
Abstract
The expression of immediate early genes (IEG) has been documented in the brain after various kinds of insults such as ischemia and hypoxia. To determine whether acute carbon monoxide intoxication (ACOI) might trigger IEG expression, adult ddY mice were subjected to carbon monoxide exposure at a rate of 30 mL/min for 35 seconds. The levels of NGFI-B, c-fos, and c-jun mRNA were determined by Northern blot analysis. A time-course study in the cerebral cortex indicated that the induction of NGFI-B, c-fos, and c-jun mRNA started as early as 15 minutes, reached a peak at 30 minutes, and returned to the basal level at 1 hour after the ACOI. In addition, the temporal feature of the induction of these IEG mRNA in the hippocampus was very similar to that in the cerebral cortex. Examination of brain regions at 30 minutes after the ACOI revealed a significant induction of NGFI-B mRNA in the cerebellum, thalamus-hypothalamus, brainstem, as well as in the cortex and hippocampus, but not in the striatum or olfactory bulb. Furthermore, the neuroanatomical distribution of c-fos mRNA at 30 minutes after the ACOI was very similar to that of the NGFI-B mRNA. The widespread distribution of these IEG in the brain, especially in the cerebellum and brainstem, indicates that the major cause for the triggering of IEG expression in the brain by the ACOI might be a diffuse hypoxia. These findings show for the first time the temporal and spatial expression of IEG in the brain after ACOI.
Carbon monoxide (CO) can cause neuronal injury and produce death in humans. (Ginsberg, 1985; Hardy and Thom, 1994; Choi, 1983). Carbon monoxide binds to heme proteins with much higher affinity, rendering them incapable of normal function (for review see Penney, 1990). Carbon monoxide competes with oxygen for binding sites on hemoglobin to form carboxyhemoglobin, which produces tissue hypoxia–especially brain hypoxia, as its major toxic effect (Killick, 1944; Ginsberg, 1985; Penney, 1990; Hardy and Thom, 1994). In addition, acute carbon monoxide intoxication (ACOI) always causes hypotension, which may impair the compensatory response of an increase in the cerebral blood perfusion to hypoxia (Okeda et al., 1981; Ginsberg, 1985; Penney, 1990). Thus, the brain hypoxia associated with ACOI is inevitably complicated by relative ischemia. Delayed neurological sequelae, such as learning and memory deficits, are frequently observed clinically after even mild ACOI (Choi, 1983; Ginsberg, 1976; Hardy and Thom, 1994). Delayed pathological findings in these brains consist essentially of neuron loss in some specific regions such as the cerebral cortex, hippocampus, globus pallidus, and substantia nigra (Sawada et al., 1983; Ginsberg, 1985; Lapresle and Fardeau, 1967). The mechanisms responsible for these delayed events are still unknown.
During the past few years, immediate early genes (IEG) have received attention because many of their protein products are transcription factors and play a crucial role in the coupling of short-term extracellular stimuli to long-term changes in the cellular phenotype within the nervous system (Robertson, 1992; Morgan and Curran, 1991). NGFI-B (also known as nur77) is an IEG and belongs to a member of the nuclear receptor superfamily (Evans, 1988). An NGFI-B response element similar to the half-site of the thyroid/estrogen hormone receptor response element has been identified and confers NGFI-B-dependent transcription activity to heterologous promoter (Wilson et al., 1991). Conversely, c-fos and c-jun belong to members of proto-oncogene family. Their products can interact via the leucine zipper to form heterodimers or homodimers and then bind specifically to the AP-1 binding sites in the promoter region(s) of target genes to regulate their expression (Chiu et al., 1988; Landschulz et al., 1988). Evidence has indicated that genes such as the prodynorphin, nerve growth factor, brain-derived neurotrophic factor, and glial fibrillary acidic protein are the potential targets of the NGFI-B, c-fos, and c-jun (for review see Pennypacker et al., 1995; Liu and Chen, 1994). Thus, activation of these IEG in the brain plays an important role in the triggering of certain complex genomic responses to extracellular stimuli.
The expression of IEG has been investigated after various kinds of insults to the brain, including electrical stimuli and kindling (Sagar et al., 1988; Dragunow and Robertson, 1987), seizure activity (Morgan and Curran, 1991), trauma (Dragunow et al., 1990), and ischemia/hypoxia (Welsh et al., 1992; Neumann-Haefelin et al., 1994; Takemoto et al., 1995; Kinouchi et al., 1994a;Dragunow et al., 1994). Recently, we have developed a novel ACOI model in mice, to which a very short duration of CO insult is delivered (Nabeshima 1991b;Hiramatsu et al., 1996). Because ACOI produces brain hypoxia or the hypoxia complicated by ischemia, it could trigger the expression of IEG in the brain. However, the difference between the induction of IEG by ACOI and by other hypoxia or ischemia is not known yet. To address this issue, we examined temporal and spatial expression of NGFI-B, c-fos, and c-jun mRNA in the brain after the ACOI in mice.
MATERIALS AND METHODS
The experiments were conducted in accordance with the Japanese Experimental Animal Research Association standards as defined in the Guidelines for Animal Experiments (1987), and the protocol was approved in advance by the Animal Research Committee at Nagoya University.
Acute carbon monoxide intoxication in mice
Male ddY mice (Nihon SLC, Shizuoka, Japan), weighing between 30 and 35 g at the beginning of the experiments, were used. The animals were housed in groups of six per cage under standard conditions (23 ± 1°C, 50 ± 5% humidity) with a 12-hour light-dark cycle (light on at 9:00 AM). Water and food were given ad libitum.
After the animals adapted to laboratory conditions for 1 week, the animals were handled daily for another week by an investigator who was responsible for the ACOI experiment. The procedure of ACOI was performed as described previously (Nabeshima et al., 1991b). Briefly, mice were individually placed into a transparent plastic vessel (3-cm radius, 10-cm high) with a pipe feeding into it. Pure CO gas was delivered at a rate of 30 mL/min for 35 seconds. After the CO exposure, the righting reflex was evaluated and the mice that did not show the loss of the righting reflex were excluded from the experiment. The animals were then returned to their prewarmed home cage at 37°C to prevent hypothermia because hypothermia reduces the extent of the neuronal damage associated with the ACOI (Ishimaru et al., 1992). For sham-intoxication, mice were placed in an identical vessel and fed with room air for the same duration. Another group, which was neither handled nor sham treated, was used as quiet control (naive). The ACOI mice were killed by decapitation immediately, 15, 30 minutes, 1, 2, 3, and 6 hours after the ACOI, and the sham-treated mice were killed at 30 minutes after the sham treatment, a time point of the maximal IEG expression in this model in our pilot experiment. The brains were quickly removed and dissected on a chilled marble plate into the following regions: the cerebral cortex, hippocampus, striatum, olfactory bulb, cerebellum, thalamus-hypothalamus, and brainstem; then frozen immediately on dry ice. The samples from every three animals were pooled and stored at −80°C until used for mRNA analysis.
Northern blot analysis
Total RNA was extracted by the method of Chomczynski and Sacchi (1987). Amount of total RNA was determined by optical absorbance at 260 nm. Levels of NGFI-B, c-fos, c-jun mRNA were determined by Northern blot analysis as described previously (Kambe et al., 1988). Briefly, 15 μg of total RNA was fractionated electrophoretically on 0.8% (weight to volume ratio) agarose gels and then transferred to GeneScreen Plus membranes (New England Nuclear, Boston, MA. U.S.A.) using a vacuum transfer apparatus (VacuGene, Pharmacia LKB Biotechnology, Uppsala, Sweden). The membranes were prehybridized in a solution containing 5 × SSPE (5 mmol/L EDTA, 900 mmol/L NaCl, 50 mmol/L sodium phosphate, pH 8.3), 5 × Denhardt's solution (0.1% Ficoll [400,000 molecular weight], 0.1% polyvinylpyrrolidone [360,000 molecular weight], 0.1% bovine serum albumin), 1% (weight to volume ratio) sodium dodecyl sulfate, 50% (volume to volume ratio) formamide (Merck, Darmstadt, Germany), and 0.1 mg/mL (weight to volume ratio) herring sperm DNA (Boehringer-Mannheim, Mannheim Germany). Hybridization was performed at 42°C for about 20 hours in the same solution containing 32P-labeled cDNA probe. The c-DNA for NGFI-B (Milbrandt, 1988), c-fos (van Straaten et al., 1983), and c-jun (Angle et al., 1988) were used as the probes. After digestion with appropriate enzymes, the selected cDNA fragments were purified by agarose gel electrophoresis and then labeled with 32P-dCTP (SA, 3,000 Ci/mmol, New England Nuclear, Boston, MA, U.S.A.) using a random primed DNA labeling kit (Boehringer-Mannheim, Mannheim, Germany). The radioactivities of the probes in the hybridization buffer were about 1 × 106 cpm/ml.
After hybridization, the membranes were washed two times each for 5 to 10 minutes in 2 × SSC solution (300 mmol/L NaCl, 30 mmol/L sodium citrate, pH 7.0) at room temperature followed by two washes each for 30 minutes in 2 × SSC solution containing 1% sodium dodecyl sulfate at 65°C, and finally washed twice each for 15 minutes in 0.1 × SSC solution at room temperature. Autoradiography was carried out by exposing the membranes to Kodak X-AR film (Eastman Kodak, Rochester, NY, U.S.A.) at −80°C for 25 to 30 days.
After autoradiography, the same membranes were reprobed with 32P-labeled glyceraldehyde-3-phosphate dehydrogenase cDNA for normalizing the relative RNA quantity. The radio-activities of bands for NGFI-B,c-fos, and c-jun, and glyceraldehyde-3-phosphate dehydrogenase were determined by computer-assisted imaging analysis (BAS 2000 Fujix Bioimage Analyzer, Fuji Photo Film Co. Ltd., Tokyo, Japan). The significant difference of each mRNA abundance between the ACOI and the sham-intoxication was determined by student's t-test.
RESULTS
No mortality was found in the mice that were subjected to the ACOI at the rate of 30 mL/min for 35 seconds, a procedure that has been previously shown to produce profound learning and memory deficits (Nabeshima et al., 1991b;Maurice et al., 1994). The loss of righting reflex was found in more than 90% of mice after the ACOI (data not shown).
Induction of NGFI-B mRNA in the cerebral cortex after acute carbon monoxide intoxication
As shown in Fig. 1, a time-course study of the induction of NGFI-B mRNA in the cerebral cortex revealed that the levels of NGFI-B mRNA increased as early as 15 minutes, reached a maximum at 30 minutes, and returned to the basal level at 1 hour after the ACOI. After that, the expression of NGFI-B mRNA maintained a lower or undetectable level up to 6 hours. Similar results were observed in four separate experiments. Figure 1 shows an autoradiograph of Northern blot analysis and Fig. 2 shows the quantitative results of the NGFI-B mRNA levels normalized with the glyceraldehyde-3-phosphate dehydrogenase mRNA level.
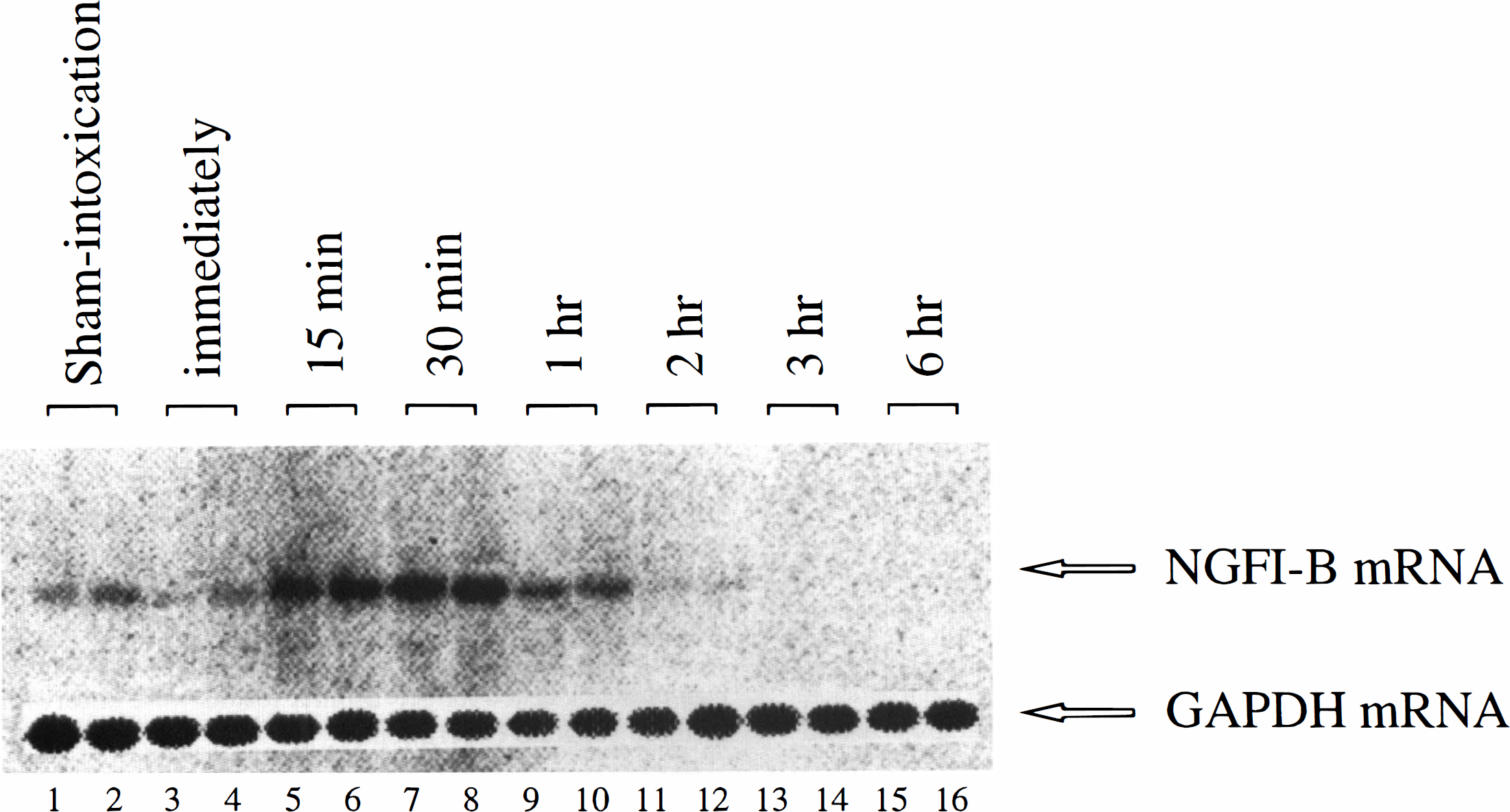
The temporal profile of accumulation of NGFI-B mRNA in the cerebral cortex of the mice after the acute carbon monoxide intoxication. Figure shows an autoradiograph of Northern blot analysis of NGFI-B mRNA expression. Lanes 1 to 2 are the duplicate samples from the sham-intoxication mice. Every two lanes from 3 to 16 are the duplicate samples from the animals that were killed immediately, 15, 30 minutes, 1, 2, 3, and 6 hours after the ACOI, respectively. The experiment was repeated four times. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
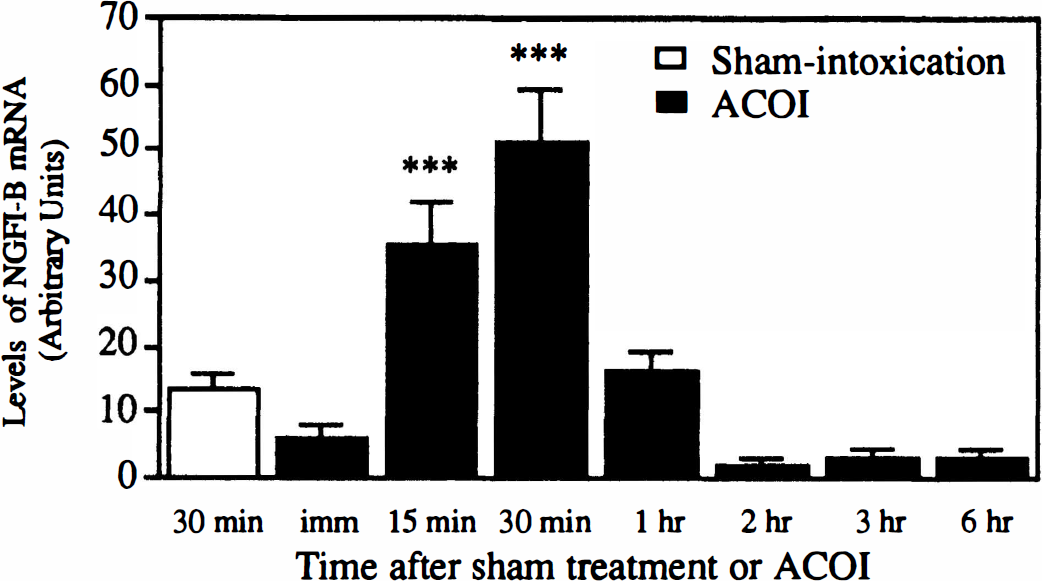
Quantitative data from the four experiments in Fig. 1 are shown. The bars represent the means ± SD of the arbitrary units of NGFI-B mRNA levels normalized with GAPDH mRNA level. ***P < 0.001, compared with those of the sham-intoxication mice, determined by Student's t-test. ACOI, acute carbon monoxide intoxication, imm, immediately.
Induction of NGFI-B mRNA in the hippocampus after acute carbon monoxide intoxication
A transient induction of NGFI-B mRNA, similar to that observed in the cerebral cortex, was also fount in the hippocampus. As shown in Figs. 3 and 4, the induction of NGFI-B mRNA was detectable from 15 minutes to 1 hour after the ACOI, during the maximal laevel was found at 30 minutes. After 1 hour, the expression of NGFI-B mRNA maintaned a lower or undetectable level up to 6 hours. Similar results were observed in four separate experiments.
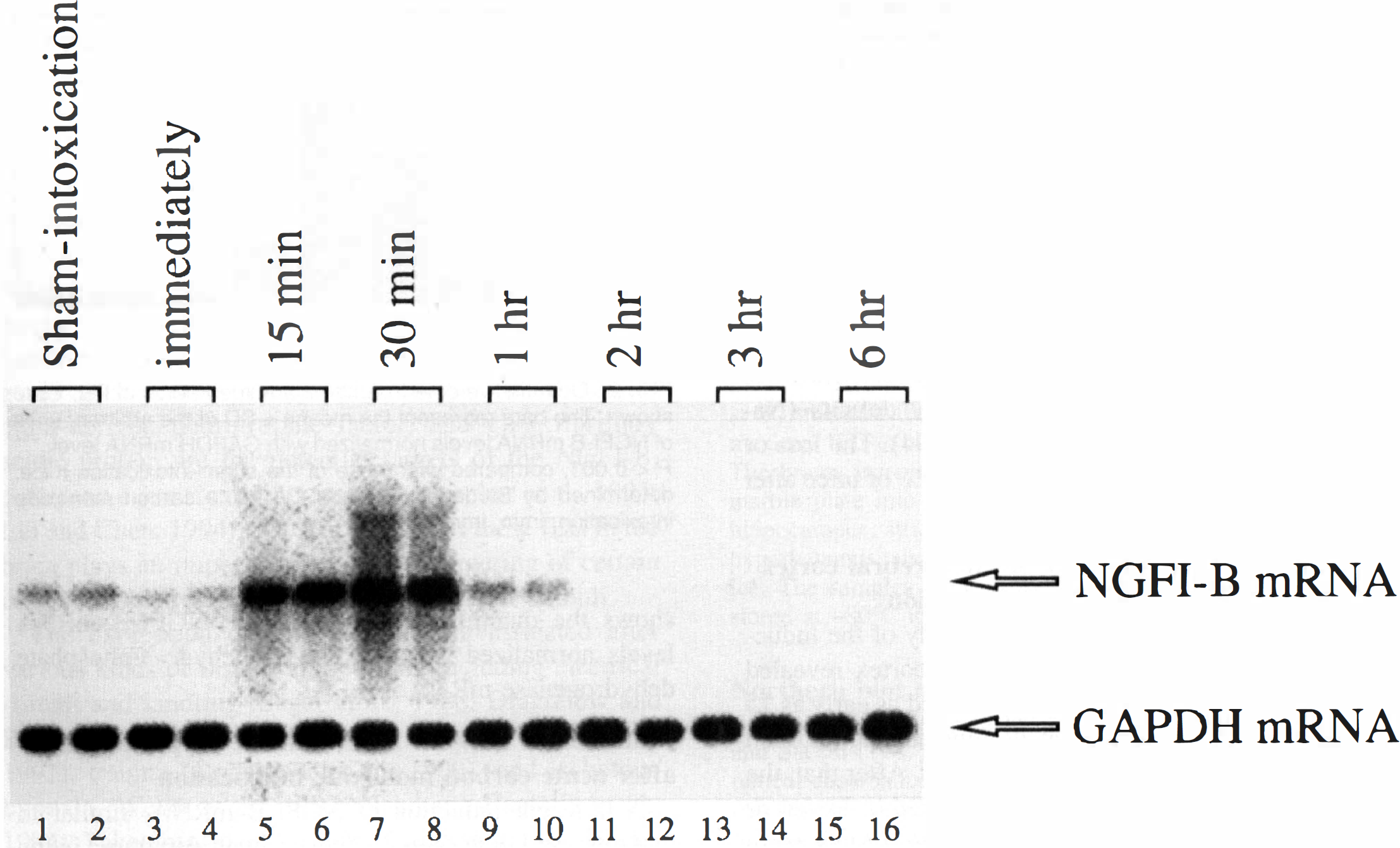
The temporal profile of accumulation of NGFI-B mRNA in the hippocampus of the mice after the ACOI. Figure shows an autoradiograph of Northern blot analysis of NGFI-B mRNA expression. Lanes 1 to 2 are the duplicate samples from the sham-intoxication mice. Every two lanes from 3 to 16 are the duplicate samples from the animals that were killed immediately, 15, 30 minutes, 1,2,3, and 6 hours after the ACOI, respectively. The experiment was repeated for four times.
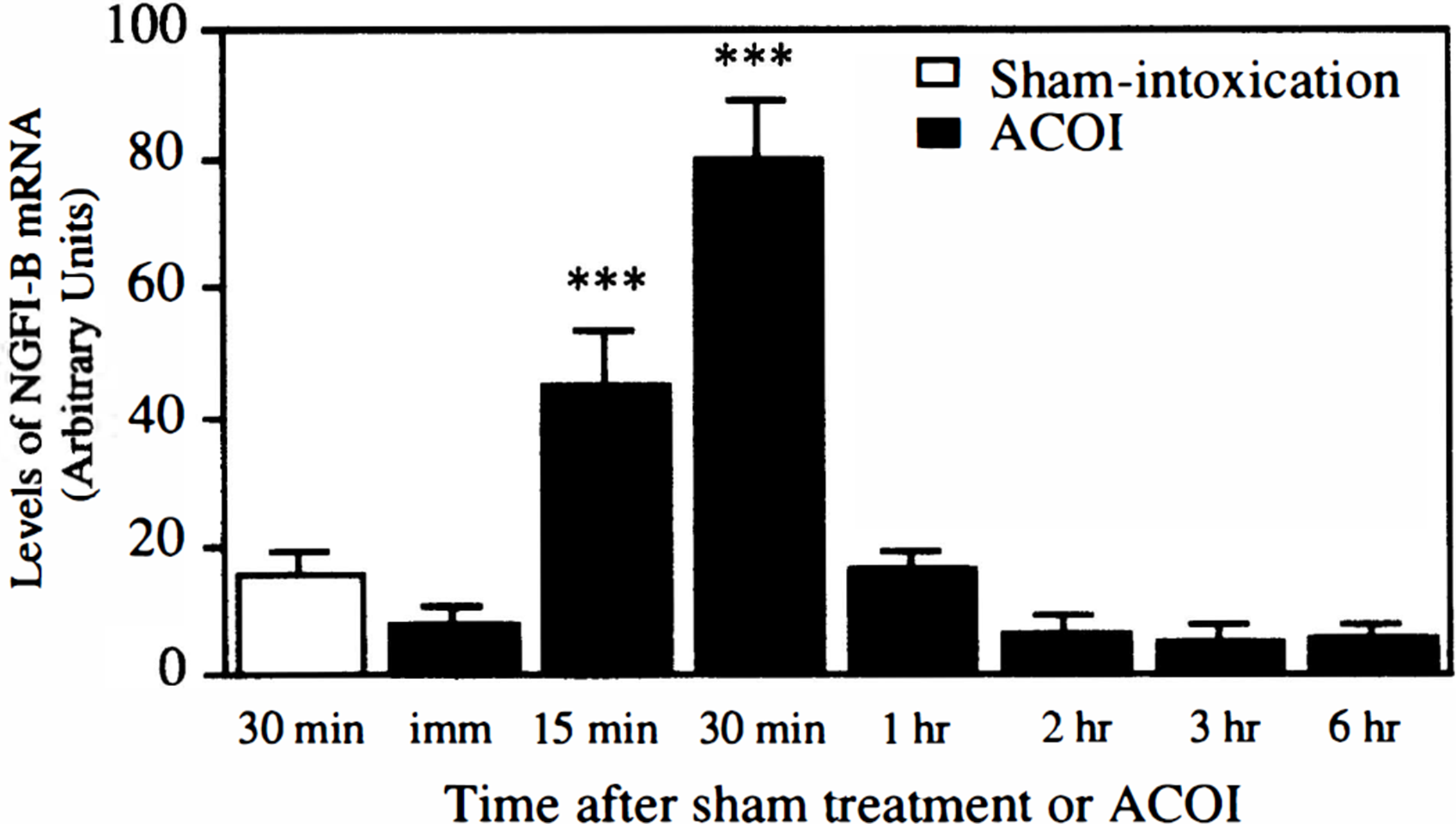
Quantitative data from the four experiments in Fig. 3 are shown. The bars represent the means ± SD of the arbitrary units of NGFI-B mRNA levels normalized with GAPDH mRNA level. ***P < 0.001, compared with those of the sham-intoxication mice, determined by Student's t-test.
Induction of c-fos and c-jun mRNA in the hippocampus after acute carbon monoxide intoxication
As shown in Fig. 5, the time-course studies of the induction of both c-fos and c-jun mRNA in the cerebral cortex indicated that their induction started as early as 15 minutes, reached a peak at 30 minutes, and returned to the basal level at 1 hour after the ACOI. After that, the expression of eitherc-fos or c-jun mRNA maintained a lower or undetectable level up to 6 hours. Similar results were observed in three separate experiments.
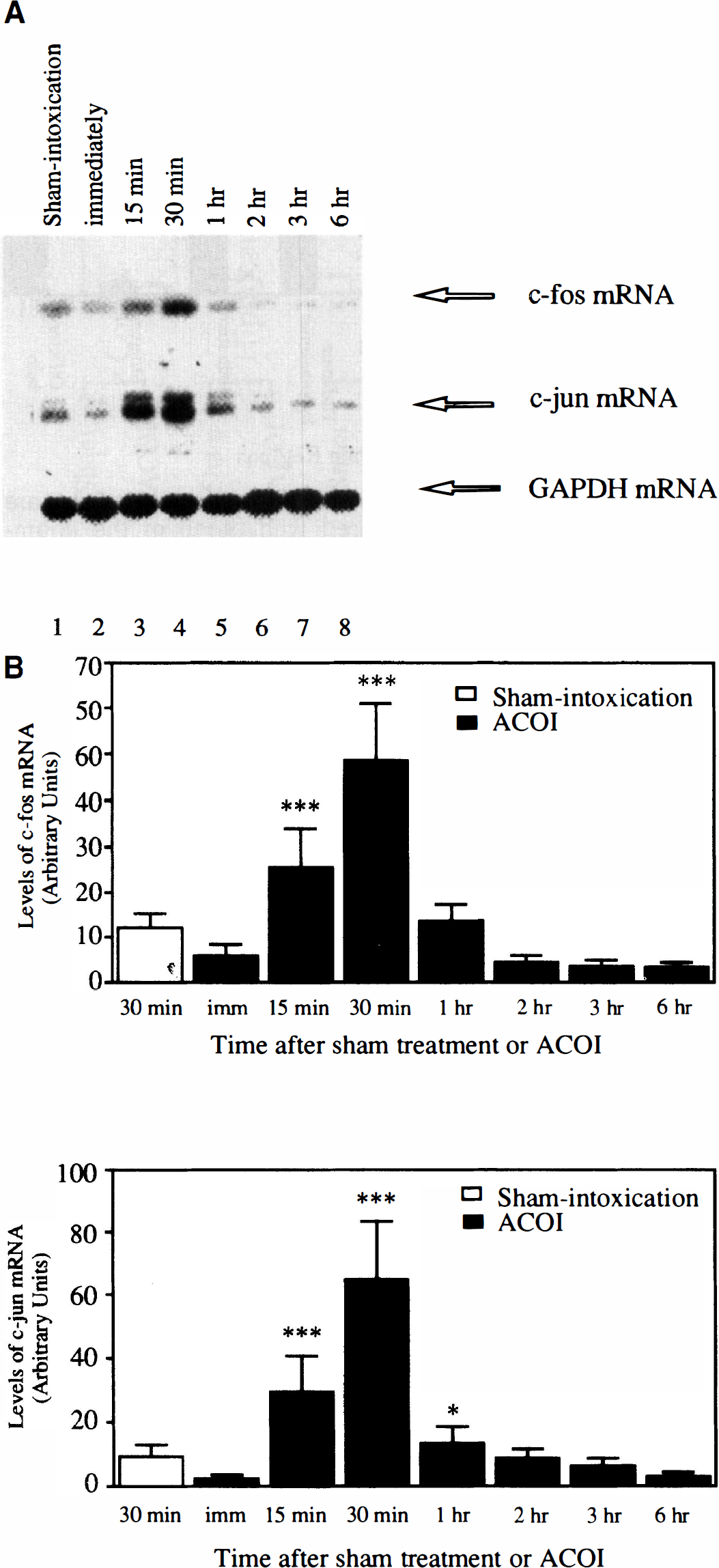
The temporal profile of accumulation of c-fos and c-jun mRNA in the cerebral cortex of the mice after the ACOI.
Induction of c-fos and c-jun mRNA in the hippocampus after acute carbon monoxide intoxication
The temporal feature of the induction of c-fos and c-jun mRNA after the ACOI in the hippocampus is shown in Fig. 6. Similar to that observed in the cerebral cortex, the induction of c-fos and c-jun mRNA started as early as 15 minutes, reached a peak at 30 minutes, and returned to the basal level at 1 hour after the ACOI. After that, the expression of c-fos mRNA maintained a lower or undetectable level up to 6 hours but the induction of c-jun mRNA lasted longer. Similar results were observed in three separate experiments.
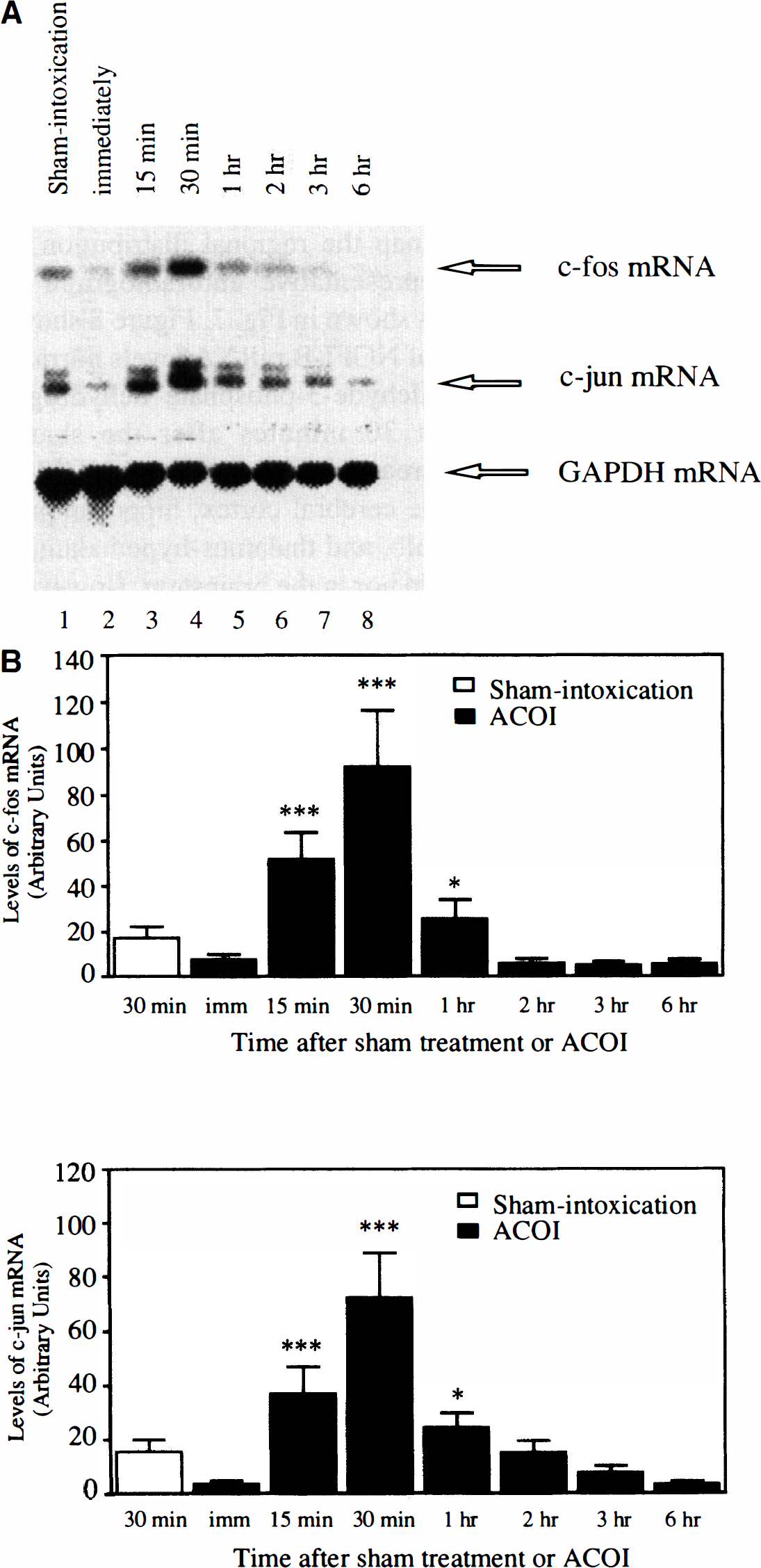
The temporal profile of accumulation of c-fos and c-jun mRNA in the hippocampus of the mice after the ACOI.
Regional induction of NGFI-B mRNA in the brain after acute carbon monoxide intoxication
Although an exhaustive mapping of NGFI-B induction after the ACOI was not attempted, our attention was given to the brain areas which are considered to be related to the neurotoxicity of CO. Basal expression of NGFI-B mRNA in the naive animals was very low or undetectable in all of the examined regions, except for the cerebral cortex, hippocampus, and olfactory bulb, in which clear NGFI-B mRNA expression was observed (data not shown). A time point of 30 minutes after the ACOI was chosen to map the regional distribution of NGFI-B mRNA. A representative autoradiograph of Northern blot analysis is shown in Fig. 7. Figure 8 shows the quantitative results of NGFI-B mRNA levels normalized with the glyceraldehyde-3-phosphate dehydrogenase mRNA level. At 30 minutes after the sham-treatment, a small increase in the level of NGFI-B mRNA was noted in the cerebral cortex, hippocampus, cerebellum, olfactory bulb, and thalamus-hypothalamus, but neither in the striatum nor in the brainstem. However, 30 minutes after the ACOI, a dramatic increase in levels of NGFI-B mRNA was observed in the cerebral cortex, hippocampus, thalamus-hypothalamus, cerebellum, and brainstem, but not in the striatum or olfactory bulb. Similar results were replicated in three separate experiments.
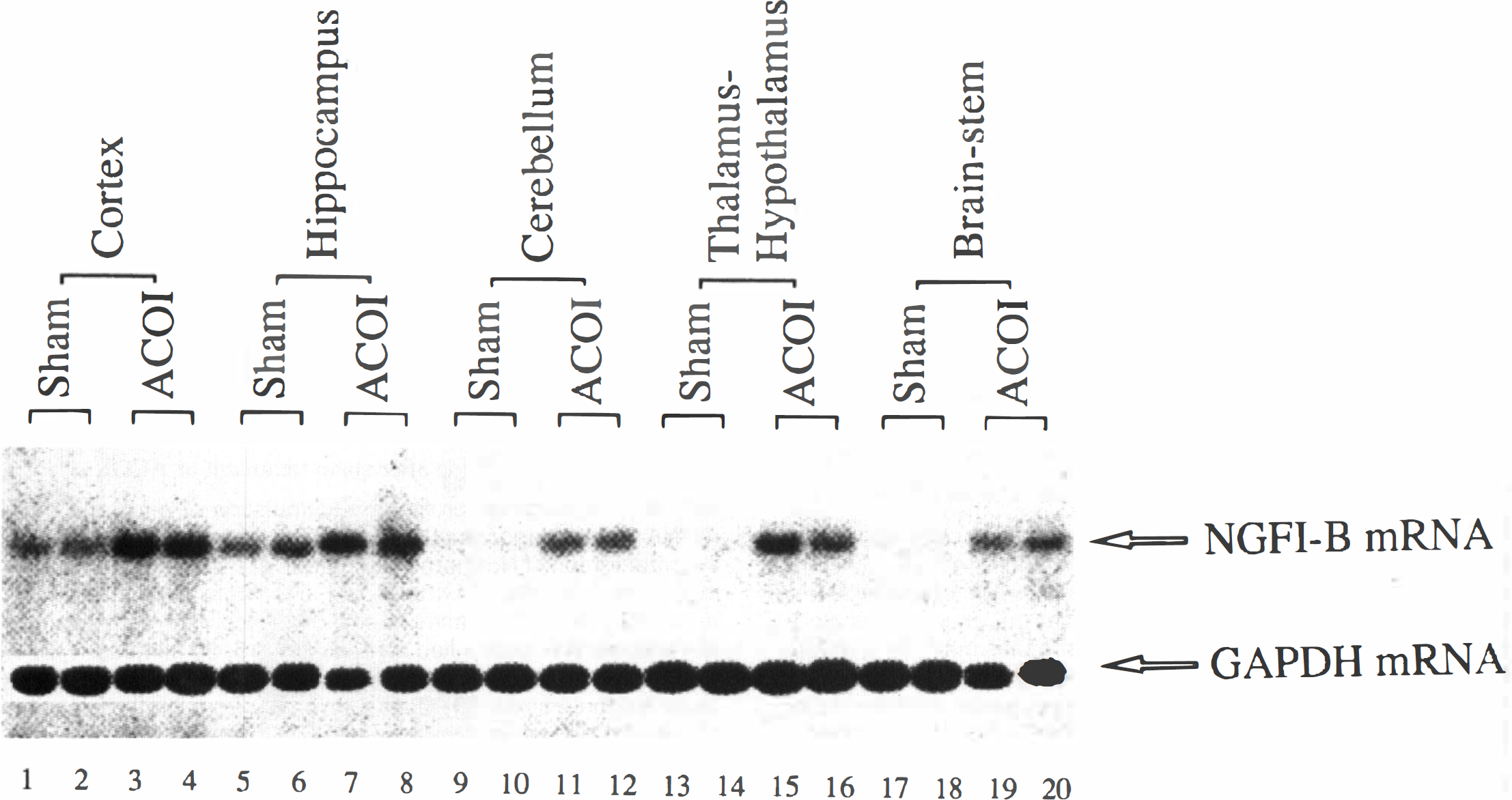
The spatial profile of accumulation of NGFI-B mRNA in the different brain regions of the mice after the ACOI. Figure shows an autoradiograph of Northern blot analysis of NGFI-B mRNA expression. All of the samples were from the animals that were decapitated at 30 minutes after the ACOI or sham-intoxication. Lanes 1 to 4, 5 to 8, 9 to 12, 13 to 16, 17 to 20 indicate the cerebral cortex, hippocampus, cerebellum, thalamus-hypothalamus, and brainstem, respectively. The first two lanes in each brain region were duplicate samples from the mice with sham intoxication and the last two from the animals with ACOI. The experiments were repeated three times. The autoradiograph for the striatum and olfactory bulb is not shown.
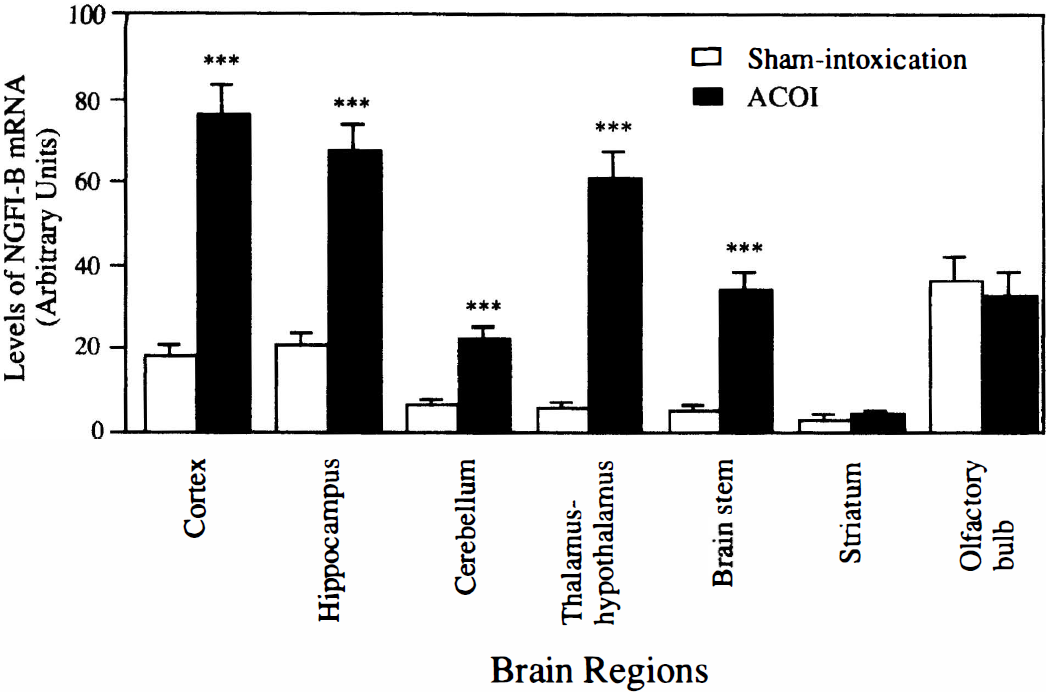
Quantitative data from the three experiments in Fig. 7 are shown. ***P < 0.001, compared with those of the sham-intoxication mice, determined by Student's t-test. ACOI, acute carbon monoxide intoxication.
Regional induction of c-fos mRNA in the brain after acute carbon monoxide intoxication
Regional examination of c-fos mRNA expression at 30 minutes after the ACOI revealed that the neuroanatomical distribution of the mRNA was very similar to that of NGFI-B mRNA. Figure 9 shows an autoradiography of Northern blot analysis. A dramatic increase in c-fos mRNA levels was observed in the cerebral cortex, hippocampus, cerebellum, thalamus-hypothalamus, and brainstem, but neither in the striatum nor in the olfactory bulb. Similar results were replicated in three separate experiments and quantitative data from the three experiments are shown in Fig. 10.
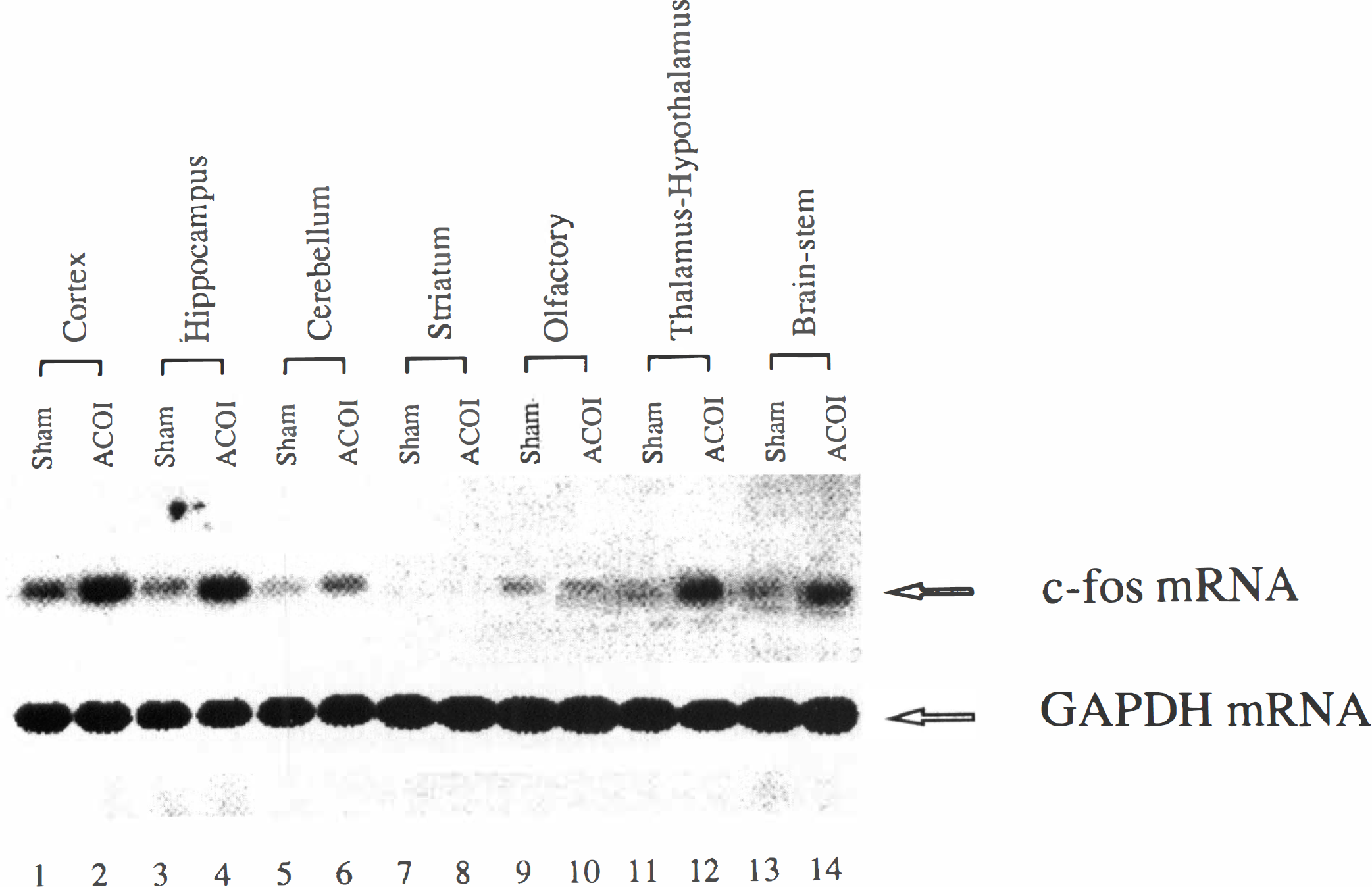
The spatial profile of accumulation of c-fos mRNA in the different brain regions of the mice after the ACOI. Figure shows an autoradiograph of Northern blot analysis of c-fos mRNA expression. All of the samples were obtained from the animals that were decapitated at 30 minutes after the ACOI or sham-intoxication. Lanes 1 to 2, 3 to 4, 5 to 6, 7 to 8, 9 to 10, 11 to 12, and 13 to 14 indicate the cerebral cortex, hippocampus, cerebellum, striatum, olfactory bulb, thalamus-hypothalamus, and brainstem, respectively. The first lane in each region was the sample from the sham-intoxication mice and the other from the ACOI mice. The experiments were performed three times.
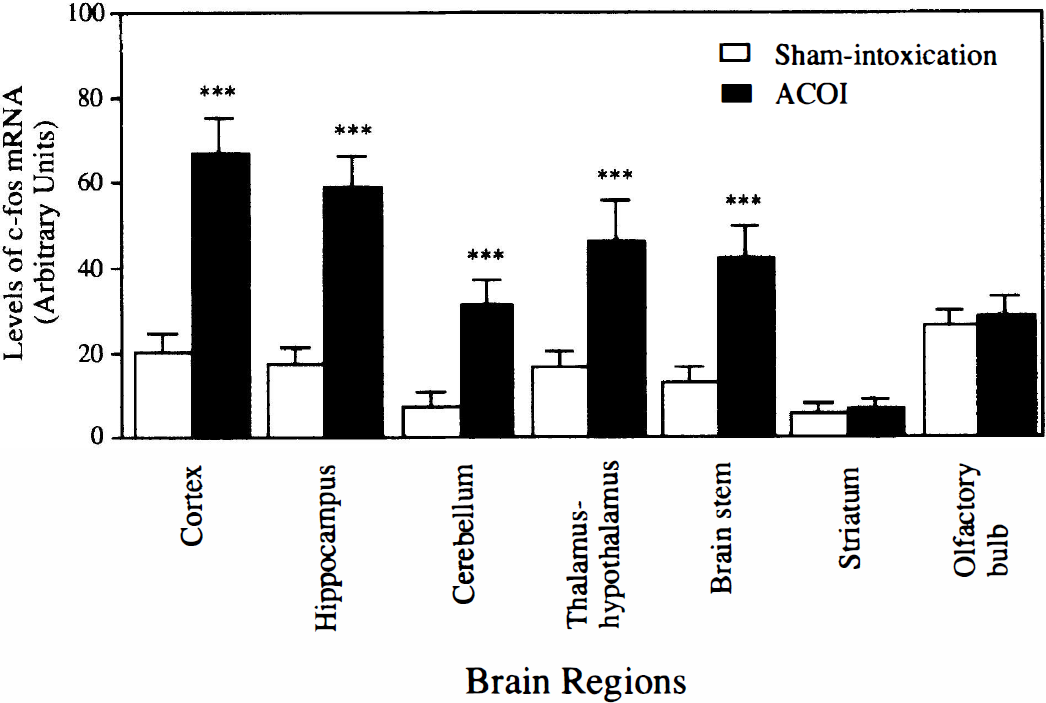
Quantitative data from the three experiments in Fig. 9 are shown. ***P < 0.001, compared with those of the sham-intoxication mice, determined by Student's t-test. ACOI, acute carbon monoxide intoxication;
DISCUSSION
Early and transient expression of IEG has been documented in the brain after various kinds of insults including hypoxia and/or ischemia, and trauma (Kinouchi et al., 1994a;Neumann-Haefelin et al., 1994; Dragunow et al., 1990). These early genomic responses have been sug-gested to be crucial in the regulation of the expression of certain target genes (Pennypacker et al., 1995; Chiu et al., 1988; Liu and Chen, 1994). Most of the previous studies pointed out that the temporal feature of the induction of IEG was directly related to the duration of insults (Kindy et al., 1991; Wessel et al., 1991; Take-moto et al., 1995). In the models of ischemia, for example, the induction of IEG mRNA for c-fos, c-jun, and NGFI-B reached a peak at 1 to 2 hours after reperfusion (Wessel et al., 1991; Neumann-Haefelin et al., 1994; Kamme et al., 1995). In the models of ischemia plus hypoxia, the maximal induction of IEG mRNA was also found at 1 to 2 hours after the insults (Dragunow et al., 1994; Munell et al., 1994; Gubits et al., 1993). In all of these models the duration of the insults lasted for more than 10 minutes to a few hours. However, in the case of brief cerebral ischemia or traumatic brain injury, the maximal induction of IEG mRNA was found at 15 to 30 minutes (Takemoto et al., 1995; Dragunow et al., 1990). Also in the present study, the induction of the IEG mRNA was most evident at 15 to 30 minutes. These results would seem to imply that brief durations of isch-emia or CO exposure would lead to early induction of IEG–independent of what the stimulus was.
An important finding of this study is the selective regional induction of IEG in the brain. The preference of induction of NGFI-B and c-fos in the cerebral cortex and hippocampus is consistent with the findings that either region is most vulnerable to ACOI (Ginsberg, 1985; Sawada et al., 1983; Nabeshima et al., 1991b). The cortex and hippocampus have been shown to be the key structures for the processes of learning and memory (Zola-Morgan and Squire, 1986; Winkler et al., 1995). Their vulnerabilities to ACOI have been considered to be involved in the development of the post-ACOI-delayed neurological sequelae (Ginsberg, 1985; Nabeshima et al., 1991b). Thus, the accumulation of the mRNA in these regions indicates a potential link to the delayed neurological manifestations as well as to the delayed pathological changes associated with ACOI. The activation of the IEG in the thalamus-hypothalamus is in agreement with the observations made after ischemia (Kinouchi et al., 1994a; Wessel et al., 1991) or other insults (Chan et al., 1993), indicating a vulnerability of these regions to the ACOI. The distribution of the IEG in these brain regions is very similar to that observed in hypoxia or ischemia models (Wessel et al., 1991; Kinouchi et al., 1994 a,1994 b; Takemoro et al., 1995), indicating that the ACOI produces a carbon monoxide tissue hypoxia or the hypoxia complicated with an ischemia in the brain. In view of the carboxyhemoglobin levels, half-life of car-boxyhemoglobin (15 minutes), and especially the very short duration of the loss of righting reflex (data not shown), it seems likely that the major cause for the triggering of the IEG expression in the brain is the carbon monoxide tissue hypoxia rather than the hypoxic ischemia. However, we can not exclude a possibility that prolonged hypotension with cerebral ischemia after ACOI might induce the expression of IEG in the brain.
It should be noted that the induction of the IEG was also found in the cerebellum. This region contributes primarily to motor coordination and control. A great number of experiments recently have indicated that the cerebellum also plays a potential role in cognitive functions (for review see Fiez, 1996). Although granular cells as well as Purkinje cells in the cerebellum are vulnerable to hypoxia, ischemia, and ACO1, the incidence of damage in this structure is low, and the damage, if any, shows a lesser severity than that in the cortex and hippocampus (Hara et al., 1995, Ng et al., 1989; Lapresle and Fardeau, 1967; for review see Ginsberg and Myers, 1976). The blood vessels to the cerebellum are different from those to the cortex or hippocampus. After ischemia, blood flow to the cerebellum recovers more rapidly than in the rest of the brain during the reperfusion phase, and thus the cerebellum endures less damage after ischemia than other regions (Ginsberg et al., 1985). In fact, previous studies show that the expression of IEG in cerebellum after ischemia, by the methods such as common carotid artery or four-vessel ligation, showed an earlier tendency to peak and to return to the basal level than that in the cortex or hippocampus (Wessel et al., 1991). However, the present study showed that the temporal feature of the induction of the IEG in the cerebellum was similar to that in the cortex or hippocampus. Thus, our results indicate that the ACOI exerts a diffuse hypoxia rather than an ischemia or hypoxic ischemia in the cerebellum.
Another interesting finding is the induction of these IEG in the brainstem. This region is associated with the regulation of respiratory and cardiovascular activities. The pathological examination indicated that the neurons in this area are particularly resistant to hypoxia, ischemia, or ACOI (for review see Ginsberg and Myers, 1976; Graham, 1992). However, studies on the neuronal activities after hypoxia in the brainstem revealed that they are not only sensitive to the changes of the local oxygen concentration, but also to the afferent stimulation from the carotid baroreceptors and chemoreceptors (Donoghue et al, 1984; Fung and St, 1995). It has been reported that electrical stimuli to these receptors can induce Fos expression in both the catecholaminergic and serotoninergic neurons in the brainstem (Erickson and Millhorn, 1994), where these neurons control the respiration and arterial pressure (Eldridge and Millhorn, 1981; Philippu, 1988). The present findings together with these previous observations suggest that IEG are inducible not only by the neuronal injury but also by the neuronal response to the extracellular stimuli such as ACOI.
The striatum is another region that is vulnerable to ischemia, hypoxia, and ACOI (Ginsberg et al., 1985; Nabeshima et al., 1991b; Ginsberg, 1985). However, in this study, significant induction of these IEG was not observed in this area. Similar results were also observed in other ischemia models (Neumann-Haefelin et al., 1994; Kinouchi et al., 1994a, b ; Welsh et al., 1992). Because the blood vessels to the striatum are relatively small and with few collaterals, the deficiency in the blood supply after ischemia or hypoxia seems to be most severe (Nagasawa and Kogure, 1989; Ginsberg et al., 1985). The pathological observations including neuron loss and changes in the neurotransmitter systems after ACOI are consistent with these features (Penney, 1990; Nabeshima et al., 1991b). It has been suggested that the severe deficiency in the energy metabolism in the neurons will render them incapable of making mRNA in response to external stimuli (Kinouchi et al., 1994a;Welsh et al., 1992). This may be a possible explanation for the absence of IEG mRNA induction in this region in the present study. In addition, the induction of the IEG mRNA was not found in the olfactory bulb. This failure may not be explained by the energy metabolism deficiency, because the olfactory bulb receives abundant blood supply. It remains to be determined whether the olfactory bulb is more resistant to hypoxic insult than the other brain areas.
The intracellular pathway that mediates the expression of these IEG in the present study is still unknown. Recently, however, it has been shown that the induction of IEG after various kinds of insults including ischemia and hypoxia can be blocked by the treatment with NMDA receptor antagonists, e.g., MK-801 (Kinouchi et al., 1994b;Bading et al, 1995; Woodbum et al., 1993). In addition, it has been shown that activation of NMDA receptors contributes to the development of the delayed cognitive deficits and delayed hippocampal neuron loss after ACOI or ischemia (Nabeshima et al, 1991a;Simon et al., 1984; Ishimaru et al., 1992). Thus, the activation of NMDA receptors could be involved in the induction of IEG mRNA after ACOI.