
Abstract
Spreading depression induces tolerance to ischemic injury, and ischemic tolerance has been associated with expression of heat shock proteins (Hsp). Here we examine Hsp27 expression after KCl-induced spreading depression. Twenty-minute cortical KCl application induced Hsp27 immunoreactivity in glial fibrillary acidic protein-positive astrocytes of the ipsilateral neocortex. Systemic administration of MK-801 (3 mg/kg) suppressed KCl-induced Hsp27 expression in the parietal cortex. Astrocytes in the posterior cingulate and retrosplenial cortex did not express Hsp27 after KCl application but did express Hsp27 after systemic administration of high dose MK-801 (9 mg/kg). Whereas Hsp27 was usually observed in all layers of the parietal cortex after 5-minute application of KCl, in 2 of 6 rats, Hsp27 was seen in clusters of astrocytes or in astrocytes in the superficial layers I to III of the parietal cortex. We conclude that (1) cortical application of KCl triggered Hsp27 astrocytic expression; (2) astrocytes in the cingulate and retrosplenial cortex responded differently compared with astrocytes of the parietal cortex; (3) Hsp27 expression progressed from small clusters of astrocytes throughout superficial layers of the cortex that joined and recruited astrocytes in deeper layers; (4) several mechanisms induced Hsp27 astrocytic expression. We propose that Hsp27 is involved in spreading depression-induced ischemic tolerance through protection of astrocyte function.
Application of potassium chloride to the cerebral cortex induces cortical spreading depression (Kraig et al., 1991; Bonthius et al., 1995) and protects against subsequent transient focal cerebral ischemia (Kobayashi et al., 1995; Matsushima et al., 1996). A similar resistance to ischemic injury has also been observed after a mild ischemic preconditioning treatment (Kitagawa et al., 1991). A mild global ischemic insult reduced the extent of hippocampal damage after a subsequent severe global ischemic insult (Kitagawa et al., 1991; Liu et al., 1992) and significantly decreased the infarct size after permanent middle cerebral artery occlusion (Simon et al., 1993). In addition, a brief focal ischemic episode has been shown to be protective against subsequent severe global ischemic injury (Glazier et al., 1994). This 'ischemic tolerance' phenomenon has been associated repeatedly with the synthesis of heat shock proteins (Kirino et al., 1991; Kitagawa et al., 1991; Nishi et al., 1993; Simon et al., 1993; Glazier et al., 1994) but conclusive evidence for an association remains elusive.
Heat shock proteins are highly conserved proteins that are induced by a wide variety of stresses, including oxidative and ischemic injury (Currie and White, 1981; Nowak, 1985; Dienel et al., 1986; Vass et al., 1988; Gonzalez et al., 1991; Sharp et al., 1991b;Simon et al., 1991). Heat shock proteins contribute to the cellular repair processes by refolding denatured proteins (Pelham, 1986) and participate as molecular chaperons in normal processes such as protein translocation and folding (Becker and Craig, 1994; Ryan and Jensen, 1995; Frydman and Hartl, 1996; Hartl, 1996; Schatz and Dobberstein, 1996).
Much of the attention in 'ischemic tolerance' and endogenous protective mechanisms has focused on the inducible 70-kd heat shock protein. The inducible member of the 70-kd heat shock protein family (Hsp70) has been suggested to participate in the protective mechanisms against ischemic injury to both the heart (Currie et al., 1988; Marber et al., 1995; Plumier et al., 1995; Plumier and Currie, 1996b) and brain (Kirino et al., 1991; Liu et al., 1992; 1993; Simon et al., 1993; Nishi et al., 1993; Glazier et al., 1994). While cortical spreading depression induced ischemic tolerance throughout the cerebral cortex, Hsp70 was only detected around the KCl application site (Kobayashi et al., 1995). It was concluded that Hsp70 was not involved in ischemic tolerance induced by cortical spreading depression. Similarly, brief repetitive middle cerebral artery occlusion decreased infarct size after a subsequent 100-minute occlusion (Chen et al., 1996). However, Hsp70 accumulation and degradation did not match exactly with the acquisition and decay of ischemic tolerance. These results did not exclude a role for other heat shock proteins such as the 27-kd heat shock protein (Hsp27) whose expression is known to increase cell resistance to oxidative injury (Mehlen et al., 1995). In fact, Hsp27 has been shown to be expressed after cerebral ischemia (Kato et al., 1994; 1995).
The purpose of this study was to examine and characterize the temporal and spatial distribution of Hsp27 expression after KCl-induced spreading depression. The effect of systemic administration of N-methyl-D-aspartate (NMDA) receptor antagonist, MK-801, a compound known to inhibit spreading depression was also examined. We conclude that KCl-induced cortical spreading depression triggered a complex astroglial response, i.e., Hsp27 immunoreactivity varied according to brain region and could be dissociated from neuronal activity as measured by Fos immunoreactivity.
MATERIALS AND METHODS
Animals
All the animals were treated in accordance to the guidelines set by the Canadian Council on Animal Care.
Potassium chloride application
Eighteen adult male Sprague-Dawley rats (250 to 300 g, Charles River, Montreal) were anesthetized with sodium pentobarbital (50 mg/kg intraperitoneally. The surface of the skull was exposed through a longitudinal incision in the skin. A rectangle of bone 3-mm wide and 5-mm long was removed from the right side of the skull. The dura mater was exposed. A cotton tip soaked in KCl (3 M) was applied to dura mater over the right frontoparietal cortex for 5 minutes (n = 6) or 20 minutes (n = 9). Then, the cranial bone was glued back using dental cement and the skin was sutured. At 2 hours (n = 3), 2 days (n = 3) or 4 days (n = 9) after surgery, the rats were killed and perfused via the aorta with 100 mmol/L phosphate buffer containing 2% paraformaldehyde and 0.2% glutaraldehyde (4°C). The brain was then removed and postfixed in 100 mmol/L phosphate buffer containing 2% paraformaldehyde. In three rats, the dura mater was exposed as described above but KCl was not applied. These sham-treated rats were killed 4 days after surgery and perfused as described above.
MK-801 treatment
Adult rats were injected intraperitoneally with 3 or 9 mg/kg of MK-801 (n = 3 for each dose) and killed 2 days later to determine the effect of MK-801 on Hsp27 expression. To reduce reactive gliosis, a dose of 3 mg/kg of MK-801 was injected intraperitoneally 30 minutes before a 20-minute cortical application of potassium chloride (Herrera and Cuello, 1992; Bonthius and Steward, 1993). At 2 days (n = 5) after surgery, MK-801-treated rats were killed and perfused as described above.
Fos and Hsp27 immunohistochemistry
Each brain was sectioned (50 μm) on a vibratome in ice-cold 100 mmol/L phosphate buffered saline (PBS; pH = 7.6). Immunohistochemistry was performed on free-floating sections. For Fos immunohistochemistry, sections were incubated in 0.3% H2O2/PBS, rinsed 3 times in PBS, and incubated 1 hour in 10% rabbit serum made in PBS. After 3 PBS washes, sections were incubated 2 days at 4°C in PBS containing a sheep polyclonal antiserum that recognizes c-Fos (1:20,000; Genosys Biotechnologies, The Woodlands, TX, U.S.A.). After three PBS rinses, sections were incubated in PBS containing a biotinylated rabbit polyclonal antibody raised against sheep IgG (1:500; Vector Laboratories Inc., Burlingame, CA, U.S.A.). Sections were then immersed 1 hour in avidin-biotin-horseradish peroxidase complex (1:1,000; Vector Laboratories Inc.). After PBS washes, sections were immersed in diaminobenzidine-tetrachloride (DAB, 0.05%; Sigma Chemical Co., St. Louis, MO, U.S.A.) made up in PBS containing 0.3 mg/100 mL glucose oxidase, 40 mg/mL ammonium chloride, and 200 mg/100 mL β-D(+)glucose (Sigma Chemical Co.).
For Hsp27, immunohistochemistry sections were rinsed 3 times in 100 mmol/L phosphate-buffered saline and then incubated 30 minutes in 3% hydrogen peroxide in PBS. After 3. rinses in PBS, sections were incubated overnight in PBS containing 2% goat serum and a primary rabbit polyclonal antibody raised against mouse Hsp25 (1:5,000; StressGen Biotechnologies Corp., Victoria, British Columbia, Canada). This primary antibody has been shown to crossreact specifically with the rat Hsp27 (Plumier et al., 1996a). The following day, after 3 PBS washes, sections were incubated for 1 hour in PBS containing a biotinylated secondary goat antibody raised against rabbit IgG (1:400; Vector Laboratories Inc.) followed by washes. Hsp27 immunoreactivity was then revealed using the avidin-biotin-DAB reaction as described above for c-Fos immunostaining.
Immunohistochemically stained sections were mounted onto coated slides, air dried overnight and coverslipped. Some immunostained sections were counter-stained with cresyl violet (0.1%).
Immunofluorescence
For glial fibrillary acidic protein (GFAP)/HSP25 double labeling, sections were incubated I hour in PBS containing 10% horse and 10% goat serum. After three PBS rinses, sections were incubated overnight in PBS containing the rabbit polyclonal HSP25 antibody and the mouse monoclonal GFAP antibody. After three PBS washes, sections were incubated with Fluorescein-conjugated horse antibody raised against mouse IgG (1:30; Vector Laboratories Inc.) and Texas Red-conjugated goat antibody raised against rabbit IgG (1:300; Vector Laboratories Inc.) to detect the primary antibodies. Immunofluorescent sections were examined using a Zeiss Axioplan microscope equipped with an incident-light fluorescence illuminator and filters suitable for viewing the fluorochromes fluorescein and Texas Red.
Image processing
Immunostained sections were digitalized using a JVC TKFF7 300U video camera attached to a dissecting scope or a orthoplan Leitz microscope. Images were captured on a Macintosh computer (Apple Inc., Cupertino, CA) using Adobe Photoshop 3.0.5 software (Adobe Systems Inc., Mountain View, CA, U.S.A.). Prints were made using a 300 dpi continuous tone dye-sublimation 8600 XLS Kodak printer (Eastman Kodak, Rochester, NY, U.S.A.).
Semiquantitative densitometry
Immunostained sections were digitalized as described above and analyzed using NIH Image 1.60 software. Densitometry of Hsp27 immunostaining was measured on three sections for each animal in the ipsilateral and contralateral parietal cortices and in the thalamus. Final optical densities for the parietal cortex were obtained by subtraction of corresponding densitometry measurements of the thalamus, used as background values. Optical densities from MK-801-treated (3 mg/kg intraperitoneally, 30 minutes before KCl cortical application for 20 minutes, 2 day-survival) and nontreated rats were compared with one-way analysis of variance. Analyses were followed by Newman-Keuls tests.
RESULTS
Hsp27 immunoreactivity after application of KCl
Unilateral cortical application of KCl-induced Hsp27 immunoreactivity in the rat ipsilateral cerebral cortex (Fig. 1). Hsp27 immunoreactivity was observed in the frontoparietal cortex, entorhinal cortex and piriform cortex, but not in the retrosplenial cortex or in the posterior cingulate cortex. No Hsp27 immunoreactivity was observed in the contralateral cortex of KCl-treated rats or in the cerebral cortex of sham-operated rats. In the ipsilateral KCl-treated cortex, most of the immunoreactivity for Hsp27 appeared to be in astrocytes. At the site of KCl application, a few Hsp27 immunoreactive microglial cells were detected. Occasionally neurons on the border of the injured area showed Hsp27 immunoreactivity.
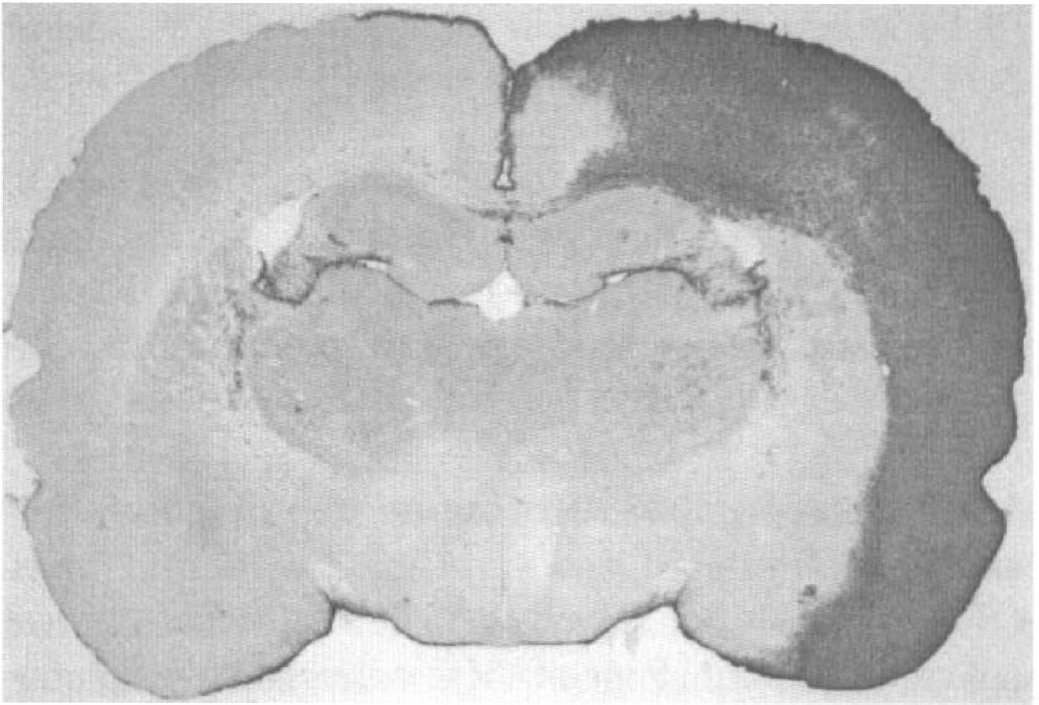
Hsp27 immunoreactivity in the rat cerebral cortex after potassium chloride application. Four days after the 20 minute application of KCl, Hsp27 immunoreactivity was observed in all cortical layers of the ipsilateral parietal, perirhinal, and piriform cortex. Note the absence of Hsp27 immunostaining in the retrosplenial cortex and in the contralateral cortex.
Hsp27- and GFAP-double immunofluorescence
Four days after unilateral cortical application of KCl, Hsp27-immunopositive cells in the frontoparietal cortex (Fig. 2A) were also GFAP-immunopositive (Fig. 2B). Similarly, all Hsp27-immunoreactive cells of the piriform cortex (Fig. 2C) were GFAP-immunoreactive (Fig. 2D). However, Hsp27-immunoreactivity distribution was restricted to the piriform cortex and was not detected in the amygdala (Fig. 2C) whereas GFAP-immunofluorescence was detected in both the piriform cortex and the amygdala (Fig. 2D). Hsp27 immunofluorescence illustrated the brushy appearance of the astrocytes with their numerous branching processes (Fig. 2E) while GFAP immunoreactivity labeled mainly cell bodies and major processes (Fig. 2F).
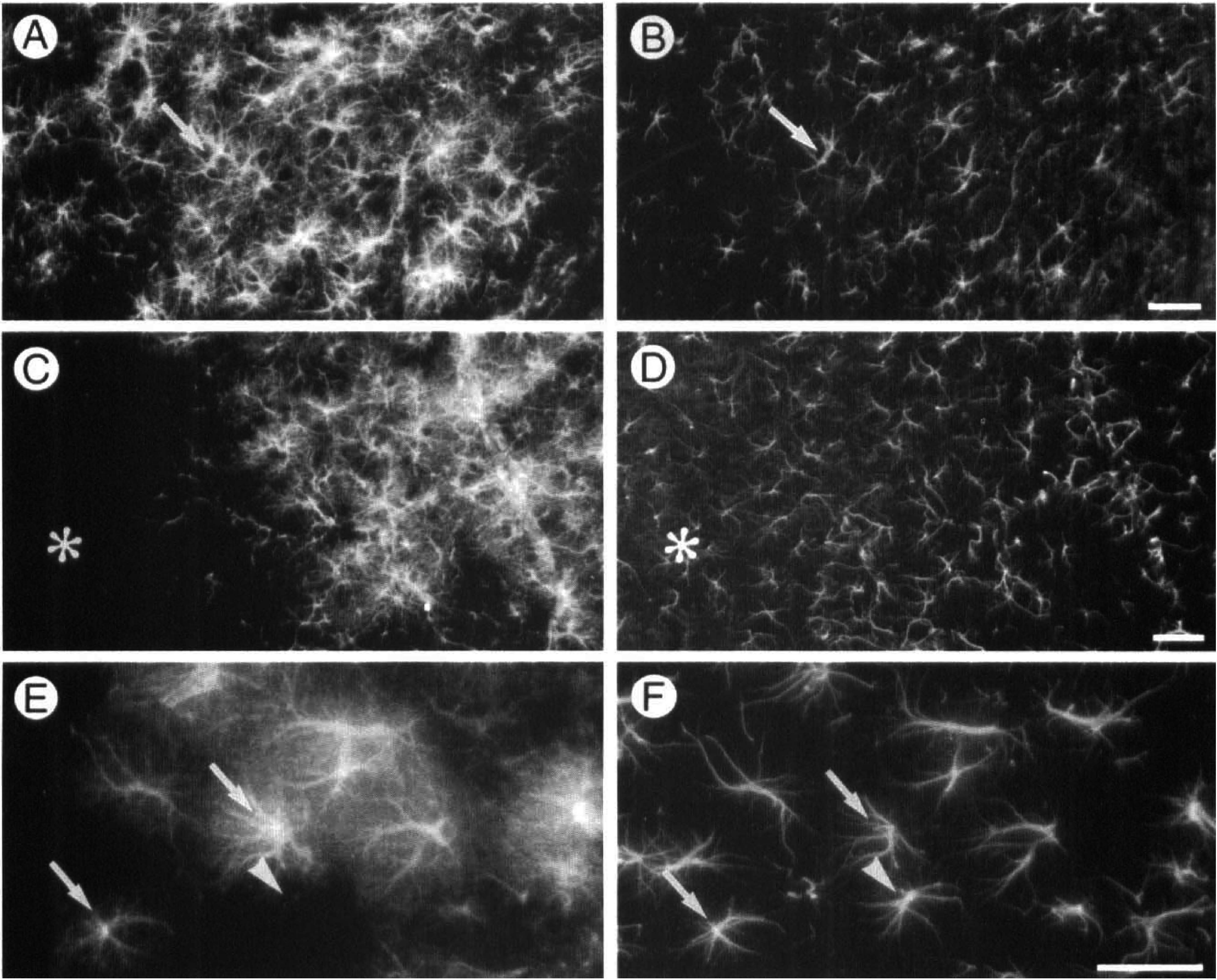
Hsp27 and glial fibrillary acidic protein (GFAP) immunoreactivity 4 days after cortical application of KCl. A Texas Red-conjugated antibody was used to detect the rabbit polyclonal antibody immunoreacting with Hsp27 (
Effect of MK-801 on KCl-induced Hsp27 immunoreactivity
Systemic administration of the NMDA-receptor antagonist MK-801 (3 mg/kg intraperitoneally) 30 minutes before cortical application of KCl reduced the extent of Hsp27 immunoreactivity in the ipsilateral parietal cortex (Fig. 3). Two days after cortical application of KCl, Hsp27 immunoreactivity in non-MK-801-treated animals was observed in all layers of the parietal cortex (Fig. 3A). Numerous astrocytes were Hsp27-immunoreactive (Fig. 3B). Systemic administration of MK-801 reduced the extent of Hsp27 immunoreactivity in the ipsilateral parietal cortex (Fig. 3C) with apparently fewer Hsp27-positive astrocytes (Fig. 3D). Semiquantitative analysis of optical densities of Hsp27 immunoreactivity in the parietal cortex (Fig. 4) revealed that (1) in non-MK-801-treated rats, KCl-induced Hsp27 immunoreactivity was significantly different between the contralateral and ipsilateral parietal cortices (16.5 ± 2.3 versus 73.3 ± 5.2 arbitrary units, respectively; P < 0.05); (2) in MK-801-treated rats, KCl-induced Hsp27 immunoreactivity was significantly different between the contralateral and ipsilateral parietal cortices (14.7 ± 5.2 versus 42.8 ± 6.6 arbitrary units, respectively; P < 0.05); (3) Hsp27 immunoreactivity was not different between contralateral cortices of non-MK-801-treated and MK-801-treated animals (16.5 ± 2.3 versus 14.7 ± 5.2 arbitrary units, respectively); (4) Hsp27 immunoreactivity was significantly different between ipsilateral cortices of non-MK-801-treated and MK-801-treated animals (73.3 ± 5.2 versus 42.8 ± 6.6 arbitrary units, respectively; P < 0.05).
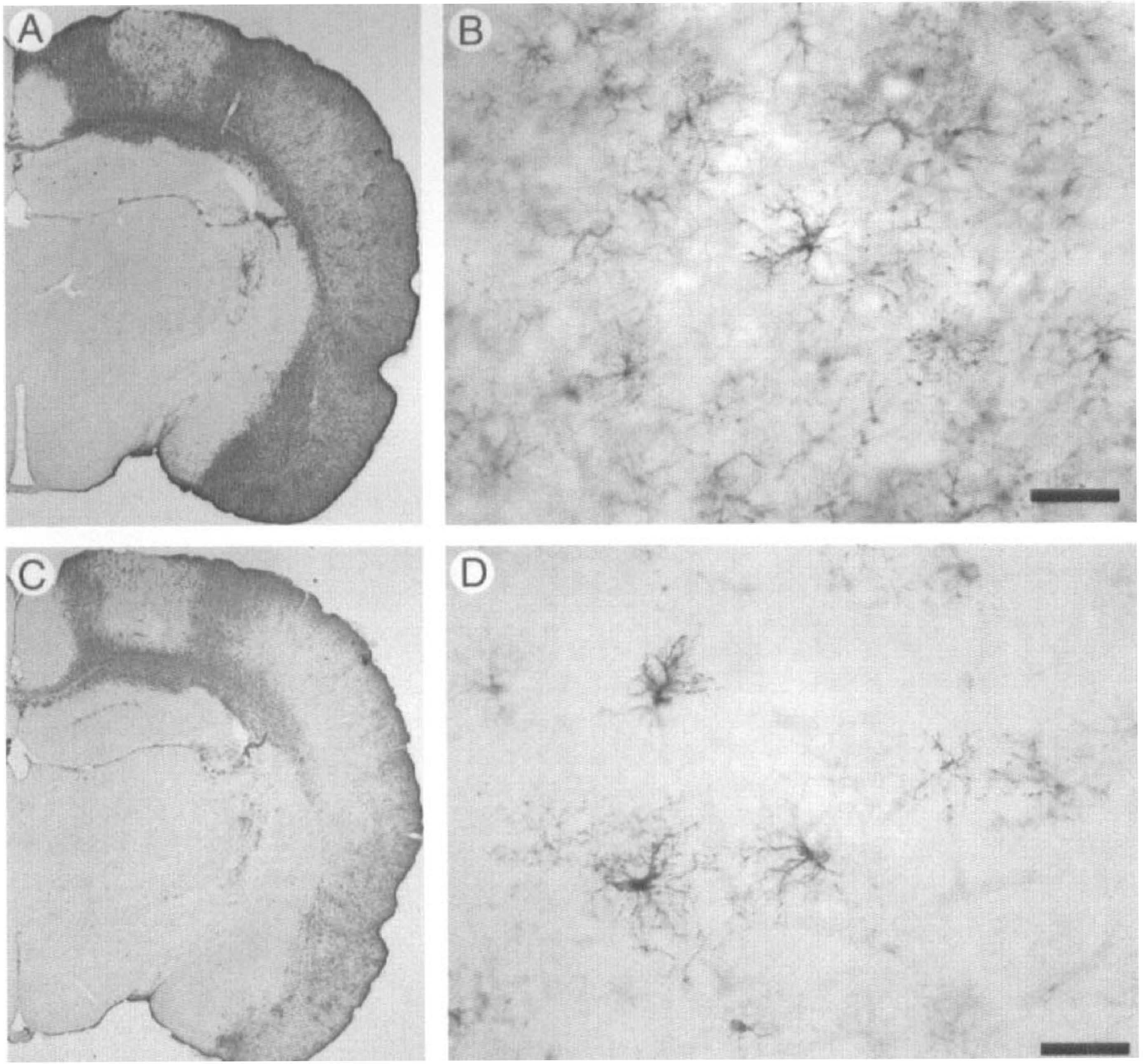
Effect of systemic administration of MK-801 (3 mg/kg) on the induction of Hsp27 immunoreactivity after KCl cortical application. In non-MK-801 treated rats, 2 days after the 20-minute cortical application of KCl, Hsp27 immunoreactivity was detected in all layers of the cortex, but not in the posterior cingulate or retrosplenial cortex
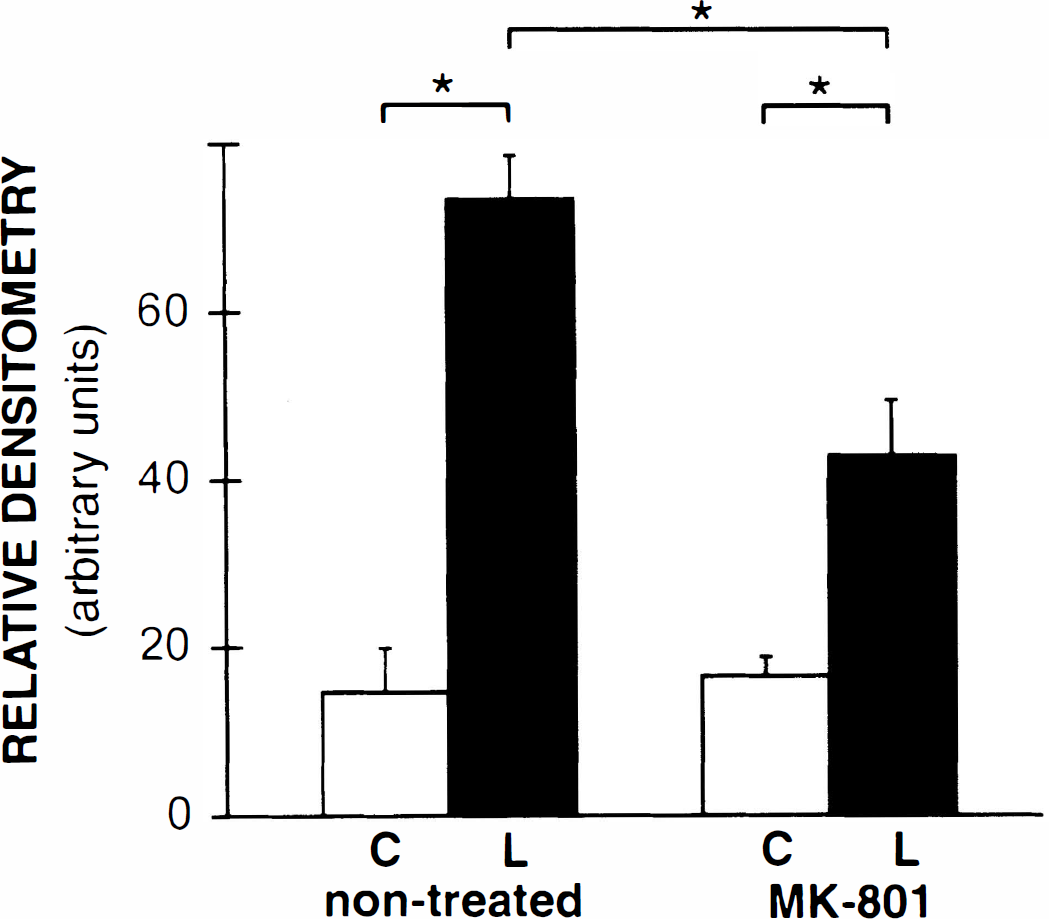
Semiquantitative analysis revealed that MK-801 treatment significantly reduced the intensity of Hsp27 immunoreactivity. Values indicated are means ± SEM (arbitrary units). C, contralateral parietal cortex; L, lesioned ipsilateral parietal cortex. *Significant differences in Hsp27 immunoreactivity between treatments using one-way analysis of variance and Newman-Keuls post hoc comparisons (P < 0.05).
Regional distribution of Hsp27 immunoreactivity
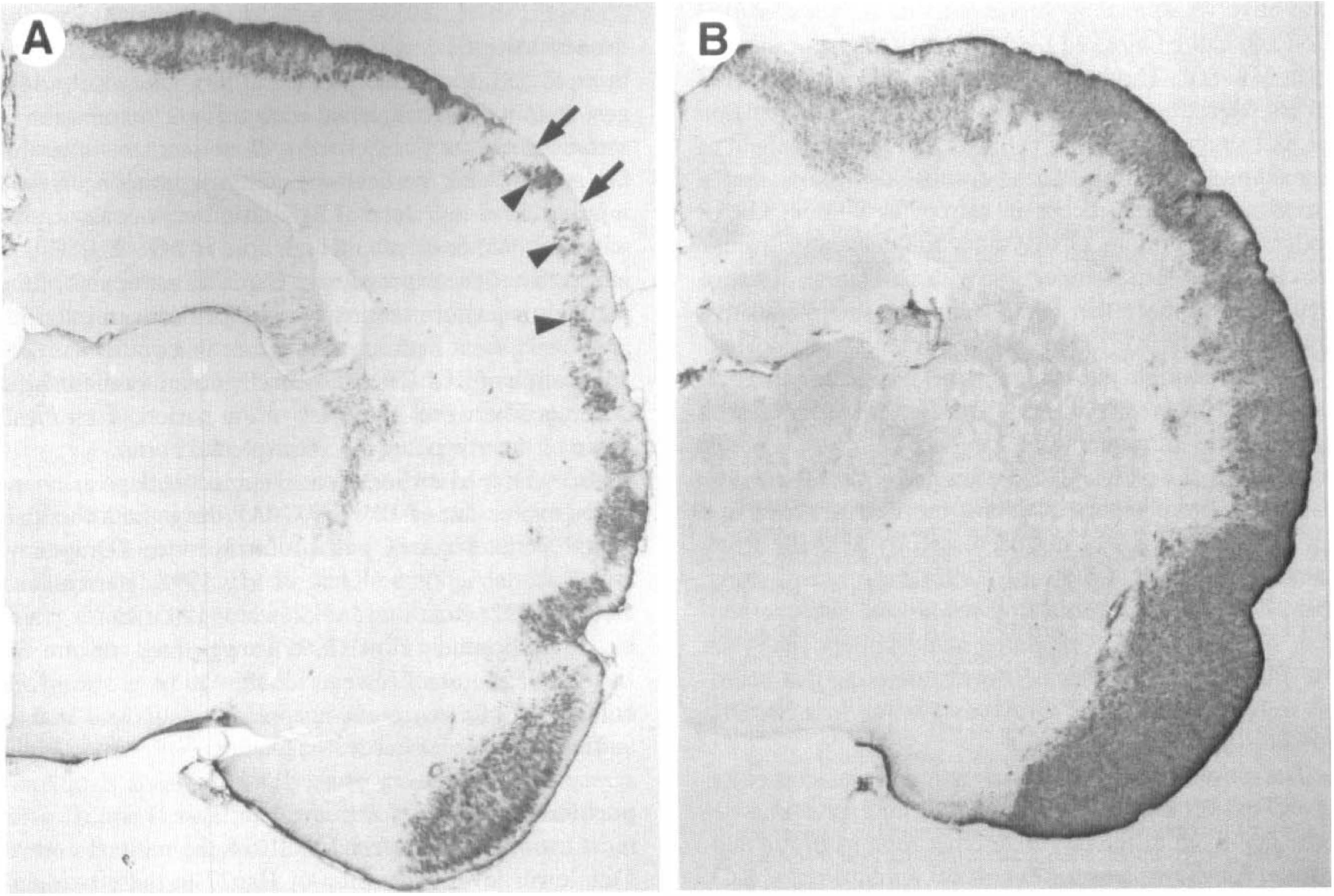
Hsp27 immunoreactivity was not observed in the cingulate cortex, except occasionally in layer I (see Fig. 1) or in the retrosplenial cortex (Fig. 5A). Fos immunoreactivity, a marker of neuronal activation, was detected in the entire ipsilateral cortex, including the cingulate cortex and retrosplenial cortex (Fig. 5B).
Hsp27 immunoreactivity in the granular retrosplenial cortex. At 2 or 4 days after cortical application of KCl, no Hsp27 immunoreactivity was observed in the retrosplenial cortex, except in the glial limitans
High doses of the NMDA-receptor antagonist, MK-801, have been reported to induce Hsp70 specifically in neurons of the posterior cingulate cortex and retrosplenial cortex (Sharp et al., 1991a). Thus we examined the posterior cingulate cortex and retrosplenial cortex for Hsp27 immunoreactivity after a higher dose of MK-801. As shown above (Figs. 1 and 5A), 2 days after systemic administration of 3 mg/kg of MK-801, no Hsp27 immunoreactivity was observed in the cingulate cortex or retrosplenial cortex. However, 2 days after systemic administration of 9 mg/kg of MK-801, Hsp27 immunoreactivity was detected in the retrosplenial cortex and in the posterior cingulate cortex (Fig. 5C). Counterstaining of immunostained sections revealed that the Hsp27-positive astrocytes were localized in layer III of the cingulate cortex (Fig. 5D).
Modulation of Hsp27 distribution by reduction of cortical injury
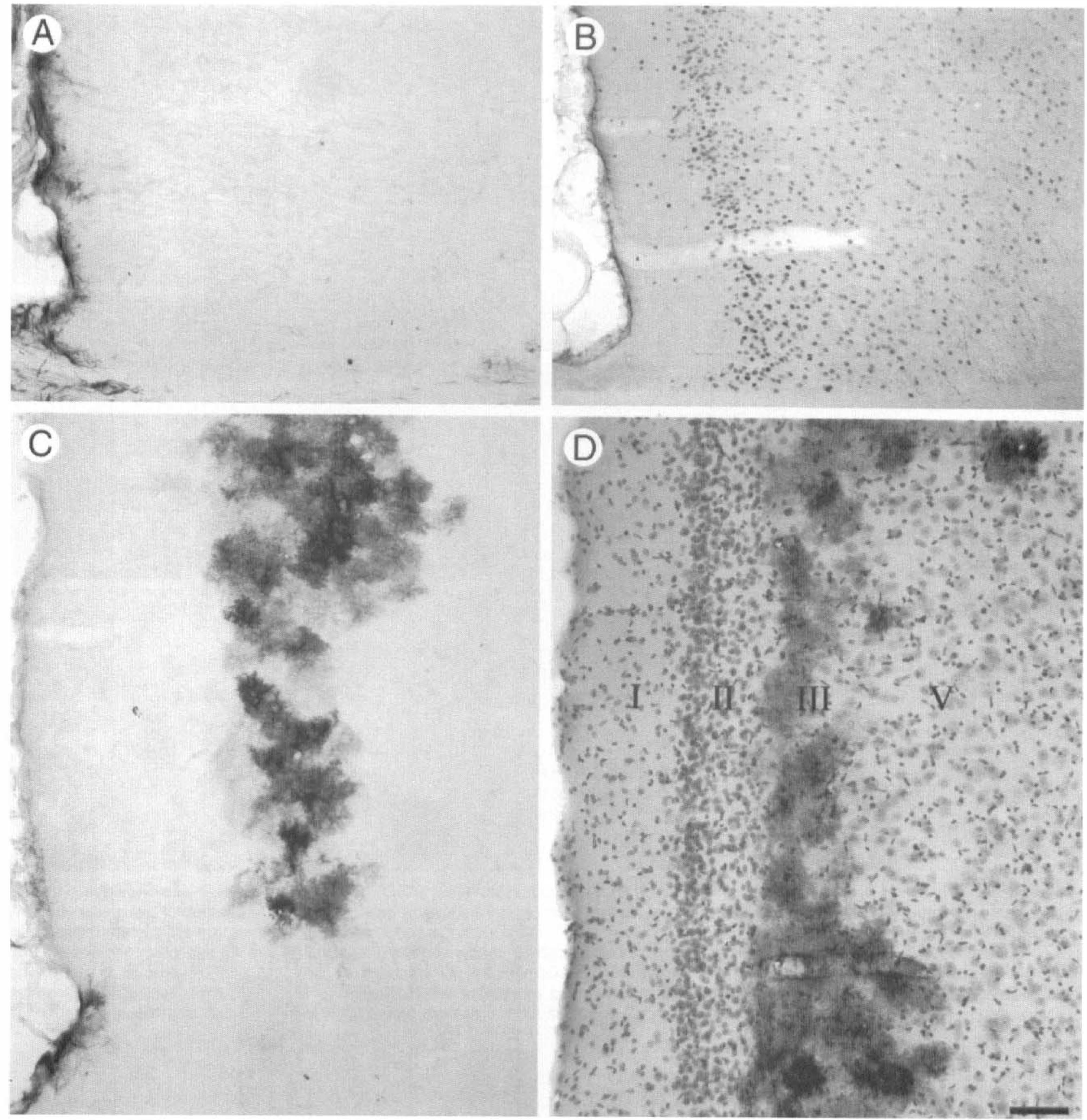
Six animals were subjected to 5 minutes of cortical application of KCl. Four of these animals had immunoreactivity for Hsp27 similar in intensity and distribution as the animals treated with KCl for 20 minutes. However, two of six animals subjected to 5 minute application of KCl showed a distribution of Hsp27 immunoreactivity different from that observed in animals after a 20-minute application of KCl. In both cases, Hsp27 immunoreactivity was not observed in all layers of the ipsilateral cortex but was observed only in the superficial layers (Fig. 6). In one rat, Hsp27-immunoreactive astrocytes were detected around the lesion site, in clusters in layers I to III throughout the ipsilateral cortex, and in layers II and III of the piriform cortex (Fig. 6A). In the second rat, Hsp27 immunoreactivity was detected in layers I to III of the ipsilateral cortex and in the entire piriform cortex (Fig. 6B).
Hsp27 immunoreactivity in the rat cortex 4 days after 5 minute cortical application of potassium chloride. While four of six rats had Hsp27 immunoreactivity similar to those with the 20-minute application of KCl, two of six rats showed less intense Hsp27 immunoreactivity. In one rat, Hsp27 immunoreactivity was observed around the lesion site, in clusters of astrocytes in layers I to III of the parietal cortex, and in layers II and III of the piriform cortex
DISCUSSION
Cortical application of KCl-induced Hsp27 immunoreactivity in the ipsilateral cerebral cortex of the rat. Hsp27 immunoreactivity was localized in GFAP-positive astrocytes. Suppression of KCl-induced cortical spreading depression with systemic administration of the NMDA-receptor antagonist, MK-801 (3 mg/kg), significantly attenuated the expression of Hsp27. While cortical application of KCl induced Hsp27 immunoreactivity in most of the ipsilateral cerebral cortex, there was no Hsp27 immunoreactivity in the posterior cingulate cortex and retrosplenial cortex. However, astrocytes in the posterior cingulate and retrosplenial cortex did express Hsp27 after injury to adjacent neurons by systemic administration of a very high dose of MK-801 (9 mg/kg). Finally, brief (5 minutes) cortical application of KCl revealed a progressive recruitment of astrocytes expressing Hsp27, from cell clusters (Fig. 6A), to superficial cortical layers (Fig. 6B) to all cortical layers (Fig. 1; seen after 20 minutes of KCl application).
In this study, we monitored the effect of KCl application on neuronal activation and spreading depression by assessing Fos expression 2 hours after KCl application. c-fos expression has been used as an indicator of spreading depression, whether induced by cortical application of KCl (Herrera and Robertson, 1990; Herdegen et al., 1993) or focal ischemia (Lindberg, et al., 1996; Welsh et al., 1992; Moskowitz et al., 1993). We have shown that cortical application of KCl and the subsequent cortical spreading depression, indicated by neuronal c-fos expression, induced Hsp27 expression in astrocytes. Similar treatment also induces expression of GFAP in astrocytes as well as Fos expression in neurons throughout the ipsilateral cortex (Herrera and Robertson, 1990; Kraig et al., 1991; Herrera and Cuello, 1992; Bonthius and Steward, 1993; Herdegen et al., 1993; Bonthius et al., 1995).
To investigate the pathways for induction of Hsp27 after cortical application of KCl, MK-801, an NMDA-receptor antagonist, was used to block cortical spreading depression (Marrannes et al., 1988). Similar MK-801 treatment before application of KCl to the cerebral cortex also blocked Fos expression (Herrera and Robertson, 1990; Jacobs et al., 1994) and the generation of astrogliosis measured by increased GFAP (Herrera and Cuello, 1992; Bonthius and Steward, 1993; Bonthius et al., 1995). In the present study, Hsp27 immunoreactivity was also affected by MK-801 treatment. A significant reduction in the intensity of Hsp27 immunoreactivity in the parietal cortex was observed. This suggests that Hsp27 is induced by a similar mechanism as that of GFAP. However, systemic administration of MK-801 at a dose that is reported to block cortical spreading depression and subsequent GFAP increase did not completely inhibit Hsp27 expression. There was still a significant difference between contralateral and ipsilateral parietal cortices of MK-801-treated animals that were examined 2 days after cortical application of KCl. This result suggests that after cortical application of KCl, Hsp27 is induced, but by a mechanism different from that of GFAP. This hypothesis is also supported by the observation that cortical application of sodium chloride (3 M) induced Hsp27 expression primarily around the application site and in only a few astrocytes in the ipsilateral cortex (data not shown). However, application of NaCl does not produce spreading depression (Kraig et al., 1991; Moskowitz et al., 1993). Thus it seems that Hsp27 may be induced through more than one pathway. One pathway involves spreading depression and neuronal activation and can be blocked by the NMDA-receptor antagonist, MK-801. The lack of complete MK-801 inhibition of Hsp27 astrocytic expression after cortical application of KCl suggests a second pathway that is independent of spreading depression. These results also show that a stress response can occur in astrocytes without GFAP induction. Therefore, GFAP induction does not correlate as closely as Hsp27 induction with activation of astrocytes. We propose that Hsp27 may be a more sensitive marker than GFAP for the study of reactive astrocytes.
A third possible alternative pathway for the induction of Hsp27 is necrosis. In this study, Hsp27 was expressed in astrocytes surrounding the necrotic cells at the site of application of KCl (and NaCl). Similarly, GFAP-positive astrocytes were reported around the necrotic area and GFAP expression was not decreased by MK-801 treatment (Bonthius and Steward, 1993). In a recent study, Hsp27 was abundant in astrocytes around necrotic neurons of the piriform cortex after status epilepticus in the rat (Plumier et al., 1996a). It seems therefore that necrosis triggered Hsp27 and GFAP expression in a NMDA-receptor-independent manner.
The posterior cingulate and retrosplenial cortex respond to cortical application of KCl and spreading depression much differently than other regions of the neocortex. As shown here, after cortical application of KCl, Hsp27 was not expressed in the posterior cingulate and retrosplenial cortex. In addition, GFAP was not expressed in the cingulate and retrosplenial cortex after cortical application of KCl (Bonthius and Steward, 1993; Bonthius et al., 1995). The absence of GFAP expression was attributed to either a lack of propagation of spreading depression or an intrinsic difference of astrocytes in these specific regions (Bonthius et al., 1995). In the present study, Fos, whose induction has been shown to be triggered by cortical spreading depression through NMDA receptor activity (Dragunow and Robertson, 1988; Herrera and Robertson, 1990; Herdegen et al. 1993), was detected in neurons throughout the ipsilateral cerebral cortex, including the posterior cingulate and retrosplenial cortex. This suggests that there were at least some changes in neuronal activity and gene expression that resulted from spreading depression into the posterior cingulate and retrosplenial cortex.
In addition, Hsp27 was induced in astrocytes in the posterior cingulate and retrosplenial cortex after systemic administration of a high dose of MK-801 (9 mg/kg) but not after a low dose (3 mg/kg). Interestingly, MK-801 treatment has been shown to induce neuronal injury in the posterior cingulate and retrosplenial cortex, measured by Hsp70 immunoreactivity (Olney et al., 1990; 1991). Neurons in rats treated with 1 mg/kg of MK-801 express higher levels of Hsp70 than neurons in rats treated with 5 mg/kg MK-801 (Allen and Iversen, 1990; Fix et al., 1993). It was concluded that 5 mg/kg dose of MK-801 was more toxic to neurons and resulted in rapid cell death (Sharp et al., 1994). Our results suggest that, in the retrosplenial cortex, Hsp27 expression in astrocytes was not associated with an increase in neuronal activity after cortical spreading depression or even injury after a low dose of MK-801, but was associated with neuronal death after a high dose of MK-801. This is in contrast to the expression of Hsp27 in astrocytes of the parietal or piriform cortex where cell death or even injury of adjacent neurons was not detected after cortical application of KCl. Thus it seems that there is an intrinsic difference between astrocytes of the parietal cortex and those of the cingulate and retrosplenial cortex.
As mentioned earlier, cortical spreading depression induces expression of Fos and GFAP throughout the ipsilateral parietal cortex and piriform cortex (Dragunow and Robertson, 1988; Kraig et al., 1991; Herrera and Cuello, 1992; Bonthius and Steward, 1993; Herdegen et al., 1993; Bonthius et al., 1995), suggesting uniform activation of neurons. However, in this study, in two of six animals, 5 minutes cortical application of KCl that is sufficient to trigger cortical spreading depression (Kraig et al., 1991; Herdegen et al., 1993) induced Hsp27 expression in clusters of astrocytes in layer II and III or in most astrocytes of layers I to III of the parietal cortex. This lower level expression of Hsp27 in these two animals suggested that the signal for Hsp27 expression in astrocytes did not progressively spread through the ipsilateral cortex, but rather appeared in small clusters of astrocytes throughout the superficial layers of the cortex that eventually joined, and recruited astrocytes from deeper cortical layers. It may be that KCl-induced cortical spreading depression has also a heterogeneous effect on neurons undetectable by Fos immunoreactivity.
At least three functions for Hsp27 have been reported in vitro. First, Hsp27 has been implicated in the regulation of actin filament dynamics (reviewed by Landry and Huot, 1996). Hsp27 inhibited actin polymerization and enhanced actin filament depolymerization in vitro (Miron et al., 1988; 1991). Overexpression of Hsp27 in transfected cells modified the cellular distribution of actin, increasing cortical F-actin and decreasing cytoplasmic stress fibers (Lavoie et al., 1993). Additional experiments showed that Hsp27 function was regulated by phophorylation. Phosphorylated Hsp27 isoforms enhanced resistance of actin filaments against depolymerization induced by cytochalasin D (Lavoie et al., 1995) or by oxidative injury (Huot et al., 1996). In astrocytes, Hsp27 might participate in the regulation of growth processes that have been shown to involve actin filament dynamics (Baorto et al., 1992). Second, Hsp27 may play a role in antioxidative mechanisms via an increase in the total levels of glutathione (Mehlen et al., 1996). Interestingly, glutathione has been detected in glial cells but not in neurons (Slivka et al., 1987; Raps et al., 1989). Finally, Hsp27 prevented protein aggregation and promoted refolding of denatured proteins (Jakob et al., 1993), suggesting that it may act as a molecular chaperon.
Whatever the specific role of Hsp27 may be, it seems that Hsp27 can generally protect cells from injury and facilitate recovery in reversibly injured cells (Landry et al., 1989; Lavoie et al., 1993; Mehlen et al., 1995). In the brain, this protective role may be even more interesting. After injury, cell-cell interactions may play an important role in the overall survival and function of the brain. Glial disfunction may be an initial cause of neuronal cell death in ischemic brain (Largo et al., 1996). In fact, astrocytes in vitro have been shown to increase neuronal survival against oxidative stress induced by hydrogen peroxide (Desagher et al., 1996). In the rat brain, cortical spreading depression induces Hsp27 in astrocytes (as we have shown here) and neuronal protection from ischemic injury (Kobayashi et al., 1995; Matsushima et al., 1996). We suggest that expression of Hsp27 increases resistance of astrocytes to ischemic injury, maintains normal function of astrocytes during and after ischemic injury, and plays a role, through astrocytes, in neuronal protection. Finally, it may be that neuronal perturbation induces a stress response in the surrounding glia and that these stress-conditioned glia improve neuronal survival after injury.
Footnotes
Abbreviations used
Acknowledgements
We thank Ms. Brenda Ross, Ms. Kathleen Murphy, and Mr. Marc Peterson for their excellent technical assistance.