
Abstract
In isolated canine middle cerebral arteries contracted with prostaglandin F2α, transmural electrical stimulation (TES), nicotine, and substance P produced relaxations. Transmural electrical stimulation- and nicotine-induced endothelium-independent responses are mediated by nitric oxide (NO) liberated from perivascular nerve, whereas substance P-induced relaxations are mediated by endothelium-derived NO. These responses were attenuated by replacement of 95% O2 and 5% CO2 gas (about 550 mm Hg of partial O2 pressure) with 95% N2 and 5% CO2 gas (about 40 mm Hg); inhibition of the response to TES was stabilized 30 minutes later. Reoxygenation partially reversed the response. Relaxations caused by exogenous NO were not influenced by hypoxia. Inhibition by hypoxia of the response to TES was not affected by superoxide dismutase. However, the inhibitory effect was prevented by amiloride and dimethyl-amiloride, Na+-H+ exchange inhibitors, or acidosis caused by the addition of HCl. The inhibition by hypoxia was reversed by amiloride. It is concluded that depression by hypoxia of the response mediated by endogenous NO is associated with impaired membrane function caused by restoration of normal intracellular pH by Na+-H+ exchanger.
Ischemia and reperfusion in the brain and heart impair cellular function and cause cell death, which are determinants for prognosis of patients with stroke and myocardial infarction. Clarifying mechanisms of the impairment and preventing the functional deterioration are matters of importance in solving severe cerebral and cardiac circulatory problems.
Chronic hypoxia interferes with vascular functions (Karamsetty et al., 1995). Hypoxia produces vasodilatation possibly associated with the release of vasoactive substances, such as nitric oxide (NO) (Armstead, 1995; Jiang et al., 1994; Park, et al., 1992) and prostanoids, from the endothelium and with a functional modulation of K+ channels (Wadsworth, 1994). Oxygen deprivation is reported to activate phospholipase A2 and inhibit NO synthase (Harrison et al., 1991; Rengasamy et al., 1996); thus, hypoxia is possible to alter neural and humoral regulations mediated by prostanoids and NO.
Cerebral arterial tone is regulated by NO derived from the perivascular nerve and endothelium (Toda et al., 1990; 1992; 1993a). Neurogenic and endothelial NO plays important roles in reducing cerebral vascular resistance and increasing blood flow (Faraci, 1992; Okamura et al., 1995; Suzuki et al., 1993; Toda, 1994; Toda et al., 1993). Hypoxia is expected to modulate the physiological actions of NO and thus impair cerebral circulation. Therefore, the present study was undertaken to determine the effect of hypoxia on the response mediated by NO from the nerve and endothelium in isolated canine cerebral arteries and to elucidate the way of antagonism to hypoxic actions in neurogenic response.
METHODS
The Studies Review Board at Shiga University of Medical Sciences approved the use of canine blood vessels in this study.
Twenty four mongrel dogs of either gender, weighing 7 to 15 kg, were killed by bleeding from the carotid arteries under pentobarbital anesthesia (50 mg/kg, intraperitoneally). The brain was rapidly removed, and middle cerebral arteries were isolated. The arteries were helically cut into strips of approximately 20-mm long. The specimen was vertically fixed between hooks in a muscle bath containing the modified Ringer-Locke solution, which was maintained at 37 ± 0.3°C and aerated with a mixture of 95% O2 and 5% CO2. The hook anchoring the upper end of the strips was connected to the lever of a force-displacement transducer. The resting tension was adjusted to 1.5 g which is optimal for inducing the maximal contraction. Constituents of the solution were as follows (mmol/L): NaCl 120, KCl 5.4, CaCl2 2.2, MgCl2 1.0, NaHCO3 25.0, and dextrose 5.6. The pH of the solution was 7.40 to 7.43 under control condition and hypoxia. Before the start of experiments, all of the strips were allowed to equilibrate for 60 to 90 minutes in the bathing media, during which time the fluid was replaced every 10 to 15 minutes.
Some of the arterial strips were placed between stimulating electrodes. The endothelium was removed by gently rubbing the intimal surface with a cotton ball. Transmural electrical stimulation at a frequency of 5 Hz for 40 seconds that produces submaximal and reproducible responses (Toda et al., 1991) was applied every 10 minutes, until the response was stabilized. Normal aerating gas (95% O2 and 5% CO2) was replaced with anoxic gas (95% N2 and 5% CO2) and then returned to the normal gas (reoxygenation); the time course of the altered responses to electrical stimulation was obtained. In some experiments, normal aerating gas was replaced with air including 5% CO2. At the end of each series of experiments, papaverine (10−4 M) was applied to attain the maximal relaxation. Effects of antagonists were evaluated in the response of strips treated for 30 minutes.
Isometric mechanical responses were displayed on an ink-writing oscillograph. The contractile response to 30 mmol/L K+ was obtained first, and the preparations were repeatedly washed and equilibrated. The endothelium was reserved for studies on nicotine, substance P, and NO. Nicotine (10−4 mol/L), substance P (10−8 mol/L), and NO (10−7 mol/L), which produced moderate relaxations, were successively applied to the bathing media and the order of drug application was randomized. Papaverine (10−4 mol/L) was applied at the end to attain the maximal relaxation. Relaxant responses to transmural electrical stimulation and the agonists relative to those caused by papaverine were presented. Preparations had been exposed for about 40 minutes to the bathing media with 95% N2 and 5% CO2 (hypoxic media) or 95% air and 5% CO2, before the response to the vasodilator agents was obtained. The partial O2 pressure in the control media (488 to 575 mm Hg) was decreased rapidly to 112 to 156 mm Hg and 30 to 52 mm Hg by exposure to air including 5% CO2 and the hypoxic media, respectively. Endothelial integrity was verified by a marked relaxation caused by substance P (10−7 mol/L).
Results shown in the text and figures are expressed as mean values ± SD. All reported n values refer to the number of strips from different dogs. Statistical analyses were made using the Student's paired and unpaired t-tests or the Tukey's test after one-way analysis of variance. Drugs used were nicotine (base) (Nacalai Tesque, Kyoto, Japan), substance P (Peptide Institute, Minoh, Japan), indomethacin, superoxide dismutase, amiloride hydrochloride, 5-(N,N-dimethyl)-amiloride (Sigma, St. Louis, MO, U.S.A.), tetrodotoxin (Sankyo Co., Tokyo, Japan), prostaglandin(PG)F2α, (Pharmacia-Upjohn Co., Tokyo), and papaverine hydrochloride (Dainippon Co., Osaka, Japan). Responses to NO were obtained by adding the NaNO2 solution adjusted at pH2, (Furchgott, 1988) and the concentrations of NaNO2 solution were expressed as those of NO. Oxyhemoglobin was prepared by the method described by Martin et al. (1985).
RESULTS
Inhibition by hypoxia of the responses to nerve stimulation and substance P
In cerebral arterial strips denuded of the endothelium partially contracted with PGF2α, the abrupt change in aerating gas from 95% O2-5% CO2 to 95% N2-5% CO2 (hypoxia) produced a slowly-developing, moderate relaxation or a transient contraction followed by a sustained relaxation. Restoration to the normal gas (reoxygenation) after about 60 minutes of exposure to the hypoxic media contracted the arterial strips to the level identical to or lower than that before the application of hypoxia.
The relaxant response to transmural electrical stimulation at 5 Hz for 40 seconds produced a moderate relaxation that was abolished by tetrodotoxin (3 × 10−7 mol/L). The neurogenic response was attenuated by hypoxia. Typical recordings are illustrated inFig. 1. The inhibition was stabilized 30 to 40 minutes later and persisted. The quantitative data concerning the time-dependent inhibition by hypoxia of neurogenic relaxation are summarized in Fig. 2. Reoxygenation partially restored the response. A decrease of partial O2 pressure to approximately 120 mm Hg by changing the gas to air including 5% CO2 tended to reduce the response to nerve stimulation; however, the difference was not statistically significant. Fig. 3 shows the effect of air and N2 gas on the stimulation-induced relaxation.

Recordings of the relaxant response to transmural electrical stimulation (5 Hz, 40 seconds) of a middle cerebral arterial strip denuded of the endothelium as affected by hypoxia. The strip was partially contracted with PGF2α, (5 × 10−8 mol/L). Dots represent the application of electrical stimulation. TTX, 3 × 10−7 mol/L tetrodotoxin; PA, 10−4 mol/L papaverine that produced the maximal relaxation.
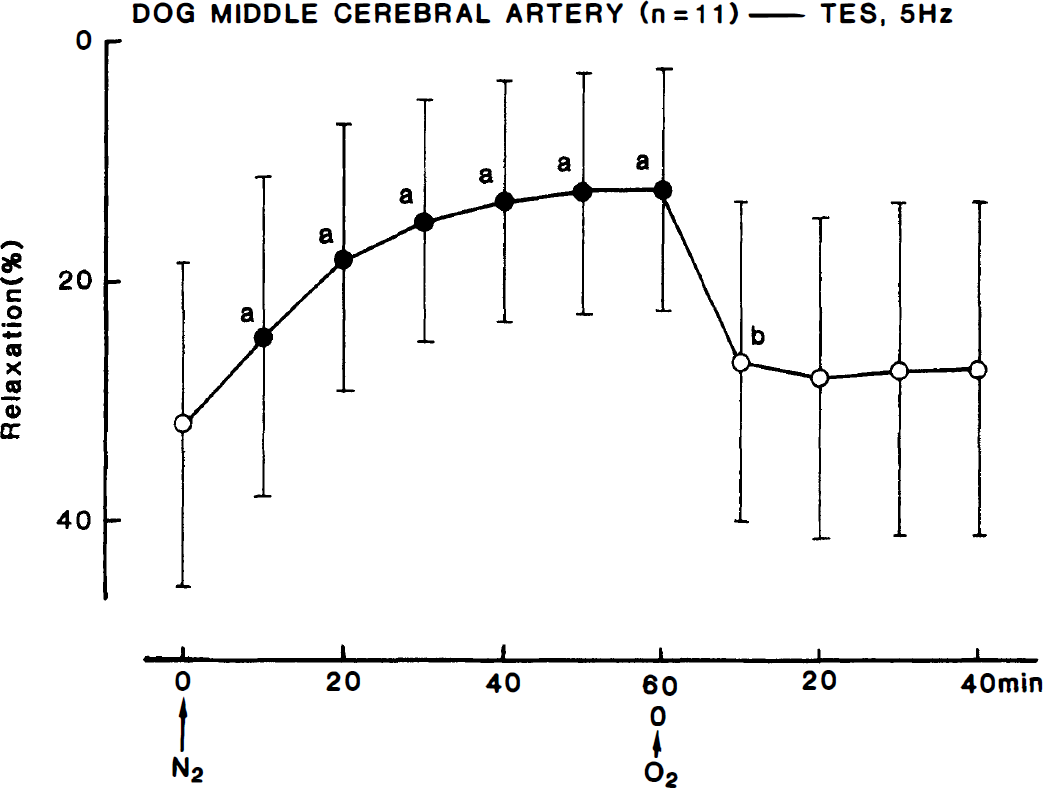
Time-course of the modification by hypoxia and reoxygenation of the relaxant response to transmural electrical stimulation (5 Hz) of middle cerebral arterial strips contracted with PG F2α. Relaxations induced by 10−4 mol/L papaverine were taken as 100%. Significantly different from zero, aP < 0.001; bP < 0.02; n denotes the number of strips from separate dogs. Vertical bars denote SD.
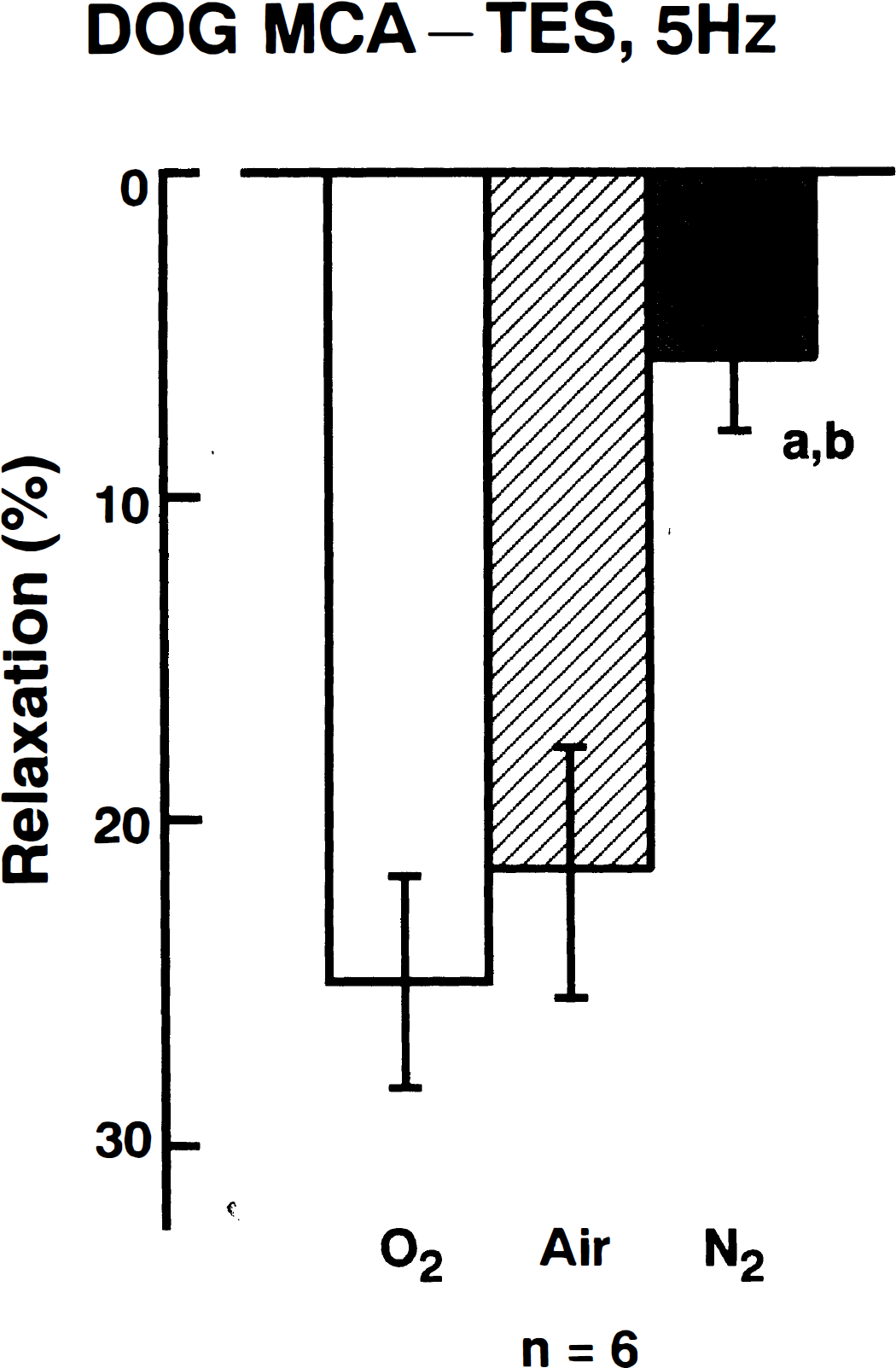
Effects of different gases (partial O2 pressures: O2, 488 to 560 mm Hg; air, 112 to 156 mm Hg, and N2, 39 to 52 mm Hg) on the relaxant response to transmural electrical stimulation (TES, 5 Hz) of middle cerebral arterial strips contracted with PGF2α. Relaxations induced by 10−4 mol/L papaverine were taken as 100%. The values were obtained at 30 to 40 minutes of exposure to the gas. Significantly different from O2 (open column), aP < 0.01; significantly different from air, bP < 0.01 (Tukey's method); n denotes the number of strips from separate dogs. Vertical bars represent SD.
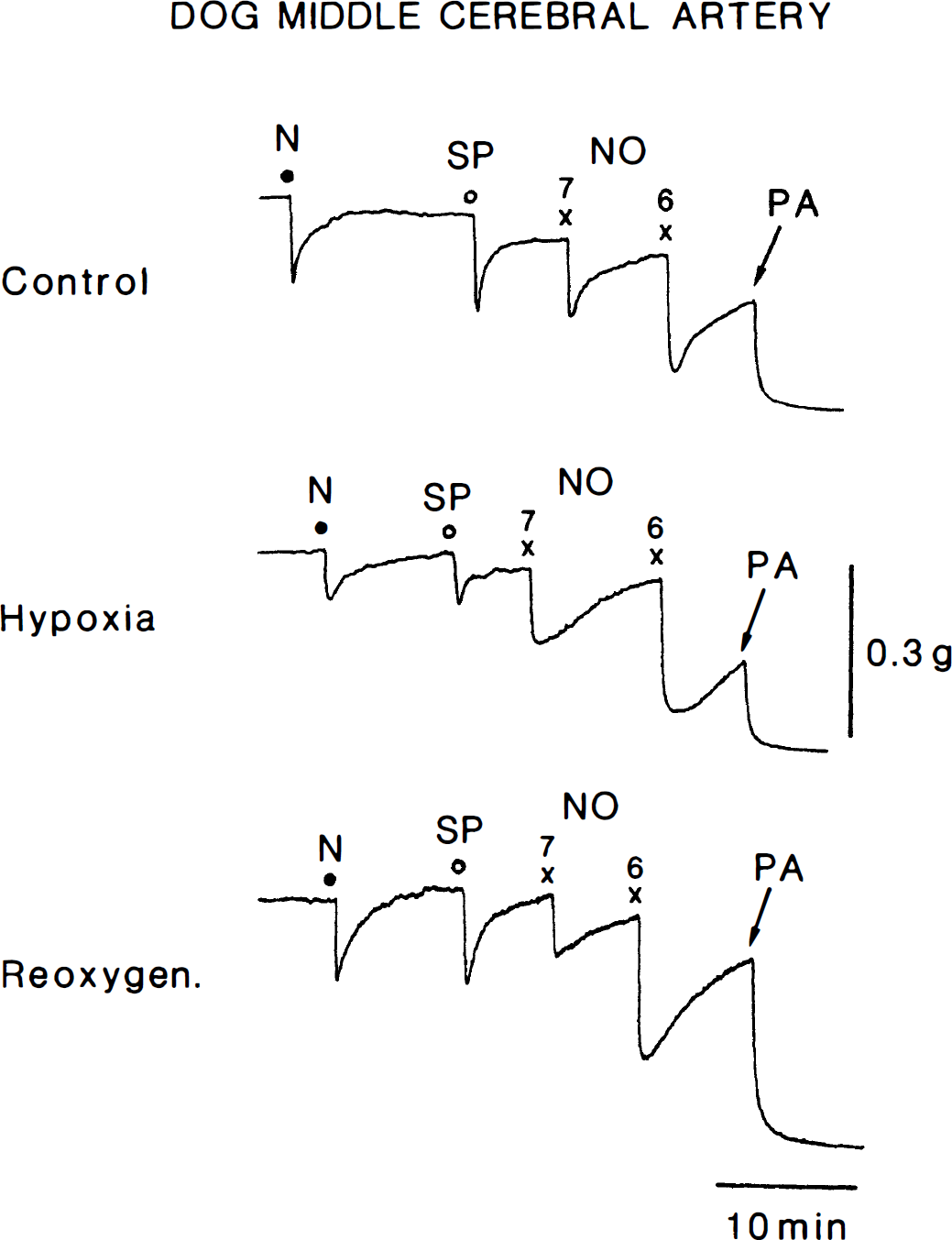
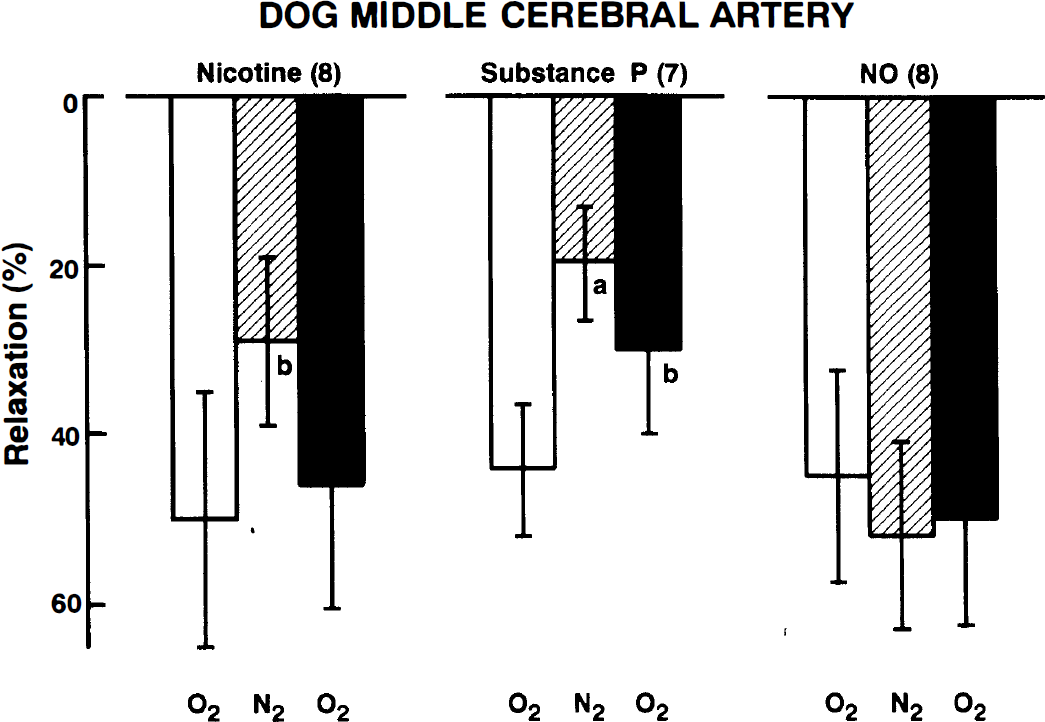
Hypoxia for about 30 minutes also attenuated the relaxant response to nicotine (10−4 mol/L) and substance P (10−8 mol/L) in middle cerebral arterial strips with the endothelium under treatment with 10−6 mol/L indomethacin (Fig. 4). Under the experimental conditions used, these agents have been reported to produce the relaxation mediated by NO derived from the perivascular nerve and endothelium (Toda et al., 1993a; 1990; 1992). Reoxygenation tended to reverse the response. Conversely, the response to exogenously applied NO (10−7 mol/L) was not influenced by hypoxia. Actual recordings of the response to these vasodilator agents in oxygenated and hypoxic media are shown in Fig. 5. These responses were abolished by treatment with 1.6 × 10−5 mol/L oxyhemoglobin (n = 3).
Effects of hypoxia (N2) and reoxygenation on the relaxant response to nicotine (10−4 mol/L), substance P (10−8 mol/L), and NO (10−7 mol/L) of middle cerebral arterial strips contracted with PGF2α. Relaxations induced by 10−4 mol/L papaverine were taken as 100%. The values were obtained at 30 to 40 minutes of exposure to hypoxia and 30 to 40 minutes after reoxygenation. Significantly different from control (open column), aP < 0.01; bP < 0.05 (Tukey's test). Numbers in parentheses indicate the number of strips from separate dogs. Vertical bars represent SD.
Relaxant responses to nicotine (N, 10−4 mol/L), substance P(SP, 10−8 mol/L), and NO (10−7 and 10−6 mol/L) of a middle cerebral arterial strip contracted with PGF2α in control, hypoxic, and reoxygenation media. PA, 10−4 mol/L papaverine that produced the maximal relaxation.
Modification by amiloride, dimethyl-amiloride, and superoxide dismutase of the effect of hypoxia
The addition of amiloride (10−4 mol/L), an inhibitor of Na+-H+ exchange, relaxed the arterial strips but did not significantly reduce the response to electrical nerve stimulation (Fig. 6). Under treatment with amiloride, the inhibitory effect of hypoxia on the neurogenic response was markedly reduced (18.8 ± 15.4% inhibition, n = 6, 30 minutes of incubation in hypoxic media, Fig. 6), compared with that in nontreated strips (55.2 ± 20.2% inhibition, Fig. 2). Conversely, in the strips in which the response to electrical stimulation was depressed by hypoxia (57.0 ± 4.1% inhibition, n = 4, middle panel of Fig. 6), the addition of amiloride significantly potentiated the response (81.3 ± 22.4% increase; n = 4, P < 0.05). Treatment with 10−5 mol/L dimethyl-amiloride, another Na+- H+ exchange inhibitor, tended to attenuate the relaxation induced by electrical nerve stimulation, the difference being statistically insignificant (right panel of Fig. 6). Dimethyl-amiloride prevented the inhibition by hypoxia of the neurogenic response (13.4 ± 28.0% inhibition, n = 5, 30 minute of incubation in hypoxia).
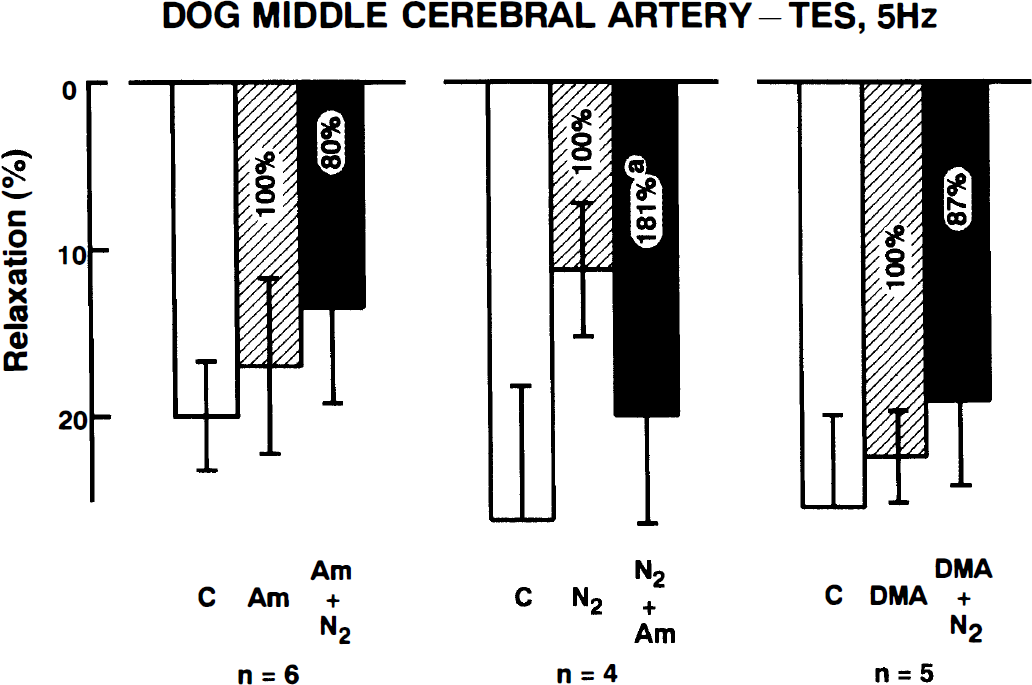
Modification by amiloride (10−4 mol/L) or dimethyl-amiloride (10−5 mol/L) of the inhibitory effect of hypoxia (N2) on the cerebroarterial response to transmural electrical stimulation (5 Hz). The ordinate represents relaxations relative to those induced by 10−4 mol/L papaverine. The neurogenic responses before and 30 minutes after exposure to hypoxia are presented. Numbers in the columns represent the data obtained by paired comparison. In the left and right panels, hypoxia did not significantly inhibit the response to nerve stimulation. C represents the response in the control media. Under hypoxic conditions in the middle panel, amiloride significantly potentiated the neurogenic response (n = 4, aP < 0. 05). n denotes the number of strips from separate dogs. Vertical bars represent SD.
Nicotine-induced relaxations were not inhibited by treatment for 30 minutes with amiloride (10−4 mol/L) (42.8 ± 8.6 to 41.2 ± 9.6%, n = 6). Under amiloride treatment, hypoxia did not inhibit the response to nicotine. (6.8 ± 12.1% inhibition, n = 6), whereas in non-treated strips, the response was depressed by 41.1 ± 21.5% (n = 8, P < 0.001; Fig. 4).
To further analyze possible mechanisms of inhibitory action of hypoxia, we evaluated effects of superoxide dismutase (200 unit/mL) on the response to electrical nerve stimulation at 5 Hz under hypoxia. Superoxide dismutase did not alter the tone of arterial strips (n = 6) nor the response to nerve stimulation under control condition (28.7 ± 6.5 versus 27.8 ± 6.4%, n = 6). In the presence of superoxide dismutase, hypoxia depressed the neurogenic response during 30 minutes incubation from 27.8 ± 6.4 to 9.0 ± 5.2% (n = 6, P < 0.001; unpaired Student's t-test), the inhibitory effect being comparable to that in the absence of superoxide dismutase (75.1 ± 14.8, n = 6, versus 55.2 ± 20.2%, n = 11; Fig. 2).
Modification by acidosis of the inhibitory effect of hypoxia
The addition of 2.6 mL of 0.1 N HCl solution reduced the pH of bathing media from 7.421 to 7.116 and 7.110 (n = 3) 40 and 120 minutes later, respectively, but did not change the partial O2 pressure under control condition and hypoxia. Acidosis relaxed the arterial strips. However, the relaxation induced by electrical nerve stimulation at 5 Hz was not significantly altered, mean values before and 40 minutes after the addition of HCl solution being 30.2 ± 13.2 and 28.0 ± 12.0%, respectively. In the acidic solutions, the neurogenic response was reduced only slightly by hypoxia, as illustrated in Fig. 7. Significant inhibition was observed only 40 minutes later. Reoxygenation completely reversed the response. Inhibitions by exposure for 40 minutes to hypoxia of the response to nerve stimulation in control and acidic media were 62.1 ± 17.8 (n = 11) and 17.6 ± 14.9% (n = 5), respectively; the difference was statistically significant (P < 0.001, unpaired Student's t-test). The typical tracings of the response under hypoxia in acidic and control fluids are shown in Fig. 8. Prevention by acidosis of the inhibitory effect of hypoxia is clearly seen in the same preparation. Similar results were also obtained in the same strips from three additional dogs.
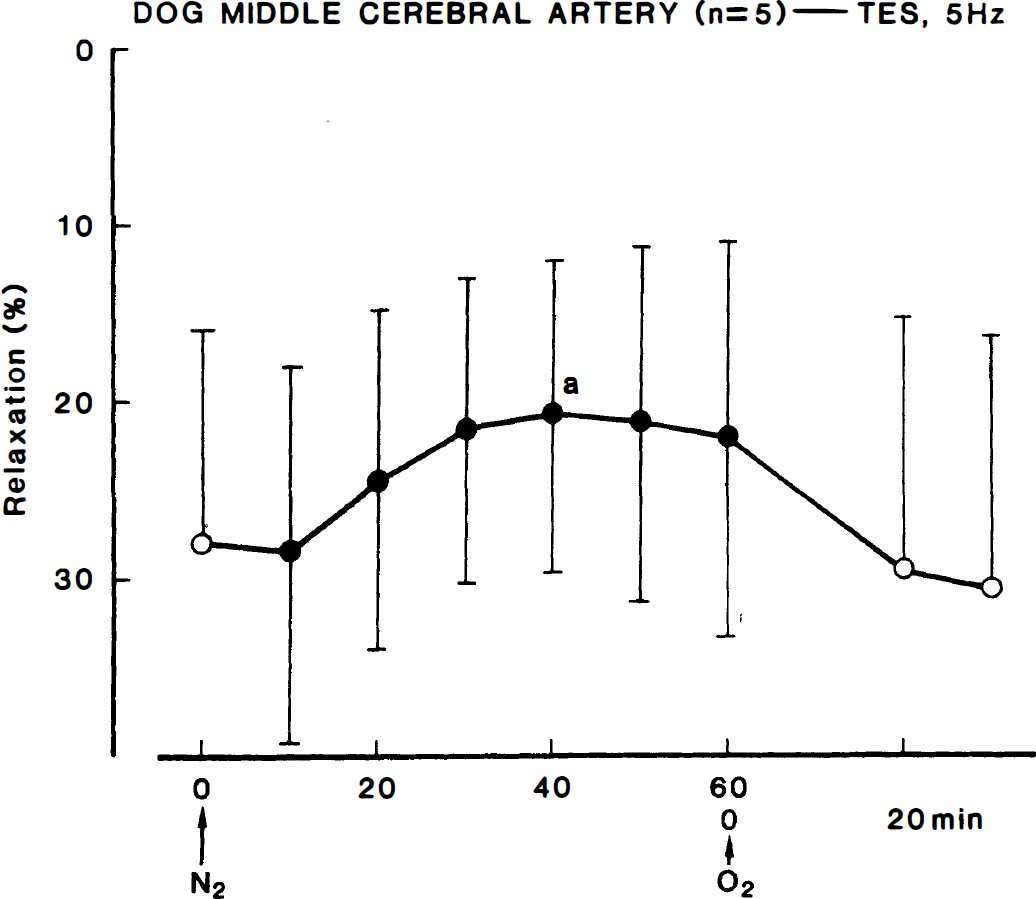
Time-course of the modification by hypoxia and reoxygenation of the response to transmural electrical stimulation (5 Hz) of middle cerebral arterial strips in acidosis. The strips were partially contracted with PGF2α. Relaxations induced by 10−4 mol/L papaverine were taken as 100%. Significantly different from zero, aP < 0.05; n denotes the number of strips from separate dogs. Vertical bars denote SD.
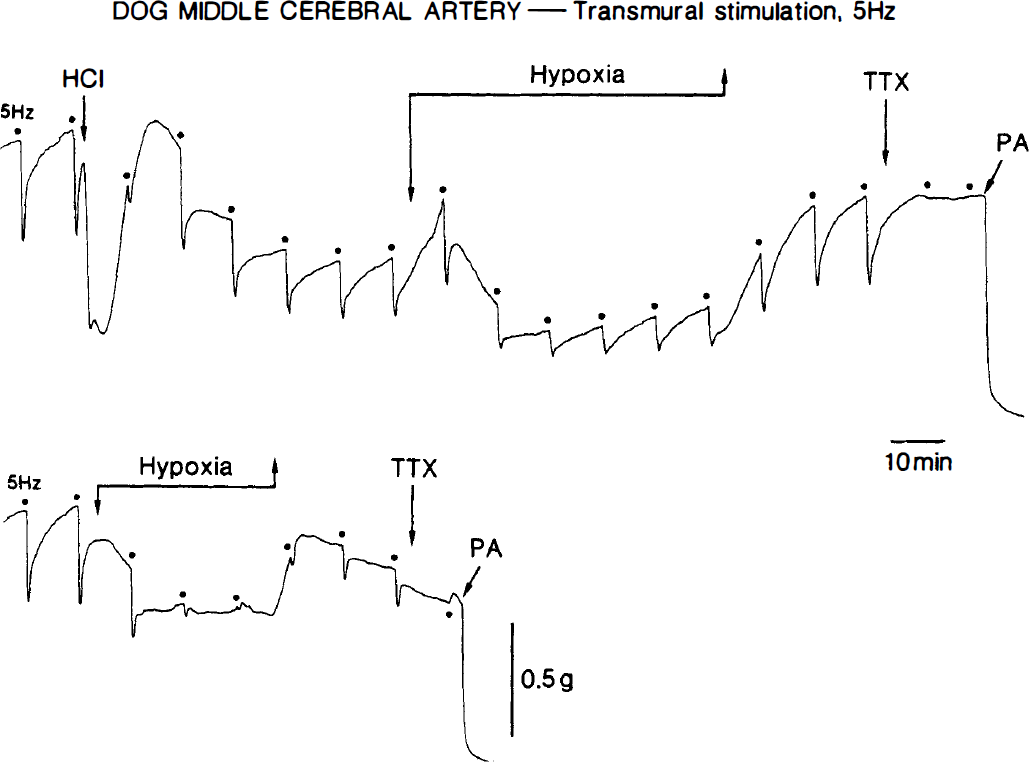
Tracings of the response to transmural electrical stimulation at 5 Hz of a middle cerebral arterial strip as affected by hypoxia and reoxygenation under acidic (HCl, upper tracing) and control media (lower). The strip was partially contracted with PGF2α (10−6 mol/L). After the relaxation caused by papaverine (PA, 10−4 mol/L) leveled off, the strip was repeatedly rinsed with control media and stabilized for 60 minutes (upper tracing). Then, the strip was contracted by PGF2α again, and the effect of hypoxia on neurogenic responses was evaluated in control media for comparison (lower tracing). TTX represents 3 × 10−7 mol/L tetrodotoxin. Dots denote the application of electrical stimulation.
DISCUSSION
The present study showed that relaxant responses to nerve stimulation, by electrical pulses or nicotine and substance P, were moderately attenuated in cerebral arterial strips exposed to hypoxia, whereas the response to exogenous NO was unaffected. The relaxation caused by nerve stimulation has been determined to be mediated by NO or NO analogs, such as S-nitrosocysteine, (Toda et al., 1992; 1996) liberated as neurotransmitter, and the substance P-induced relaxation is associated with NO derived from the endothelium (Toda et al., 1993a). Similar inhibition of the relaxant response to electrical stimulation of autonomic dilator nerves has been observed in isolated human and rabbit corpus cavernosum strips exposed to hypoxic media (Kim et al., 1993). Endothelium-dependent, NO-mediated relaxations in porcine pulmonary venous rings are abolished by hypoxia (Feletou et al., 1995).
Studies on isolated tissues have usually been performed in oxygenated media with high partial O2 pressure, because O2 delivery to the tissue without hemoglobin in vitro is less effective compared with in vivo condition. To determine whether the difference in the responses to nerve stimulation under control and hypoxic conditions was attributable to either higher or lower partial O2 pressure as compared with that under physiological conditions in vivo (about 100 mm Hg), additional studies were performed in the strips exposed to control media (about 550 mm Hg) and those aerated with 95% air + 5% C O2 (about 120 mm Hg) and 95% N2 + 5% CO2 (about 40 mm Hg). The results indicated that neurogenic relaxation was not significantly affected by lowering the partial O2 pressure from 550 to 120 mm Hg but markedly reduced by a pressure decrease to 40 mm Hg. These findings suggest that hypoxia interferes with the synthesis/release of NO or promotes the degradation of NO.
Hypoxia-reoxygenation increases the efflux of superoxide anion, which injures cultured bovine pulmonary artery endothelial cells (Terada, 1996). However, in the present study, treatment with superoxide dismutase did not prevent the inhibition of the response of canine cerebral arteries to nerve stimulation by hypoxia, suggesting that accelerated inactivation of NO by superoxide anions is not involved. In the rabbit corpus cavernosum? hypoxia prevents cyclic guanosine monophosphate accumulation induced by nerve stimulation and inhibits NO synthase activity (Kim et al., 1993). On the basis of Km values for O2 of neuronal and endothelial NO synthases, Rengaswmy and Johns (1996) have indicated that the reduced production of NO from L-arginine under pathophysiological conditions involves a decrease in tissue O2 concentration and that the neuronal enzyme is more susceptible to reduced O2 concentration than the endothelial enzyme. However, this may not be the case for the hypoxia-induced inhibition of neurogenic response in the present study, because O2 concentration in the hypoxic media is still higher than Km for oxygen of neuronal NO synthase (Rengasamy et al., 1996).
The hypoxia-induced inhibition of the response to electrical nerve stimulation was prevented by treatment of the arterial strips with amiloride or dimethylamiloride, inhibitors of Na+-H+ exchange (Kleyman et al., 1988; Meng et al., 1991), and the neurogenic response depressed by hypoxia was reversed by amiloride. Hypoxia decreases intracellular pH. Production of H+ by anoxic cells is suggested to be a protective feedback mechanism (Penttila et al., 1974). It has been reported that restoration of normal pH by Na+-H+ exchange activation enhances ischemia and reperfusion injury in hepatocytes and ventricular cells (Bond et al., 1994; Gores et al., 1988; More et al., 1995). Monensin, a Na+-H+ ionophore (Lake et al., 1987; Reed, 1979), potentiates hepatocyte killing by chemical hypoxia with KCN (Gores et al., 1988). Intracellular acidosis caused by Na+-H+ exchange inhibition is suggested to delay a loss of cell viability and a release of arachidonic acid (Harrison et al., 1991). Activation by hypoxia of phospholipase A2 may cause alterations of plasma membrane phospholipid composition and impair cellular functions, including the decreased Ca2+ influx, reduced NO synthase activation, etc. Hypoxia depolarizes the membrane of pulmonary artery endothelial cells and decreases intracellular Ca2+ (Stevens et al., 1994). The Ca2+ influx is inhibited by hypoxia in rabbit basilar arteries (Pearce et al., 1992). Whether this is also the case in vasodilator nerves has not been determined. However, the fact that the neurogenic response depressed by hypoxia was reversed only partially by reoxygenation suggest the involvement of impaired nerve functions.
Conversely, Meng and Pierce (1991) have suggested that an increase in intracellular Na+ by reperfusion in the isolated rat ventricular wall exposed to ischemia represents a crucial or primary step for the development of cardiac contractile dysfunction, because the ischemia-reperfusion injury was protected by treatment with dimethyl-amiloride. In the present study, acidosis clearly eliminated the inhibitory effect of hypoxia on the neurogenic response of cerebral arteries, as seen in hepatocytes and ventricular cells (Bond et al., 1994; Gores et al., 1988; More et al., 1995). Therefore, the ability of vasodilator nerve innervating the cerebral artery to synthesize and liberate NO is protected from hypoxia-induced impairment possibly by intracellular acidosis rather than interference with elevated intracellular Na+.
Ischemia and reperfusion injury in the brain may be associated with depressed vasodilator function of cerebral arteries and arterioles. The present study provided evidence suggesting that the vasodilator response to NO derived from the nerve is reserved by Na+-H+ exchange inhibition or extracellular acidosis under hypoxia. It seems that hypoxia-induced inhibition of the response to nerve stimulation is associated with an impaired membrane function caused by restoration of normal pH by Na+-H+ exchange. Further study is required to determine whether similar mechanisms underlying increased viability and restored function by treatment with amiloride or intracellular acidosis in ventricular, pulmonary, and hepatic cells exposed to hypoxia and reoxygenation (Gores et al., 1988; Harrison et al., 1991; Karmazyn, 1988; Meng et al., 1991; More et al., 1995) are also involved in perivascular neurons of the cerebral artery.