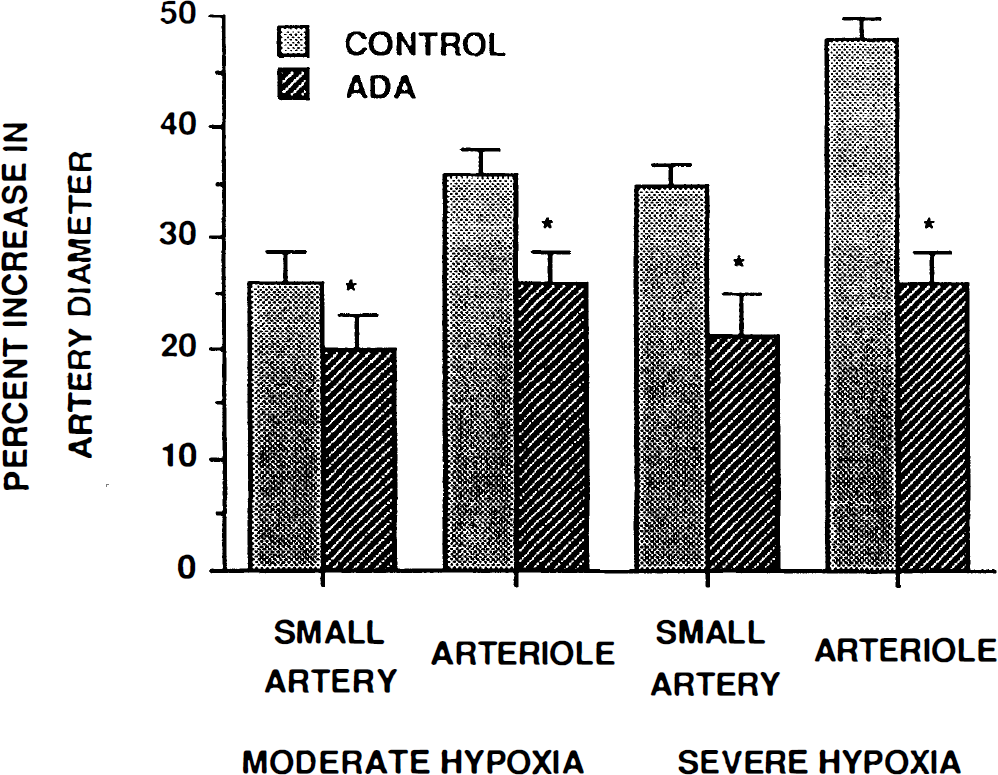
Influence of moderate (Pa
Influence of 8-phenyltheophylline and adenosine deaminase on adenosine-induced pial artery vasodilation
Adenosine (10–8, 10–6, 10–4 M) elicited reproducible vasodilation in pial small arteries and arterioles (Table 2, n = 5). These increases in vessel diameter were blunted in the presence of either 8-phenyltheophylline or adenosine deaminase (Figs. 3 and 4; n = 5). These antagonists had no effect on pial small artery diameter (129 ± 12 vs. 132 ± 11 μm for 8-phenyltheophylline and 142 ± 12 vs. 146 ± 16 μm for adenosine deaminase, respectively, n = 5). Similarly, these antagonists had no effect on pial arteriole diameter (61 ± 4 vs. 62 ± 4 μm for 8-phenyltheophylline and 58 ± 30 vs. 59 ± 4 μm for adenosine deaminase, respectively, n = 5). Finally, these antagonists had no effect on the dilation produced by SNP (10–8, 10–6, 10–4 M) (data not shown).
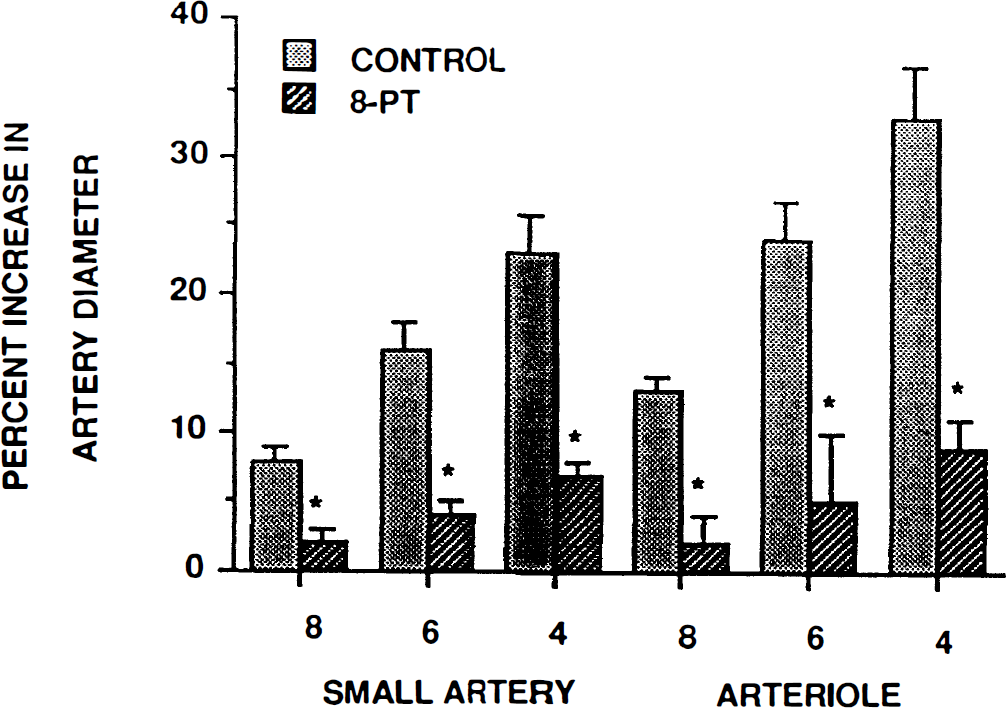
Influence of adenosine (10–8, 10–6, 10–4 M) on smal pial arteries and arterioles, in the absence (control) and presence of 8-phenyltheophylline (8-PT, 10–5 M), n = 5. *p < 0.05 compared with corresponding control.
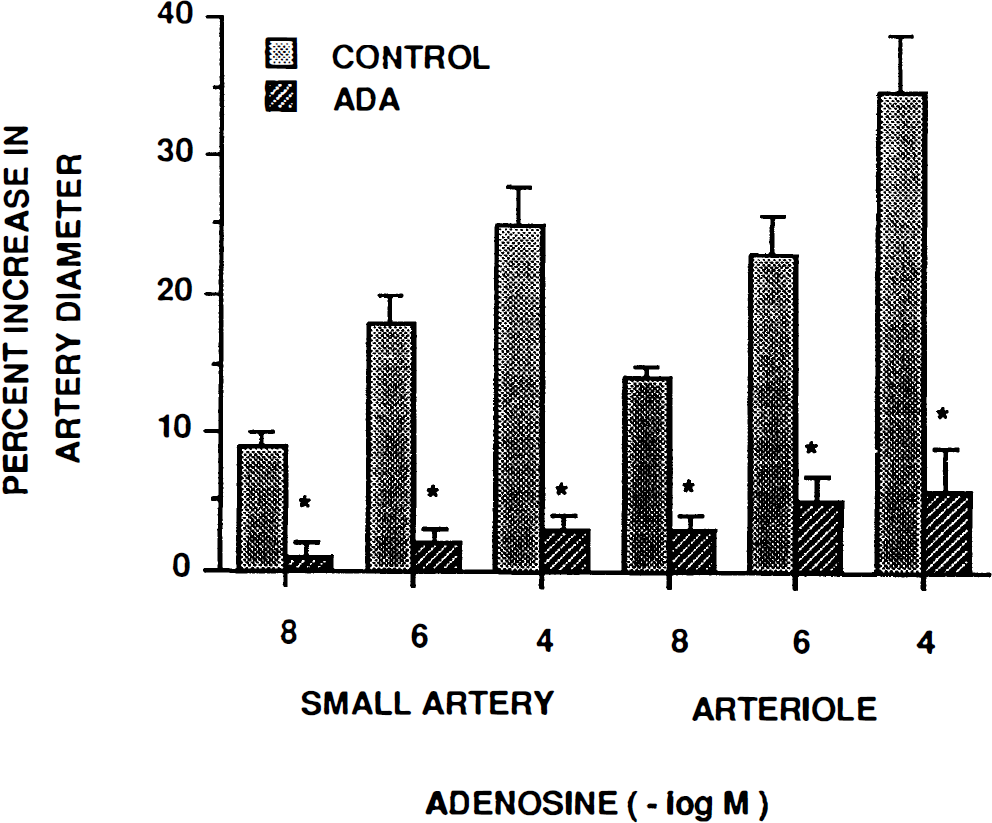
Influence of adenosine (10–8, 10–6, 10–4 M) on smal pial arteries and arterioles in the absence (control) and presence of adenosine deaminase (ADA, 20 U/ml), n = 5. *p < 0.05 compared with corresponding control.
Role of NO in adenosine-induced pial artery dilation
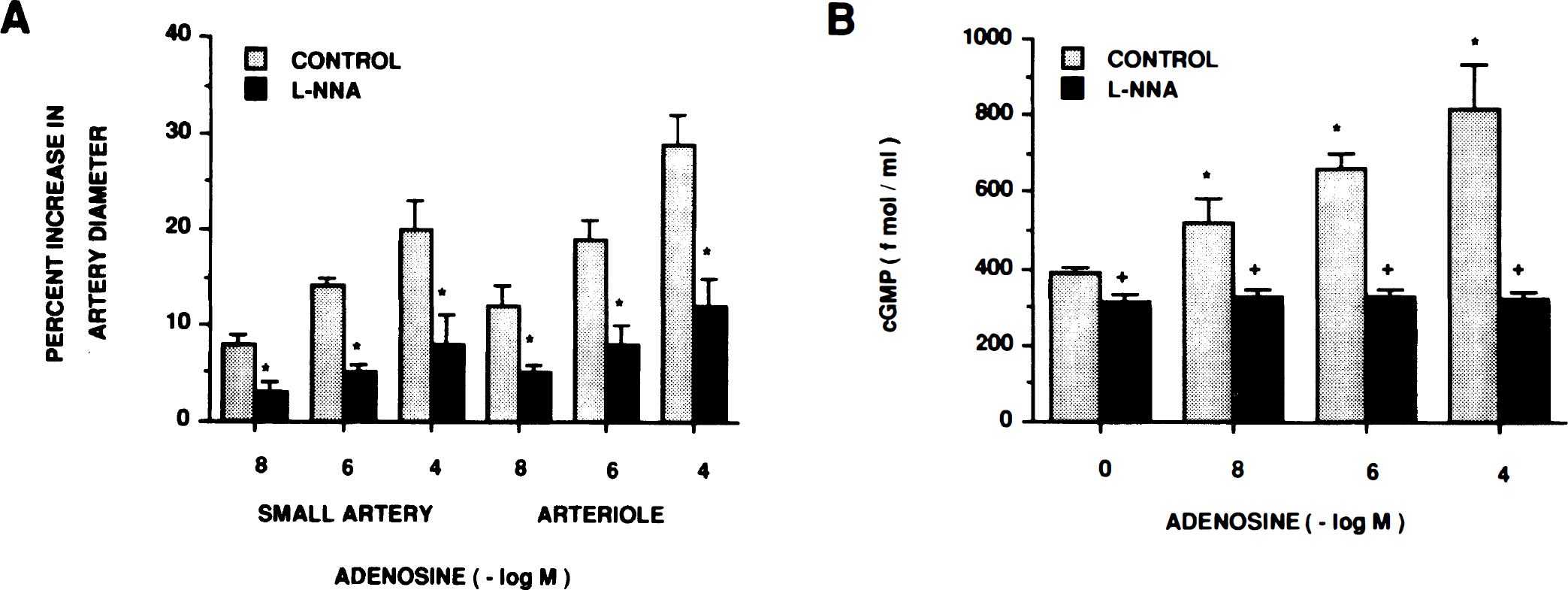
Adenosine-induced pial vasodilation was attenuated by L-NNA (10–6 M) (Fig. 5a, n = 6). Adenosine (10–8, 10–6, 10–4 M) was associated with 1.3 ± 0.1-, 1.7 ± 0.1-, and 2.1 ± 0.3-fold increases in cortical periarachnoid CSF cGMP, which L-NNA attenuated (Fig. 5b, n = 6). L-NNA decreased resting CSF cGMP concentration (Fig. 5b) and decreased pial artery diameter from 170 ± 8 to 160 ± 8 μm, n = 5.
Role of cGMP and cAMP in adenosine-induced pial artery dilation
Adenosine-induced pial dilution was associated with increased CSF cGMP concentration, as described above. Additionally, vascular responses to adenosine were attenuated by Rp 8-bromo cGMPs (10–5 M), a cGMP antagonist (Fig. 6, n = 5). Similarly, adenosine also increased CSF cAMP, and vascular responses to adenosine were attenuated by the cAMP antagonist, Rp 8-bromo cAMPs (10–5 M) (Fig. 7a, b ). Neither Rp 8-bromo cGMPs nor Rp 8-bromo cAMPs had any effect on pial artery diameter (149 ± 14 vs. 145 ± 18 μm for Rp 8-bromo cGMPs and 150 ± 10 vs. 151 ± 11 μm for Rp 8-bromo cAMPs, respectively, n = 5).
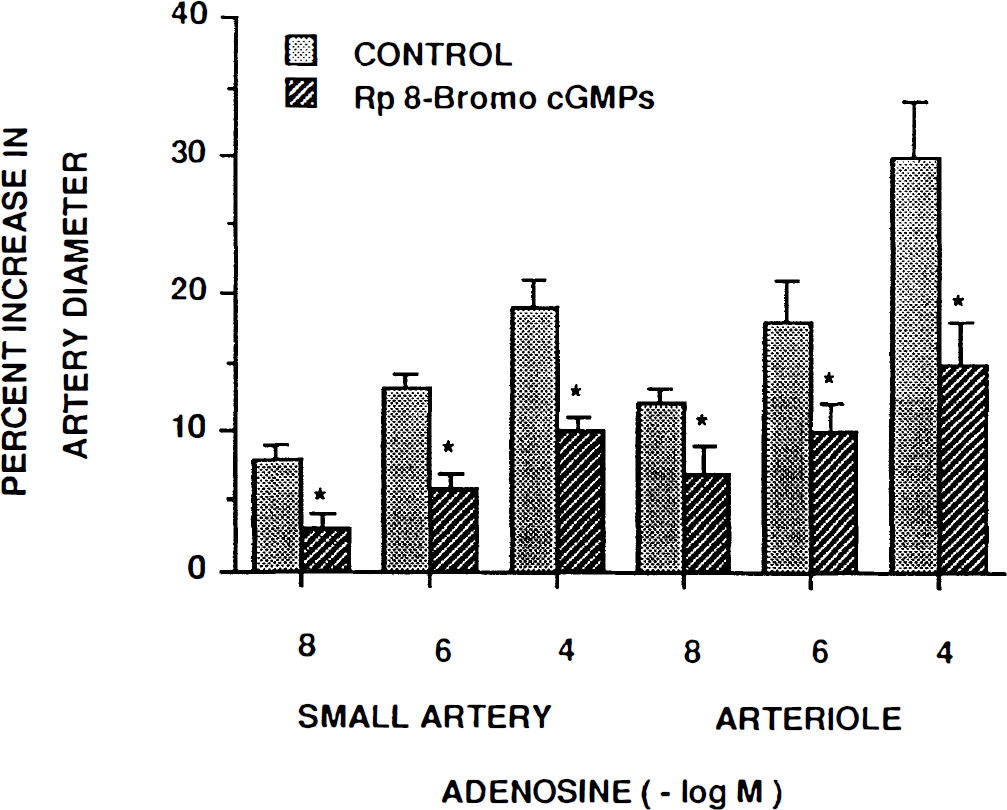
Influence of adenosine (10–8, 10–6, 10–4 M) on small pial arteries and arterioles in the absence (control) and presence of Rp 8-bromo cGMPs (10–5 M), n = 5. *p < 0.05 compared with corresponding control.
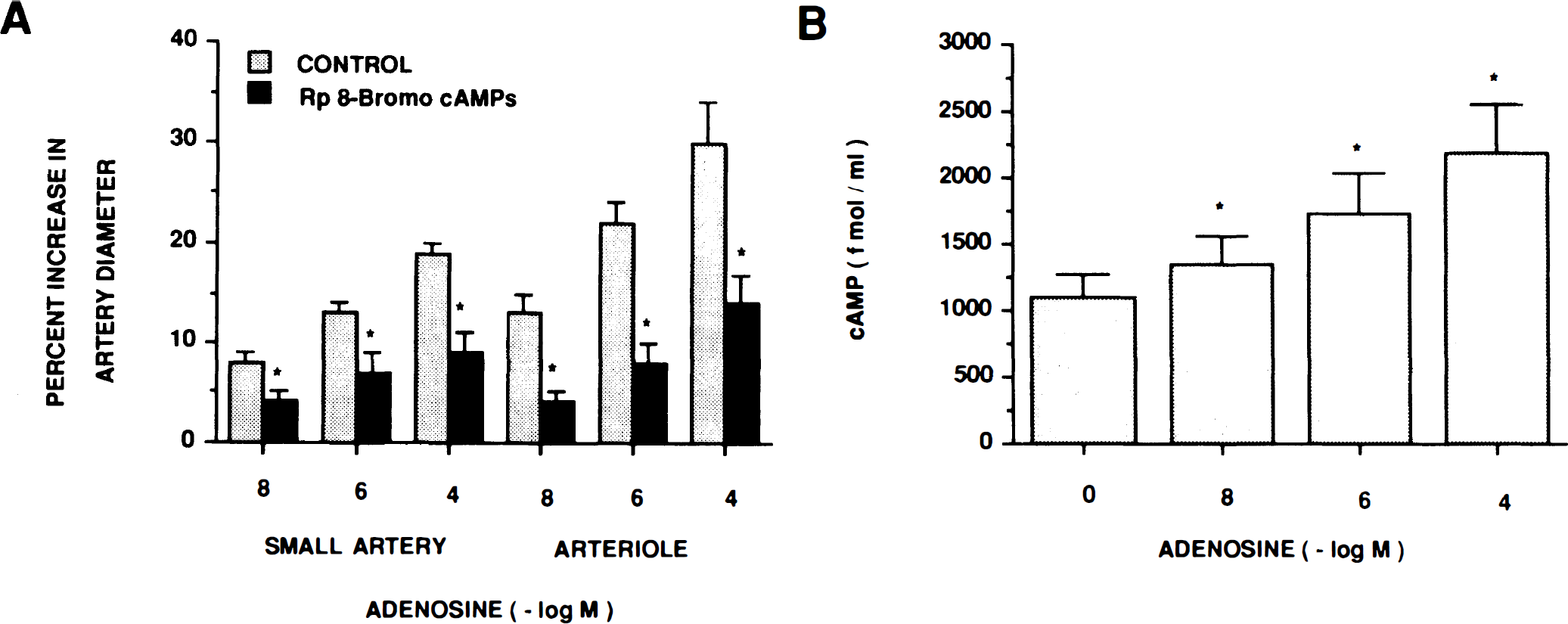
Influence of glibenclamide on adenosine-induced pial artery dilation
Adenosine-induced pial artery dilation was attenuated by glibenclamide (10–6 M) (Fig. 8, n = 5). Glibenclamide itself had no effect on pial artery diameter (146 ± 19 vs. 148 ± 20 μm, n = 5).
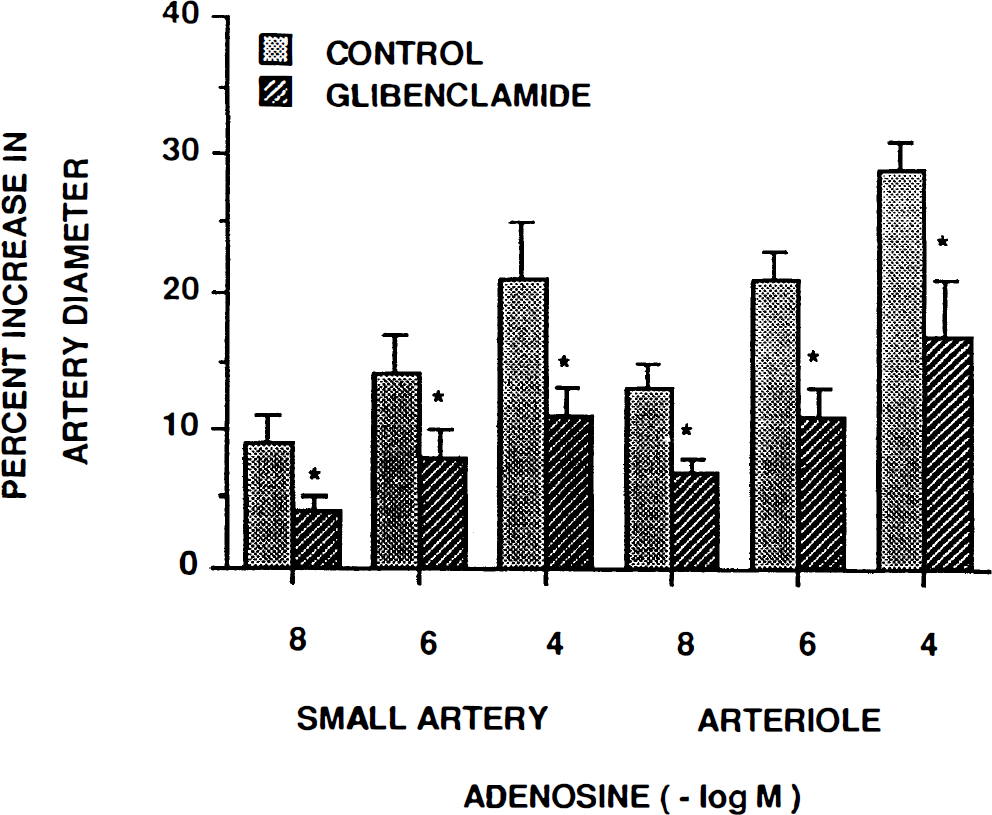
Influence of adenosine (10–8, 10–6, 10–4 M) on small pial arteries and arterioles in the absence (control) and presence of glibenclamide (10–6 M), n = 5. *p < 0.05 compared to corresponding control value.
Influence of adenosine on CSF opioid concentration
Topical adenosine had no effect on CSF methionine enkephalin or leucine enkephalin concentration (Table 3, n = 6).
Influence of adenosine on CSF methionine enkephalin and leucine enkephalin concentration

Values are means ± SD; n = 6.
Blood chemistry and mean arterial blood pressure
Blood chemistry and mean arterial blood pressure values were obtained at the beginning and end of all normoxia experiments as well as during normoxia and hypoxia in experiments designed to characterize the role of adenosine in hypoxia-induced pial artery dilation. Hypoxia decreased Pa
Blood chemistry and mean arterial blood pressure

pH, –log H; PaCO2, arterial partial pressure of CO2; PaO2, arterial partial pressure of O2.
Values are means ± SD in mm Hg; n = number of pigs.
p < 0.05 compared with corresponding normoxia values.
DISCUSSION
Results of the present study show that hypoxic pial dilation was attenuated by the antagonists of adenosine action 8-phenyltheophylline and adenosine deaminase. Both probes blocked adenosine-induced pial dilation. These data indicate that adenosine contributes to pial artery dilation during moderate and severe hypoxia. When compared to other mechanisms that contribute to hypoxic pial dilation in the newborn pig, however, the role of adenosine appears to be rather modest. For example, L-NNA decreased moderate hypoxia-induced small pial artery dilation by 47 ± 4% (Armstead, 1995a), while β-funaltrexamine, a μ opioid antagonist, decreased this response by 38 ± 4% (Armstead, 1995a). Additionally, moderate hypoxic pial artery dilation was decreased by 43 ± 2% in the presence of the δ1 opioid antagonist, BNTX or 64 ± 7% in the presence of the δ2 antagonist, naltrindole (Armstead, 1995b). In contrast, in the present study the adenosine receptor antagonist, 8-phenyltheophylline, decreased the pial response to moderate hypoxia by only 27 ± 10%. Moreover, this adenosine antagonist did not further decrease the pial response during severe hypoxia (24 ± 9%), a possibility observed with antagonists of other vasoactive systems (i.e., BNTX, 72 ± 12 vs. 43 ± 2%). While differences between the present and previous studies concerning the role of adenosine in hypoxic cerebrovasodilation could be due to age (Dora et al., 1984; Reid et al., 1995), such differences using the same age and species (McPhee and Maxwell, 1987) could also be due to choice of adenosine antagonist, concentration, and route of administration.
The present study was also designed to determine the second messengers involved in the elicitation of adenosine's vascular response and relate these mechanisms to the contribution of adenosine to hypoxic pial dilation. Results of this study show that adenosine's dilation is associated with increased cortical periarachnoid CSF cGMP concentration. Additionally, L-NNA and Rp 8-bromo cGMPs, a cGMP antagonist (Nazakawa and Imai, 1994), attenuated adenosine-induced dilation. Previous studies in the pig show that L-NNA blocked responses to acetylcholine, while responses to SNP were unchanged, indicating that L-NNA is a selective NO synthase inhibitor (Armstead et al., 1994). Since NO is thought to elicit dilation via activation of soluble guanylate cyclase (Ignarro, 1990), these data indicate that NO contributes to adenosine pial dilation via release of cGMP. Recently it has been observed that activation of KATP channels contributes to cGMP-mediated vasodilation of pial arteries (Armstead, 1996). Results of this study show that adenosine pial dilation was attenuated by the KATP channel antagonist, glibenclamide, in a concentration previously observed to block responses to the KATP channel agonist, cromakalim (Shankar and Armstead, 1995). Therefore, these data suggest that adenosine elicits pial dilation, at least in part, via release of NO, which activates KATP channels through a cGMP-dependent mechanism. Since adenosine dilation was also associated with elevated CSF cAMP concentration and that vascular response was attenuated by RP 8-bromo cAMPs, a cAMP antagonist (Wang et al., 1991), data from this study indicate that cAMP also contributes to adenosine pial dilation. Taken together, then, these data indicate that the mechanism for adenosine dilation in the cerbral circulation is multifactorial, involving NO, cGMP, cAMP, and activation of KATP channels.
In the context of previous studies, however, results of the present investigation indicating that NO and cGMP are involved in the pial dilation response to adenosine are somewhat surprising. For example, responses to adenosine were unchanged after endothelial layer removal from cat middle cerebral arteries in vitro (Conde et al., 1991) or after methylene blue application to cat pial arteries in vivo (Haberl et al., 1990). Furthermore, dilation to adenosine was unchanged by the NO synthase inhibitor, L-NMMA, in isolated rat intracerebral arterioles (Ngai and Winn, 1995) and in pial arteries of the cat in vivo (Wei et al., 1992). However, the suggested role of NO in adenosine cerebrovascular dilation is not without precedent since Dirnagl et al. (1994) have observed that another NO synthase inhibitor, L-NNA, reduced the cerebral blood flow response to adenosine in the rat. In the coronary circulation, similar observations indicate that NO was also involved in the vascular response to adenosine (Kuo and Chancellor, 1995; Vials and Burnstock, 1993). Therefore, although controversial, there is evidence for the role of NO in adenosine-induced dilation. Reasons for such divergent observations are uncertain but could be due to differences in methodology, type and dose of NO synthase inhibitor used, species, and/or age differences.
Having identified mechanisms involved in adenosine pial dilation, an attempt was then made to relate these data to the contribution of adenosine to hypoxic pial dilation. Since activation of KATP channels has been observed to contribute to hypoxic pial dilation (Shankar and Armstead, 1995), data from the present study suggest that, at least in part, adenosine's role in hypoxic dilation involves the release of NO, which activates KATP channels through a cGMP-dependent mechanism. It was initially hypothesized that opioids could serve as intermediates in this process since it was observed that the opioids methionine enkephalin and leucine enkephalin contribute to hypoxia-induced pial dilation through activation of KATP channels (Shankar and Armstead, 1995). Additionally, cGMP has recently been observed to increase CSF methionine enkephalin and leucine enkephalin concentration (Wilderman and Armstead, 1996). Therefore, adenosine could activate KATP channels via the sequential release of cGMP and opioids. However, results of the present study show that adenosine does not alter CSF methionine enkephalin or leucine enkephalin concentration. The concentrations of adenosine used for investigation in this study were chosen based on their physiologic relevance. Using microdialysis, it has been observed that interstitial fluid adenosine concentrations in the frontal cortex of the newborn pig increased from 10–7 to 10–6 M during hypoxia while cisterna magna CSF levels increased from 10–8 to 10–7 M (Park et al., 1987). Since concentrations of adenosine observed in response to hypoxia did not have any effect on CSF opioid concentration, these data indicate that adenosine and opioids contribute to hypoxic pial dilation independent of one another.
Adenosine-induced elevation of CSF cGMP and cAMP concentrations may suggest an interaction between the two cyclic nucleotides in the elicitation of the vasodilator response. Thus, the increase in cortical cGMP observed during adenosine receptor activation could be an indirect consequence of elevated cAMP as a result of cross-talk between cyclic nucleotide-metabolizing systems at the level of their synthesis or degradation (Vigne et al., 1994). For example, it has been suggested that an increase in cAMP levels would competitively prevent cGMP breakdown at the level of phosphodiesterase (Hernandez et al., 1994; Vigne et al., 1994).
Furthermore, it has been recently observed that forskolin, a direct activator of adenylate cyclase, induced pial arterial dilation and increased CSF cGMP; the vascular and biochemical changes being attenuated by L-NNA (Rebich et al., 1995). These data suggest an association between cAMP elevation and the consequent activation of NO synthase. It is presently uncertain if this possibility could account for the dual involvement of cGMP and cAMP in adenosine-induced pial arterial vasodilation. While these two cyclic nucleotides could, therefore, interact in the mediation of adenosine's contribution to hypoxic pial dilation, they may also contribute to hypoxic dilation separately.
The mechanism by which hypoxia results in cerebral vasodilation has not been fully established. Studies in the past have suggested a role for the endothelium, nitric oxide, prostanoids, opioids, and vasopressin (Armstead, 1995a, b ; Coyle et al., 1993; Eisenach et al., 1992; Fredricks et al., 1994; Iwamoto et al., 1992). The contribution of some of these systems to hypoxic vasodilation is controversial, however, For example, NO has been observed to both contribute to hypoxic pial dilation and not contribute to hypoxic hyperemia in the newborn pig (Armstead, 1995a; Ichord et al., 1994). Such controversial findings could be due to differences in methodology or types and dose of No synthase inhibitor used. Recently, the KATP channel inhibitors tolbutamide and glibenclamide have been observed to attenuate hypoxia-induced hyperemia and pial artery dilation, suggesting a role for KATP channel activation in that vascular response (Reid et al., 1993; Taguchi et al., 1994). Additional studies have attempted to link the activation of KATP channels to other vasoactive systems as contributing factors to hypoxia-induced vasodilation. For instance, adenosine has been observed to contribute to hypoxia-induced vasodilation of the isolated perfused rabbit heart through activation of KATP channels (Nakhostine and LaMontagne, 1993). In contrast, adenosine does not appear to contribute to hypoxia-induced pial artery dilation via activation of KATP channels in the adult rabbit (Taguchi et al., 1994). In another study, an endothelium-derived cyclooxygenase product has been suggested to link KATP channel activation to hypoxia-induced dilation of isolated rat middle cerebral arteries (Fredricks et al., 1994). Results of the present study are the first to link adenosine to KATP channel activation in its contribution to hypoxia-induced pial artery dilation in the newborn. Differences between this study and observations made in the adult rabbit (Taguchi et al., 1994), may be due to species-and/or age-dependent variables.
In conclusion, results of this study show that NO, cGMP, cAMP, and activation of KATP channels all contribute to adenosine-induced pial dilation. Furthermore, these data suggest that adenosine contributes to hypoxia-induced dilation via cAMP and activation of KATP channels by NO and cGMP.
Footnotes
Acknowledgment:
The author thanks Joseph Quinn for technical assistance in the performance of the experiments. This research was supported by grants from the National Institutes of Health and the American Heart Association. W. M. Armstead is an Established Investigator of the AHA.