
Abstract
Deep prepiriform cortex has an important role in modulating neurotransmission during limbic seizures. We used pharmacologic blockade of non-N-methyl-D-aspartate (NMDA) receptors to study excitatory circuitry from the deep prepiriform cortex to the hippocampus during global ischemia in rat. NBQX, a potent non-NMDA glutamate receptor antagonist, was microinjected stereotactically into the deep prepiriform cortex before global ischemia for 10 min. Neuronal cell death in the hippocampus was evaluated quantitatively 72 h after ischemia. The NBQX-injected rats had a greater number of surviving cells in CA1 sector of hippocampus than did saline-injected controls or rats that received NBQX injections 1 mm from the target. Thus, excitatory amino acid-mediated circuitry emanating from deep prepiriform cortex modulates ischemic neuronal injury in the hippocampus.
Brain ischemia induces subsequent neuronal cell death in selectively vulnerable neurons of the hippocampus, cortex, and thalamus. The mechanism of this delayed neuronal necrosis is still uncertain. Elevated extracellular levels of excitatory amino acids (EAA), however, may have an initiating role in such cell death. NMDA (N-methyl-
A region in the deep prepiriform cortex is a modulatory site for seizure propagation (Piredda and Gale, 1985) and has been referred to as the area tempestas (AT). Injection of picomolar doses of bicuculline, a γ-aminobutyric acid (GABA) receptor antagonist, into the AT induces limbic seizures. Although the circuitry from the AT is currently uncertain, immediate early gene (c-fos) or heat-shock protein (HSP72) expression in limbic structures, including the hippocampus, thalamus, and amygdala, may indicate that the circuitry involves these structures (Shimosaka, et al., 1992; Maggio et al., 1993).
Local application of an NMDA receptor antagonist into the AT inhibits limibic seizures (Piredda and Gale, 1986). Seizure-induced cell injury in limbic structures is also modulated through this anatomic system. Microinjection of an NMDA antagonist into the deep prepiriform cortex attenuates hippocampal injury in kainate-induced seizures, independent of seizure duration (Shimosaka et al., 1994). Also, injection of the non-NMDA receptor antagonist NBQX into the AT attenuates HSP expression in the hippocampus 24 h following global ischemia (Simon and Kawaguchi, 1996). Therefore, we studied the influence of NBQX injection into the AT on neuronal cell death 72 h after global ischemia. NBQX injection 1 mm away from the AT was examined to confirm the specificity of this effect.
MATERIALS AND METHODS
Implantation of guide cannulae
Twenty-nine male Sprague–Dawley rats weighing 300 to 350 g were used and divided into three groups: In Group I (n = 10), saline was injected into the AT: in Group II (n = 8), NBQX (Novo Nordisk) was injected into the AT; and in Group III (n = 11), NBQX was injected into a site 1 mm from the AT. The rats were anesthetized with 10% chloral hydrate (400 mg/kg i.p.) and mounted on a stereotaxic apparatus to implant two stainless-steel guide cannulae (22G) for microinjection of NBQX or saline through internal-injection cannulae (28G) into the AT and one cannula for monitoring the brain temperature. Tips of the injection cannulae and a thermocouple probe for monitoring brain temperature were projected 1.0 mm beyond the guide-cannulae tips. The coordinates of the AT were 3.8 mm anterior to bregma, 3.5 mm lateral to midline, and 6.5 mm below the dura with the incisor bar positioned 5 mm above the interaural line (Groups I and II). The coordinate of the site away from the AT was 1 mm medial to AT (Group III). The site for monitoring brain temperature was in right striatum: 1 mm anterior to the bregma, 2.5 mm lateral to the midline, and 5 mm below the dura. The cannulae were attached to the skull with dental acrylate.
Four-vessel occlusion
Three days after implantation of the guide cannulae, four-vessel occlusion (Pulsinelli and Brierley, 1979) was performed. The rats were anesthetized initially by face mask with 3% halothane in a mixture of 67% N2O and 30% O2. Atropine sulfate (150 mg/kg) was administered intraperitoneally. After endotracheal intubation, the rats were ventilated mechanically by a rodent respirator with a mixture of 30% O2, 69% N2O, and 1% halothane. Rectal temperature was monitored and maintained at 37.0 ± 0.5°C with a heating pad throughout the experiments. The brain temperature was monitored by thermocouple probe in the right striatum and maintained at 37.0 ± 0.2°C by heating lamp. The left femoral artery was cannulated to monitor the MABP continuously and to provide serial measurements of arterial oxygen and carbon dioxide tensions, pH, and blood glucose. The left femoral vein was cannulated to administered pancuronium bromide (0.2 mg/kg). Under an operating microscope, an incision was made behind the occipital bone directly overlying the first two cervical vertebrae. The paraspinal muscles were separated from the midline, and the bilateral vertebral arteries were exposed and electro-coagulated between the first and the second vertebrae. The common carotid arteries and external carotid arteries then were exposed bilaterally and the external carotid arteries ligated. Halothane was discontinued before drug injection into AT. NBQX or saline was microinjected into bilateral AT (Groups I and II), or NBQX was injected 1 mm away from the AT bilaterally (Group III) using a syringe pump 15 min before the induction of ischemia. NBQX was dissolved in 0.1 ml of 0.1 N NaOH, and 0.8 ml of 5.5% glucose was added; the pH adjusted to 8.0 with 0.1 N HCl. This solution was diluted to 3.7 mg/ml (10 nmol/μl) with saline. The pH of the saline also was adjusted to 8.0 with 0.1 N NaOH. Saline and NBQX microinjection rates were 0.1 μl/min; the total volume equaled 0.5 μl (NBQX total dose was 5 nmol). The rats were subjected to 10 min of global ischemia (confirmed by EEG flattening) by clamping the common carotid arteries bilaterally using microarterial clips.
Histology
The rats were sacrificed 72 h after four-vessel occlusion with 10% chloral hydrate (400 mg/kg i.p.) and perfused transcardially with saline (100 ml) and 4% paraformaldehyde (400 ml, 0.1 M phosphate buffer at pH 7.4). The brains were removed, kept in the same fixative for 10 days at 4°C, embedded in paraffin, and cut to 6-μm sections. The sections were stained with cresyl violet. The site of drug injection was confirmed microscopically in each brain. The number of surviving cells in the 0.41-mm length of CA1b layer on bilateral hippocampal sides was counted by a blinded investigator.
Statistical analysis
Cell counts and physiological variables were evaluated statistically with one-way analysis of variance (ANOVA) followed by Scheffe's post hoc test. All values were expressed as mean ± SD; p < 0.05 was regarded as significant.
RESULTS
Physiological variables
There were no significant differences in MABP, PaO2, PaCO2, pH, blood glucose, or brain temperature in the three groups (Table 1).
Physiological variables a
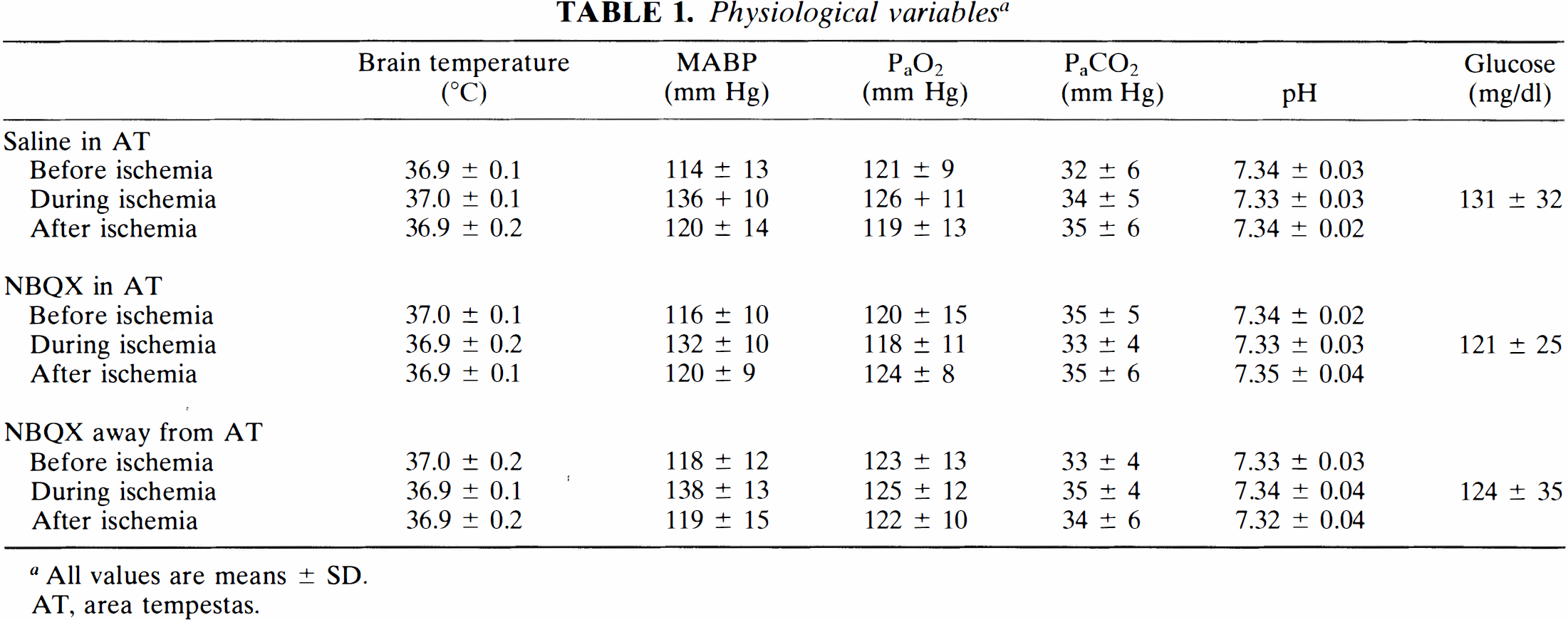
All values are means ± SD.
AT, area tempestas.
Histology
In the saline-injected group (Group I), the number of surviving cells in CA1 was decreased compared with controls (Fig. 1A, B). In contrast, animals pretreated with NBQX injection into the AT (Group II) showed a large number of surviving cells in CA1 (Fig. 1C, D). Animals in which NBQX was injected 1 mm from the AT (Group III) showed cell loss in CA1, similar to the saline group (Fig. 1E, F). The number of surviving cells in Group II was significantly greater than in Groups I and III (Fig. 2).
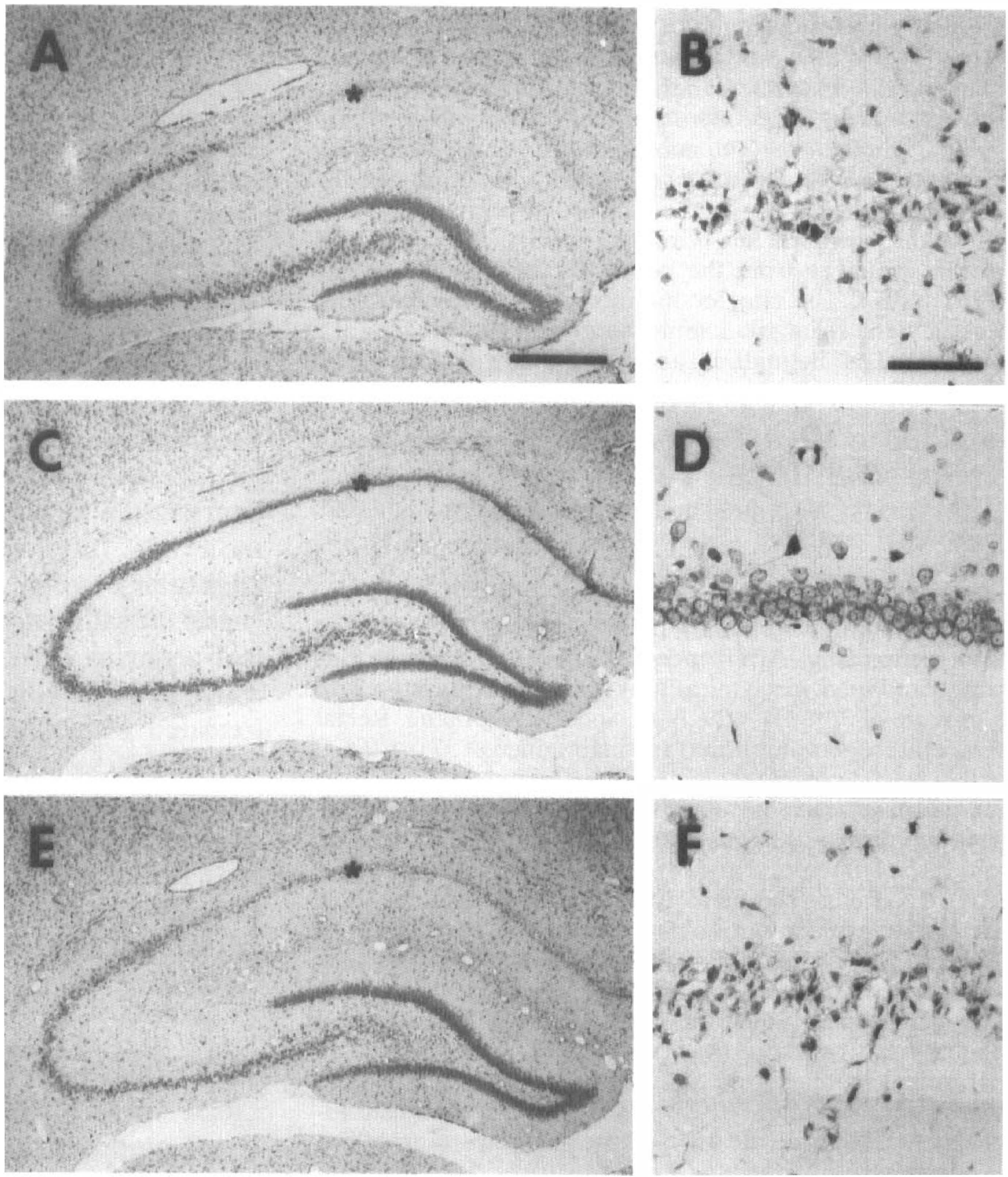
Effect of NBQX injected into the area tempestas (AT) bilaterally on the number of surviving cells in the hippocampus 72 h after 10 min of global ischemia (cresyl-violet staining).
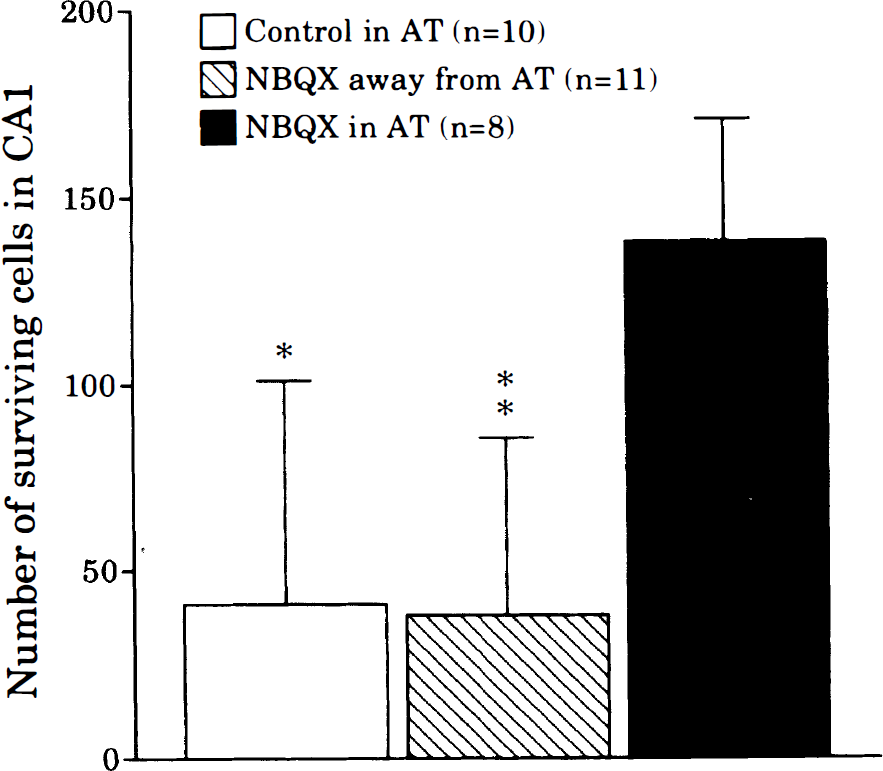
The number of surviving cells in hippocampal CA1b subsector 72 h after 10 min of global ischemia. *p < 0.005; **p < 0.001 (relative to NBQX in the area tempestas (AT), one-way analysis of variance ANOVA followed by Scheffe's test). Error bars indicate SD.
DISCUSSION
The results presented here suggest that pharmacologic blockade of EAA neurotransmission at the deep prepiriform cortex, a site that modulates neurotransmission in the limbic system, can provide neuroprotection at a remote site during ischemia. In brain ischemia, elevation of extracellular EAA concentrations activates target receptors, including NMDA, non-NMDA, and metabotropic receptors, which leads to increases in intracellular free calcium levels and neuronal cell injury (Choi, 1987; Siesjö and Bengtsson, 1989). Pharmacologic blockade of postsynaptic EAA receptors (Gill et al., 1987; Swan et al., 1988) or inhibition of EAA release (Meldrum et al., 1992; Leach et al., 1993) prevents hippocampal neuronal cell death in global ischemia or decreases infarct volume in focal ischemia. Physiological inhibition of EAA release using hypothermia also attenuates ischemic neuronal injury (Busto et al., 1989). Anatomical approaches that target this neuronal network in the brain have protective effects on ischemic neuronal injury. For example, lesions of the CA3 subsector (Benveniste et al., 1984), the dentate granular cell layer (Johansen et al., 1986), and the entorhinal cortex (Jørgensen et al., 1987) protect against CA1 pyramidal cell death in global ischemia. Further, nigral lesions reduce striatal infarct volume in focal ischemia (Buisson et al., 1992). Thus, previous experiments using lesioning techniques have demonstrated protection against hippocampal ischemic injury by interruption of limbic circuitry. We now provide a pharmacologic correlate and suggest a critical site of action.
The deep prepiriform cortex contains a site, the AT, that modulates neurotransmission in the limbic system during seizures (Piredda and Gale, 1985). Bicuculline (a GABA-receptor antagonist) or kainate (a non-NMDA agonist) injected into the AT induces limbic seizures. Microinjection of AP7, an NMDA-receptor antagonist, into the AT prevents bicuculline-induced limbic seizures (Piredda and Gale, 1986). Immediate early gene expression (c-fos) following bicuculline-induced seizure propagation from AT suggested that the pathway responsible for these effects involved the piriform cortex, thalamus, and hippocampus (Maggio et al., 1993). Previously, the AT was shown to modulate neuronal hippocampal injury. NMDA-receptor blockade with the competititve antagonist AP7 injected into the AT decreased neuronal damage in the hippocampus resulting from limbic seizure induced by systemic kainate (Shimosaka et al., 1994), implying that blockade of excitatory neurotransmission at the AT resulted in neuroprotection at a remote site (hippocampal pyramidal cells). This protective effect occurred without attenuation of the seizure duration.
We therefore applied this concept to another system in which neuronal injury is induced by EAA toxicity: global ischemia. Pretreatment with NBQX, a non-NMDA antagonist, at the AT attenuates HSP72 expression, a marker of cell injury (Gonzalez et al., 1989), in both CA1 and CA3 subsectors 24 h following global ischemia (Simon and Kawaguchi, 1996). Thus, excitatory circuitry from the AT affects both CA1 and CA3 during brain ischemia. The data presented here extend this observation by demonstrating that non-NMDA blockade in AT attenuates neuronal cell death as well as cell injury in the hippocampal CA1 subsector after global ischemia. Further, injection of NBQX 1 mm from the AT had no effect on hippocampal cell death, which implicates a specific site in the drug's action. Thus, the AT is an attractive site for modulating neuronal cell death in the hippocampus during global ischemia. The mechanism of such neuroprotection is not demonstrated by these experiments. Candidate mechanisms being explored include the induction of antiapoptotic gene expression, attenuation of death promotor gene product, and attenuation of hippocampal glutamate release in the setting of ischemia. The study also suggests a new target for neuroprotective compounds in ischemia. Perhaps neuroprotective drug binding to AT might predict neuroprotective efficacy in global ischemia.
Footnotes
Acknowledgment:
This work was supported in part by NIH grant NS 24728 (RPS).