
Abstract
Effects of nicotine treatment (4.5 mg/kg of nicotine-free base/day administered s.c. by osmotic minipumps for 14 days) on focal ischemic stroke and expression of tissue plasminogen activator (t-PA) and plasminogen activator inhibitor-1 (PAI-1) in cerebral microvessels were studied in rats in vivo using a reversible (1 h) middle cerebral artery occlusion model. Plasma levels of nicotine and its major metabolite cotinine after 14 days of treatment were 88 and 364 ng/ml, respectively. Nicotine treatment resulted in 35–40% (p <0.001) decrease in the blood flow in the periphery of the ischemic core during reperfusion, an increase in the neurologic score of 2.6-fold (p <0.01), and 36% (p <0.05) and 121% (p <0.01) increases in the injury and edema volume in the pallium, respectively. A free pool of brain microvascular t-PA antigen was completely depleted by nicotine, while the expression of the PAI-1 antigen and/or PAI-1-t-PA complexes remained unchanged. The relative abundance of cerebromicrovascular t-PA mRNA transcript versus β-actin mRNA transcript did not change with nicotine. It is concluded that chronic nicotine treatment impairs the restoration of blood flow, worsens the neurologic outcome, and enhances brain injury following an ischemic insult. These nicotine effects are associated with depletion of brain microvascular t-PA antigen.
Cigarette smoking has been accepted as a major factor that increases the risk of coronary disease, peripheral vascular disease and stroke (Gill et al., 1989). Although epidemiological studies of smokers may not directly translate into equivalent effects of nicotine (a major constituent of cigarette smoke), few recent clinical studies have indicated that application of nicotine patches alone could precipitate ischemic stroke in chronic smokers (Jackson, 1993; Pierce, 1994). It has been suggested that smoking products and/or nicotine may predispose to cerebral thrombosis, possibly by causing an imbalance between brain vascular coagulation and fibrinolytic systems (Allen et al., 1985; Folts, 1988; Hashimoto et al., 1988). Altered coagulation and abnormal fibrinolysis have been shown to be present in the blood of ischemic stroke patients (Fisher and Francis, 1990), including chronic smokers (Folts, 1988; Hashimoto et al., 1988; Ozdemir et al., 1992). In spite of this, evidence demonstrating a direct link between chronic exposure to nicotine and stroke, on the one hand, and altered brain endothelial thrombotic potential, on the other, is still limited.
Tissue plasminogen activator (t-PA) and plasminogen activator inhibitor-1 (PAI-1) play key roles in the control of fibrinolysis within the vascular system (Collen, 1980; Loskutoff et al., 1993). Injured or stimulated endothelium releases t-PA, which preferentially converts fibrin-bound plasminogen into plasmin—an active serine protease involved in a broad spectrum of extracellular proteolytic processes, including fibrinolysis and thrombolysis (van Hinsbergh et al., 1991). Much of t-PA in normal plasma and in tissues is inactive and complexed to PAI-1, which is considered to be a primary endogenous inhibitor of t-PA in plasma and tissues (Loskutoff et al., 1993). It has been suggested that t-PA and PAI-1 expression may vary among species and tissues, and with the type of stimulation (Emeis et al., 1995 versus Biemond et al., 1995; Levin and del Zoppo, 1994 versus Zlokovic et al., 1995b; Padro et al., 1994; Kollros et al., 1994 versus Kittaka et al., 1996). Expression of t-PA and PAI-1 has been shown recently in bovine (Kollros et al., 1994) and human (Shatos et al., 1995) brain microvascular endothelial cells in vitro. Recently, we reported that t-PA is normally expressed in vivo in cerebral capillaries in rodents, and that both t-PA antigen and t-PA mRNA are preserved in microvessels isolated immediately ex vivo from rodent brain (Zlokovic et al., 1995b; Zlokovic et al., 1995a; Kittaka et al., 1996). In addition to proposed fibrinolytic function and vascular effects of t-PA (Zlokovic et al., 1995b), other roles for t-PA in “mature” brain include mediation of activity-dependent synaptic plasticity—as, for example, during motor learning (Seeds et al., 1995) and long-term potentiation (Qian et al., 1994)—and a role in some types of neuronal degeneration induced by excitotoxins (Tsirka et al., 1995).
Nicotine can mediate t-PA release from bovine aortic endothelial cells in vitro (Kuo et al., 1989), and acute smoking may cause a transient increase in plasma t-PA antigen in human nonsmokers (Allen et al., 1985). However, in habitual smokers with chronically elevated plasma nicotine levels, both plasma t-PA activity and the fibrinolytic activity of systemic vessel walls are significantly reduced in comparison to those in nonsmokers (Allen et al., 1985; Kjaelgaard and Larsson, 1986; Haire et al., 1989; Ozdemir et al., 1992). In the present study, we used a reversible middle cerebral artery (MCA) occlusion model in rats to examine the effects of chronic nicotine administration on focal ischemic stroke in relation to brain microvascular t-PA and PAI-1 expression. Preliminary results have been reported elsewhere (Sun et al., 1995; Zlokovic et al., 1996).
METHODS
Male Sprague–Dawley (Charles River Breeders, Hollister, CA, U.S.A.) rats weighing 260–300 g each were housed under diurnal light conditions, with unlimited access to food and water, and allowed a minimum of 3 days for acclimation. All procedures were done in accordance with the Animal Care Guidelines at the University of Southern California as approved by the National Institutes of Health.
Nicotine administration
Rats were implanted with osmotic minipumps (Alzet 2002, Alza Corporation, Palo Alto, CA, U.S.A.) s.c. for continuous delivery of either nicotine tartrate (n = 24) in 0.9% NaCl (4.5 mg/kg) of nicotine-free base/day) or vehicle (n = 24) only 0.9% NaCl) for 14 days, according to published procedure (Murrin et al., 1987). Pumps were implanted under strictly aseptic conditions. No sign of inflammation or infection was observed in the area of pump placement. By comparing 14-day sham-operated animals with animals implanted with osmotic minipumps and vehicle only, it was confirmed that the pump placement procedure does not have any effect on the parameters in the present study including physiologic variables, blood flow and head temperature, neurologic scores, or neuropathologic outcome in a model of reversible MCA occlusion. Pump placement does not affect the expression of several antigens in brain including α1, α2, α3, β1 and β1 subunits of the sodium pump and γ-glutamyl-transpeptidase (Wang et al., 1994) and the t-PA in the present study. Levels of nicotine and its major metabolite cotinine were determined in plasma (Haley et al., 1983).
Focal ischemia study
Rats were deprived of food for 12 h prior to surgery. Reversible MCA occlusion was performed in nicotine-treated (n = 12) and control (n = 12) rats using an intraluminal thread technique (Zea Longa et al., 1989), as recently modified in our laboratory (Kittaka et al., 1996). The procedure involved initial anesthesia with metofane and maintenance with 50 mg/kg i.p. pentobarbital. Atropine methyl nitrate (0.18 mg/kg i.p.) was given as premedication to prevent airway obstruction by mucus formation. Animals were allowed to breath spontaneously. Rectal temperature was maintained at 37°C by a thermostatically regulated heating pad. A PE-50 catheter was introduced into the right femoral artery for continuous monitoring of MABP, as well as for repeated sampling of blood for serial measurements of arterial oxygen (Pa
Blood flow and head temperature measurements
Cortical CBF was monitored before and during occlusion and within 1 h of reperfusion by laser–Doppler flowmetry (LDF) (n = 12/group). LDF probes (0.8 mm diameter) positioned at 0.1 mm above the dura over the cortical surface were connected to a tissue perfusion monitor (Transonic BLF21, Ithaca, NY, U.S.A.). In the hemisphere ipsilateral to the MCA occlusion, coordinates were: point A, 1 mm posterior to the bregma and 5.4 mm lateral to the midline; point B, 1 mm posterior to the bregma and 2.1 mm lateral to the midline; and point D, 1 mm anterior to the bregma and 3.4 mm lateral to the midline. In the contralateral hemisphere, point C was 1 mm posterior to the bregma and 5.4 mm from the midline. At each point, a small burr hole was drilled in the skull, and the bone carefully removed to prevent damage to the cortex. Steady-state baseline values were recorded before occlusion; CBF during occlusion and reperfusion was expressed as a percentage of the baseline values. Head temperature was monitored with a 36-gauge thermocouple temperature probe in the temporalis muscle connected to a digital thermometer/thermoregulator, model 9000 (Omega, Stanford, CT, U.S.A.).
Neurologic examination
Neurologic examination (n = 12/group) was performed 24 h after reperfusion, just prior to killing. Neurologic outcome was scored on a six-point scale, as described (Kittaka et al., 1996): a score of 0 indicated no neurologic deficit (normal); a score of 1 (failure to extend left forepaw fully) indicated a mild focal neurologic deficit; a score of 2 (circling to the left) indicated a moderate focal neurologic deficit; and a score of 3 (falling to the left) indicated a severe focal deficit. Rats with score 4 did not walk spontaneously and had a depressed level of consciousness; a score of 5 was stroke-related death (not observed in the present study).
Measurement of volume of injury
The brain was placed into ice-cold phosphate-buffered saline and, after chilling for 20 min, sliced into 2-mm coronal sections. The area of injury (n = 12/group) was delineated by incubation of unfixed 2-mm coronal brain slices in 2% triphenyltetrazolium chloride in 0.173 M sucrose, 50 mM K+ phosphate buffer (pH 7.4) for 20 min at 37°C and then stored in 10% formalin. Serial coronal sections were displayed on a digitizing video screen using an imaging system (Jandel Scientific, San Rafael, CA, U.S.A.), and the area of nonstaining tissue was determined in each section. The volume of injury was calculated by summing up affected areas from each coronal section and multiplying by the thickness of each section. The volumes of the control and lesioned hemispheres were calculated, and the amount of injury was expressed as a percent of total cerebral volume (% injury volume) and in absolute terms (mm 3). The injury volume of gray matter structures (i.e., the pallium and striatum) was corrected for brain edema in the lesioned hemisphere by subtracting the volume of normal tissue in the lesioned hemisphere from the volume of the control (normal) hemisphere, based on a described method (Swanson et al., 1990). The edema volume was calculated by subtracting the volume of the normal gray matter in the control hemisphere from the volume of gray matter in the lesioned hemisphere (Swanson et al., 1990). Measurement of the volume of injury was done separately for the pallium and striatum. A coronal section from each brain was obtained at the level of the optic chiasm. Representations of the injury areas of individual sections from each animal in the control and nicotine-treated groups were superimposed in order to gain an impression of the topography and incidence of infarction. The boundary of infarction was redrawn for each rat on the corresponding coronal section, and the following areas delineated: the infarct area in which 100% of rats were affected, the infarct area in which ≥50% of rats were affected, and the infarct area in which <50% of rats were affected.
Isolation of brain capillaries
Brain capillaries were isolated from nicotine-treated (n = 12) and control (n = 12) rats using a modified mechanical homogenization technique (Zlokovic et al., 1993). Briefly, brains were immediately removed from the skull and immersed in ice-cold buffer B (103 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 12 mM KH2PO4, 1.2 mM MgSO4, and 15 mM HEPES, pH, 7.4). The cerebral cortical mantles were rapidly freed of meninges (i.e., pial vessels); the arteries of the circle of Willis, veins, and choroid plexi were discarded; and cerebral cortices were used to isolate the capillaries. Brain homogenization in buffer A (buffer B + 25 mM HCO3, 10 mM glucose, 1 mM sodium pyruvate, and 1 g/100 ml dextran, molecular mobility (Mr) = 64,000), using a hand-held Teflon homogenizer, was followed by dextran density centrifugation at 5,800 × g at 4°C. The pellet was resuspended in buffer A and passed over a 85 μm nylon mesh. Arterioles and venules remained on top of the mesh, while capillaries, red cells, nuclei, and other debris were collected in the filtrate passing through the mesh. This filtrate was then passed over a column containing 3 × 4-cm glass beads (0.45-mm diameter) with 44-μm nylon mesh at the bottom, and the column washed with buffer B. Brain capillaries adhere to the glass beads while the other contaminants pass unimpeded. Capillaries were recovered by repeated gentle agitation of the glass beads in buffer A, and the supernatant with capillaries was decanted and spun at 500 × g for 5 min to obtain the final pellet. The purity of the cerebral capillary preparation was checked by light and phase microscopy. The cerebral capillaries were free of adjoining brain tissue, and preparations consisted primarily of capillaries, but also contained minor amounts (5–10%) of small arterioles, as described (Zlokovic et al., 1993; Zlokovic et al., 1995b). Capillary-depleted brain homogenates were centrifuged a second time at 5,800 × g at 4°C to obtain a final capillary-free brain supernatant. The presence of no more than minimal detectable activity of a specific cerebrovascular marker, γ-glutamyl transpeptidase, was used to confirm the absence of contamination of capillary-depleted brains with microvessels, but not to demonstrate that the microvascular preparations were capillaries only. A more detailed description of the capillary isolation procedure and the capillary (microvessels) preparation, as well as possible pitfalls in data interpretation, may be found elsewhere (Joo, 1993; Zlokovic, 1993).
Western blot analysis
Brain capillaries and capillary-depleted brains from nicotine-treated and control rats were homogenzied using a motor-driven Potter-Elvehjem teflon homogenizer in a buffer containing 5% sorbitol, 5 mM histidine, 25 mM imidazole, 1 mM EDTA at pH 7.5 with inclusion of proteolytic enzyme inhibitors, 0.5 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, and 1 mM amino-benzamidine dihydrocloride, as reported (Zlokovic et al., 1993; Zlokovic et al., 1995b). A rabbit polyclonal anti-mouse t-PA IgG fraction that detects t-PA only, and not PAI-1 complexes (Seeds et al., 1995; Zlokovic et al., 1995b) was obtained from Dr. N. Seeds. The specificity of this t-PA antibody was confirmed previously in our laboratory on Western blots containing conditioned medium from developing mouse cerebellar cells, which actively secrete t-PA, 1 ng human melanoma t-PA (World Health Organization international standard), and rat kidney extracts rich in t-PA (Seeds et al., 1995; Zlokovic et al., 1995b; Kittaka et al., 1996). Preabsorption of this t-PA antibody with mouse t-PA resulted in loss of immunoreactivity in t-PA-rich kidney tissue, and abolished detection of the material on Western blots (Seeds et al., 1995; Zlokovic et al., 1995b). A rabbit polyclonal anti-rat PAI-1 IgG fraction that immunoprecipitates PAI-1 and PAI-1-t-PA complexes was obtained from American Diagnostic Inc. (Greenwich, CT, U.S.A.) (product no. 1062). Gels were blotted onto diazotized paper and incubated overnight with polyclonal anti-t-PA (1:600 dilution) and anti-PAI-1 (1:200 dilution) antibodies. The enhanced chemiluminescence Western blotting detection system (Amersham, Arlington Heights, IL, U.S.A.) was used to detect t-PA and expose it to hyperfilm. Migration of t-PA, PAI-1, and PAI-1-t-PA complexes from tissue samples was compared with the relative mobilities of molecular weight standards run on the same gel. Chemillumigrams were scanned with a Hoefer GS 300 scanning densitometer. Each sample was analyzed at least two times and each lane was scanned three times. The linearity of the system for studied antigens (standard curves) was demonstrated to be 1–10 μg of total capillary proteins for t-PA (Zlokovic et al., 1995b), as well as for the PAI-1 antigen and PAI-1-t-PA complexes (not shown).
Reverse transcription (RT)-polymerase chain reaction (PCR) analysis
Total RNA (∼4 μg) was extracted with guanidium isothiocyanate from pooled capillary samples obtained from either four nicotine-treated or four control rats, and from capillary-depleted brains (Chomoczynski and Sacchi, 1987). This experiment was repeated three times (n = 12/group). The RNA was reverse transcribed using 1 μg oligo (dT)15 in 20 μl reaction buffer containing 1 mM each dNTP, 25 mM Tris-Cl (pH 8.3), 25 mM KCl, 5 mM MgCl2, 5 mM dithiothreitol, 0.25 mM spermidine, 10 u RNasin (Boehringer Mannheim, Indianopolis, IN, U.S.A.), and 10 u avian myeloblastosis virus (AMV) reverse transcriptase (Promega, Madison, WI, U.S.A.). The mixture was incubated at 42°C for 1 h, then at 52°C for 40 min followed by the addition of 160 μl of 10 mM Tris-HCl (pH 7.4), 1 mM EDTA. PCR was performed with 10 μl of the RT reaction mixture, 50 mM KCl, 10 mM Tris-Cl (pH 9.0), 0.1% Triton X-100, 1 mM MgCl2, 400 nM of each t-PA primer, 200 μM each dNTP and 5 u Taq DNA polymerase (Boehringer Mannheim) in a final volume of 50 μl. The sample was subjected to 30 cycles (94°C, 1 min; 58°C, 1 min; 72°C, 3 min) in a DNA Thermal Cycler (Perkin Elmer, Norwalk, CT, U.S.A.), and 5 μl of the PCR product was analyzed by electrophoresis on a 1% agarose gel. Primers for t-PA were: sense (5′-TGGCACCCACAGCTTTACCACATCCAAGG-3′) and anti-sense (5′-CTCCTGAGTCACCTGGCACGCGTCATGG-3′) corresponding to nucleotides 699–727 and 1,540–1,568, respectively, of the published sequence for rat t-PA (Ny et al., 1988). Amplification of β-actin by PCR was used as a nonchanging control for gene expression. Mouse β-actin primers were obtained from Stratagene (La Jolla, CA, U.S.A.). The RT-PCR analysis was semiquantitative as the relative change in t-PA mRNA expression was compared to β-actin. The number of cycles used (n = 30) was within the linear range of the amplification response for both genes.
Statistical analysis
Physiological variables, infarction, and edema volumes were compared between groups using Student's t-test. Non-parametric data (neurologic outcome scores) were subjected to the Kruskal-Wallis test. A p < 0.05 was considered statistically significant.
RESULTS
Physiologic variables
In nicotine-treated rats, plasma levels of nicotine and cotinine were significantly elevated after 14 days of treatment (Table 1). Nicotine and cotinine were undetectable in the control group. There was no significant difference in body weight between experimental and control groups, as previously observed with this dosage regimen (Murrin et al., 1987). No difference in physiologic variables (including temporalis muscle temperature) were noted between nicotine and control groups before MCA occlusion, during occlusion, or during reperfusion (Table 2).
Plasma levels of nicotine and cotinine in nicotine treated rats

Data are mean ± SE (n = 18). Rats were administered nicotine tartrate (4.5 mg/kg of nicotine-free base/day).
Physiologic variables
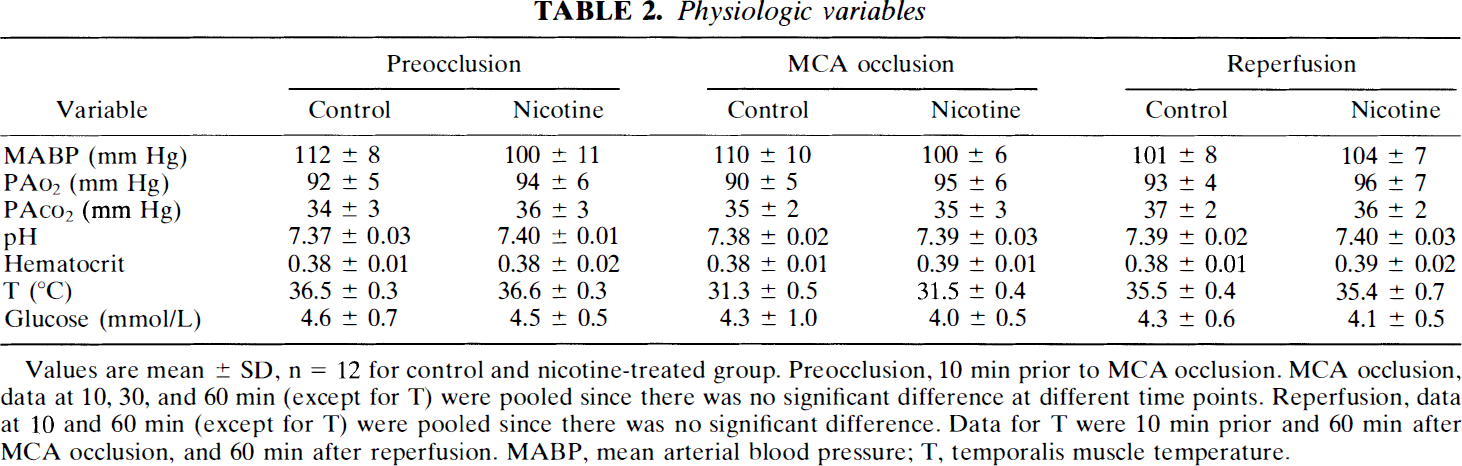
Values are mean ± SD, n = 12 for control and nicotine-treated group. Preocclusion, 10 min prior to MCA occlusion. MCA occlusion, data at 10, 30, and 60 min (except for T) were pooled since there was no significant difference at different time points. Reperfusion, data at 10 and 60 min (except for T) were pooled since there was no significant difference. Data for T were 10 min prior and 60 min after MCA occlusion, and 60 min after reperfusion. MABP, mean arterial blood pressure; T, temporalis muscle temperature.
Figure 1 illustrates changes in CBF during MCA occlusion and reperfusion. No significant difference in baseline tissue perfusion units were observed between nicotine and control rats, indicating similar preocclusion CBF values. Reductions in CBF during occlusion were comparable between the two groups, i.e., 17–19% (p < 0.001) and 15–16% (p < 0.001) of baseline in the ischemic core of control and nicotine-treated rats, respectively, and 34–37% (p < 0.001) and 31–35% (p < 0.001) of baseline in the periphery of the ischemic core of control and nicotine-treated rats, respectively. The trend towards CBF restoration during reperfusion was significantly impaired in the periphery of the ischemic core of nicotine-treated rats, and an increase from 60 to 65% (p < 0.01) of baseline was recorded 10–60 min of reperfusion. In contrast, during the same period of reperfusion, the CBF in control rats rose from 77 to 86%. In the ischemic core of both control and nicotine-treated rats, CBF was restored to ∼50% (p < 0.001) of preocclusion values.
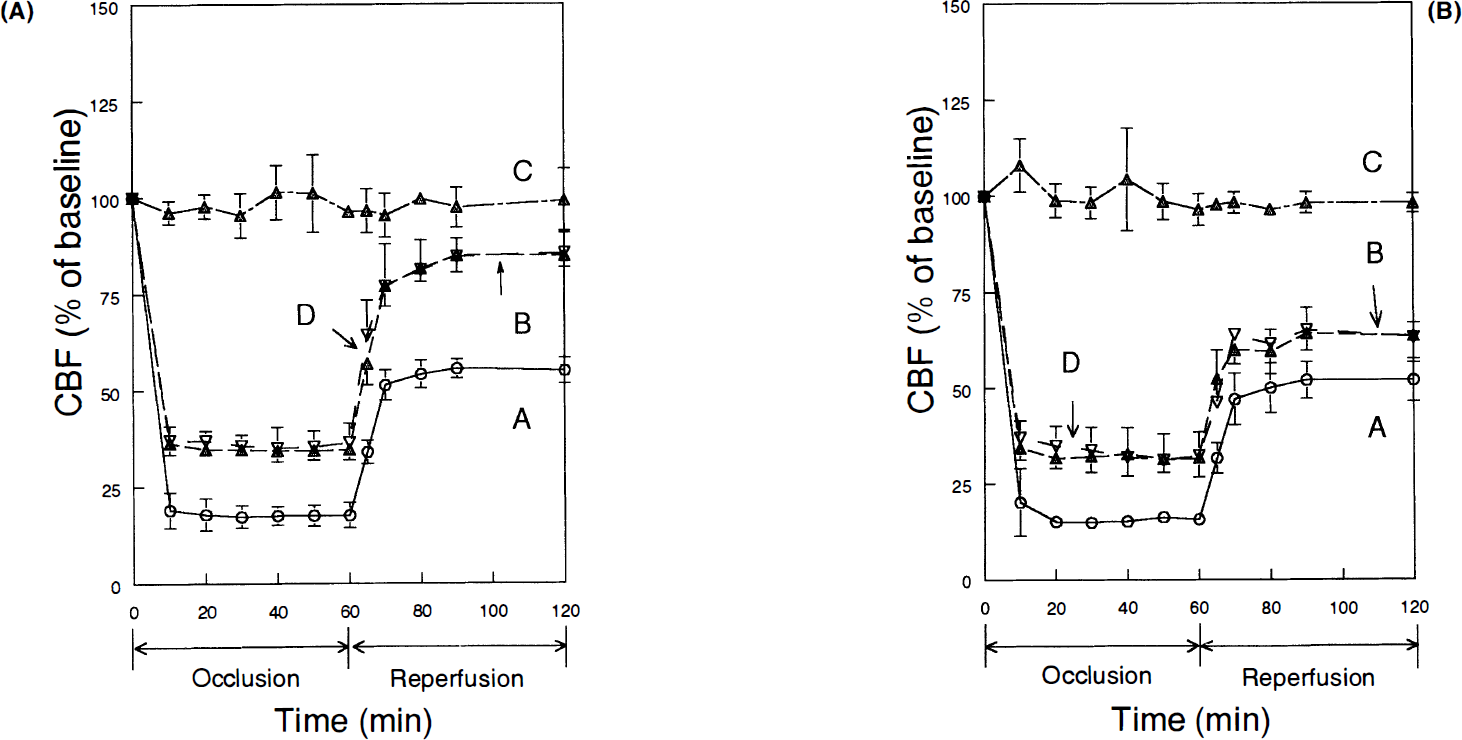
Changes in cerebral blood flow during middle cerebral artery occlusion and reperfusion in control (
Neurologic outcome
There was a marked difference in outcome between control and nicotine-treated rats (Table 3). In the control group, five of 12 rats had no neurologic symptoms, in contrast to the nicotine-treated group, in which 10 of 12 rats had some neurologic deficit. A typical score of 2 was found in 50% of nicotine-treated rats, and only in one of 12 control rats. A severe focal neurologic deficit (scores 3 and 4) was observed in two nicotine-treated rats and in none of the control rats.
Neurologic scores
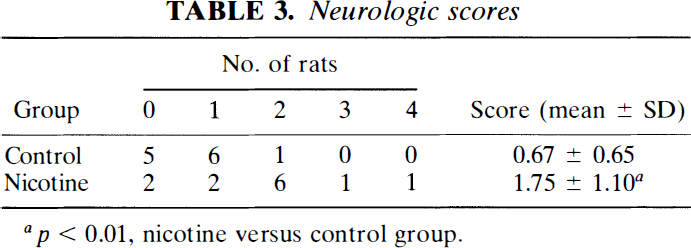
p < 0.01, nicotine versus control group.
Neuropathology
Total volume of injury corrected for edema volume was significantly increased in the nicotine group by 21% relative to that in control rats (Table 4). Total edema volume was also increased in nicotine-treated rats by 110%. These increases were caused by significant increases in injury volume (36%, p < 0.05) and edema volume (121%, p < 0.02) in the pallium indicating expansion of injury into cortical regions. Injury volume in the striatum was not affected by nicotine, while there was a significant 83% increase in edema volume. Variability of injury volume was larger in the nicotine-treated group. Total area of brain injury, determined at the level of the optic chiasm (anterior coronal block) and expressed as a percentage of the coronal sectional area, was increased in nicotine-treated rats by 35% compared to a 38% increase in brain injury expressed as a percentage of the total hemispheric volume. Figure 2 illustrates that 100% of control rats had the injury only in the lateral striatum; ≥50% exhibited changes in dorsolateral, lateral, and ventrolateral cortexes, and <50% showed changes in the medial cortex and striatum. All nicotine-treated rats exhibited changes in the dorsolateral, lateral, and ventrolateral cortexes.
Neuropathologic outcome
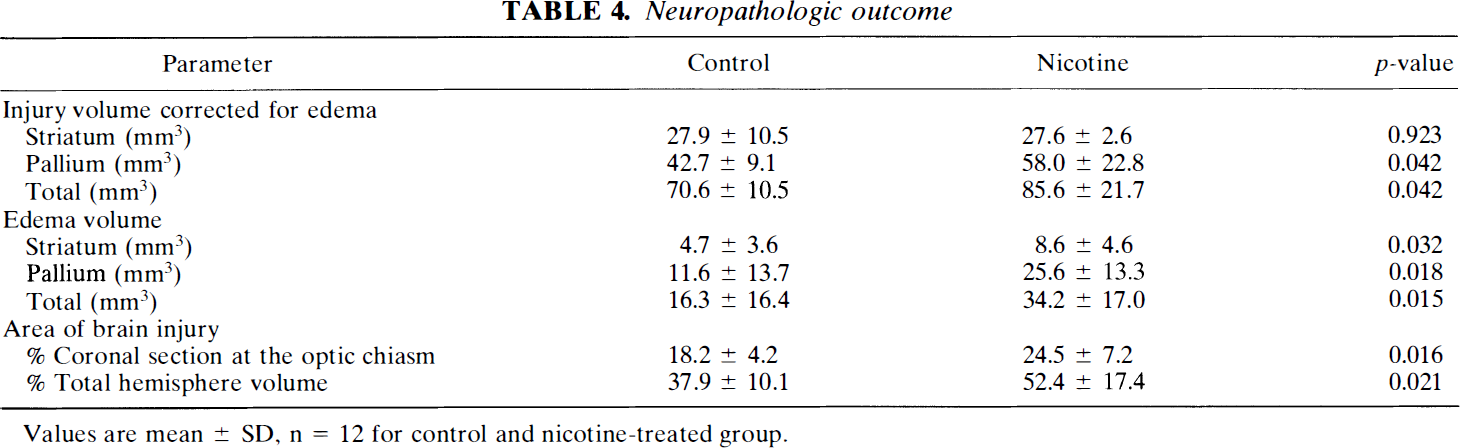
Values are mean ± SD, n = 12 for control and nicotine-treated group.
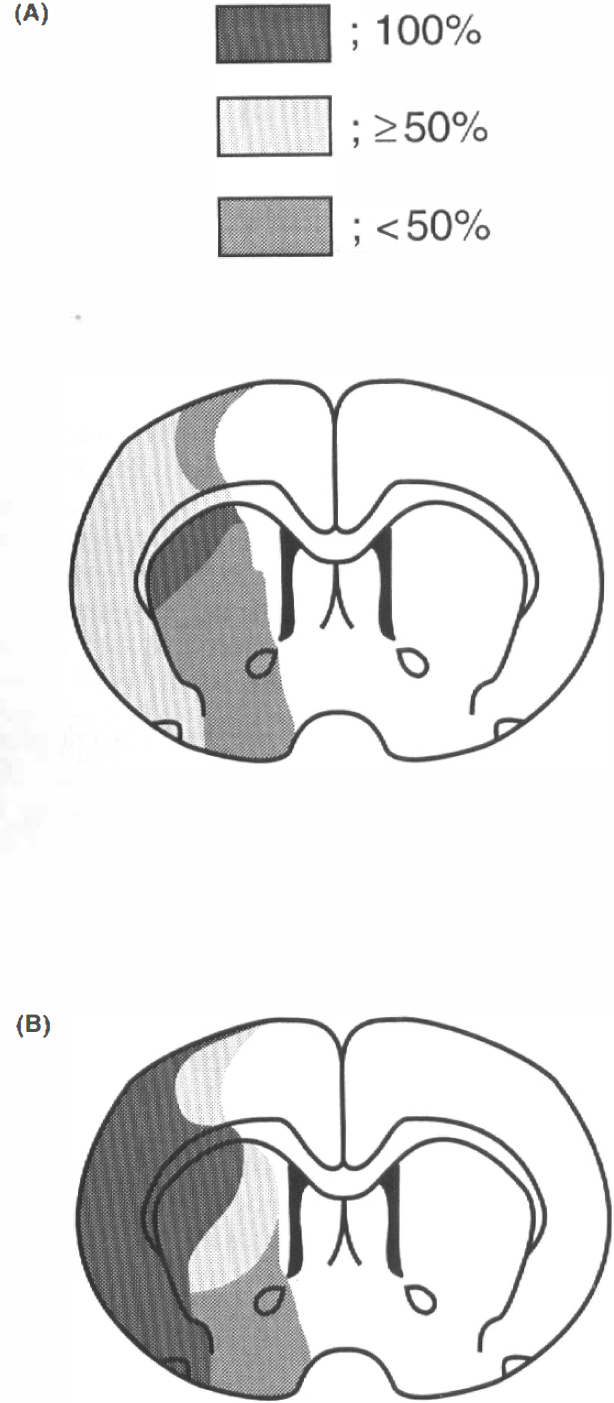
Incidence and topography of the infarction at the level of the optic chiasm during reversible MCA occlusion in control (
Expression of t-PA and PAI-1
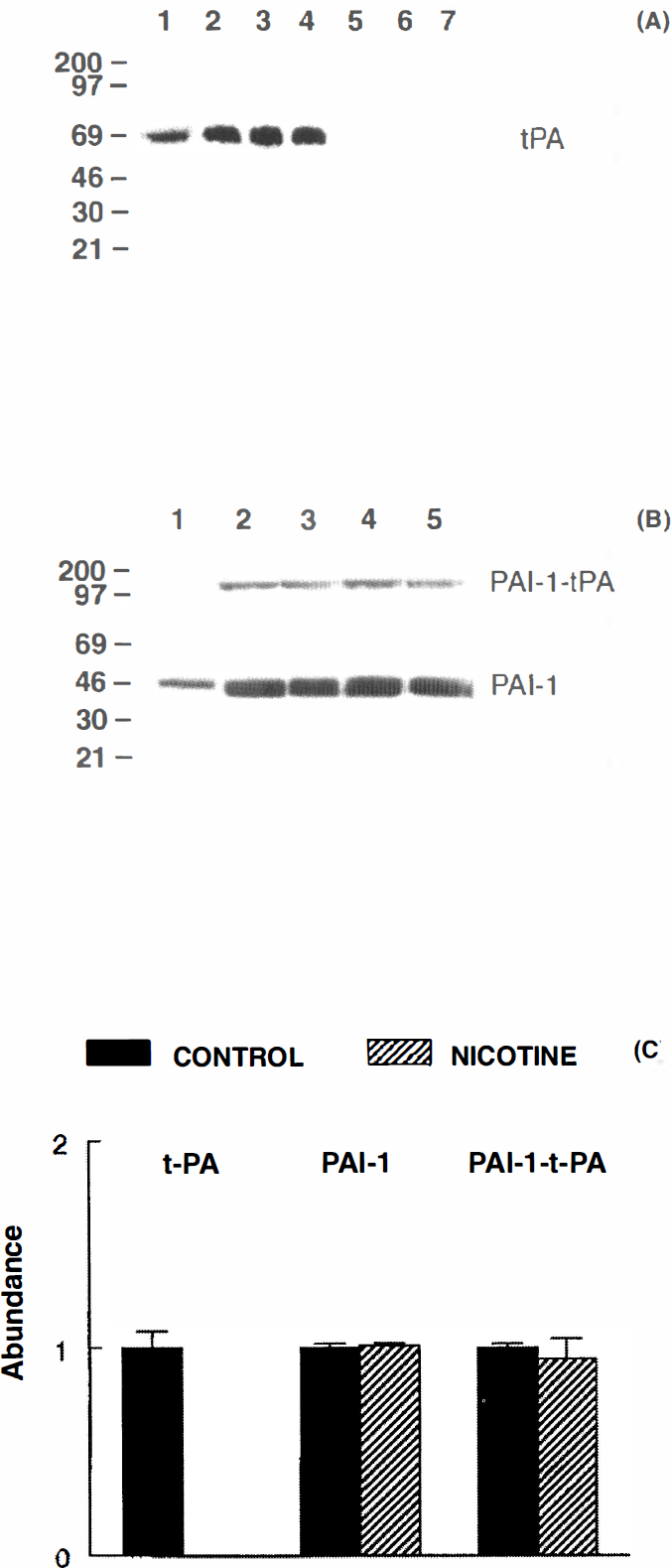
After 14 days, the presence of a single t-PA band of ∼67 kDa was detected only in brain capillaries of control rats (n = 9), and the t-PA signal was absent from capillaries of nicotine-treated (n = 9) rats (Fig. 3A). Complete depletion of t-PA antigen was found in all studied nicotine-treated rats. In both groups, the t-PA signal was absent from capillary-depleted brains (data not shown), as previously demonstrated (Zlokovic et al., 1995b). Omission of primary anti-t-PA antibodies resulted in loss of t-PA signal (data not shown); preincubation of this anti-t-PA antibody with immobilized t-PA on Affigel beads resulted in loss of immunoreactivity in t-PA-rich tissues, such as kidney glomeruli, and abolished detection of the material on Western blots (data not shown), as previously reported (Seeds et al., 1995; Zlokovic et al., 1995b). The PAI-1 antigen (45 kDa) and PAI-1-t-PA complexes (∼110 kDa) were detected in both control (n = 6) and nicotine-treated (n = 6) rats (Fig. 3B). In contrast to t-PA antigen, there was no change in the PAI-1 antigen in brain capillaries of nicotine-treated rats, and only a minimal (6%) decrease in PAI-1-t-PA complexes was found as a result of nicotine treatment. Results of scanning densitometry of t-PA, PAI-1, and PAI-1-t-PA complex immunoblots are shown in Fig. 3C.
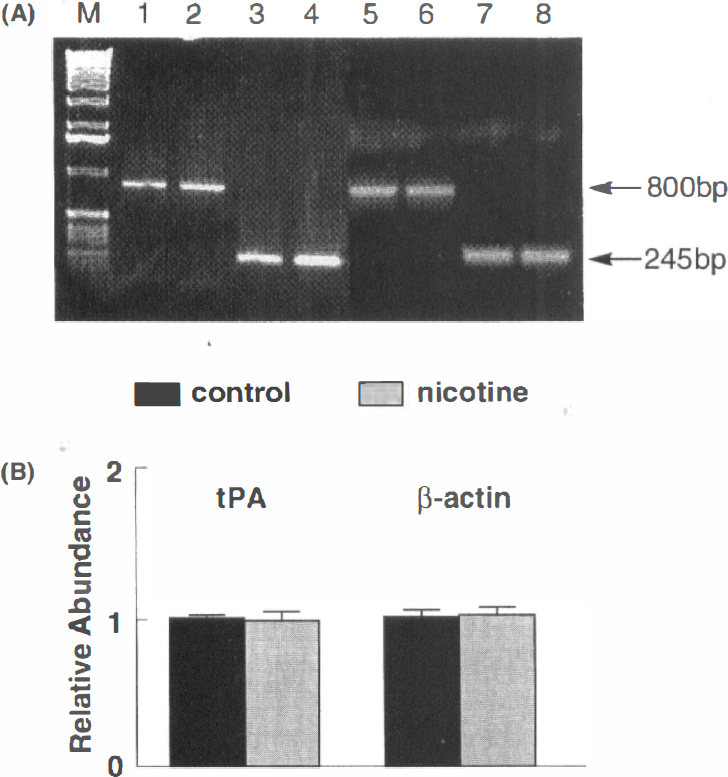
RT-PCR analysis revealed the presence of t-PA cDNA in samples derived from brain capillaries of both control and nicotine-treated rats (Fig. 4A). Scanning densitometry results indicated that the relative abundance of t-PA mRNA in the two groups (n = 12/group, i.e., 3 × 4 pooled samples/group) was comparable, using β-actin as a control (Fig. 4B). As indicated in the methods section, RT-PCR analysis showed relative changes in t-PA expression with respect to a housekeeping gene. In preliminary studies, the number of cycles varied and there was a direct relationship between cycle number, up to 35, and band intensity. Thus, the number of cycles used in the present study (n = 30) was within the linear range of the amplification response. Following RT-PCR, β-actin band density was similar in capillary samples from control and nicotine-treated rats. All capillary-depleted brains in both groups exhibited minimal or no expression of t-PA mRNA (data not shown).
DISCUSSION
The present study demonstrates that chronic nicotine administration in rats that approximates the amount of nicotine intake via cigarette smoking in humans, equivalent to ∼2–3 pack/day (Murrin et al., 1987), results in impaired restoration of blood flow following reversible MCA occlusion, poor neurologic outcome, and enhanced focal ischemic brain injury in comparison to control rats. We confirmed that both t-PA antigen and t-PA mRNA remain displayed in microvessels isolated from rodent brain (Zlokovic et al., 1995b; Kittaka et al., 1996), and that PAI-1 antigen and PAI-1-t-PA complexes in cerebral microvessels can also be demonstrated. These findings suggest that the rat may be a suitable model in which to study changes within the cerebromicrovascular t-PA/PAI-1 system in vivo in relation to ischemic stroke. Notably, the effect of nicotine on focal ischemic stroke was associated with complete depletion of the free pool of brain capillary t-PA antigen in chronically treated rats, whie PAI-1 antigen and PAI-1-t-PA complexes remained unaffected by this treatment.
In vitro studies have demonstrated that production, release, and synthesis of t-PA and its rapid inhibitor PAI-1 in systemic endothelial cells is regulated by various physiologic and pathologic stimuli. For example, coordinated induction of both t-PA and PAI-1 has been reported with thrombin or basic fibroblast growth factor, while histamine, activated protein C, and phorbol ester induce t-PA alone, and lipopolysaccharide (LPS), transforming growth factor-β, interleukin-1 (IL-1), or tumor necrosis factor (TNF-α) induce PAI-1 alone (Hanss and Collen, 1987; Saksela et al., 1987; Sakata et al., 1985; Levin and Santell, 1988; Schleef et al., 1988; Emeis and Koistra, 1986; Sawdey et al., 1989). It has been shown that human systemic endothelial cells in vitro secrete more t-PA when exposed to arterial levels of shear stress (Diamond et al., 1989) and pulsatile stretch (Iba et al., 1991), while PAI-1 secretion remains unaffected. It has been also demonstrated that t-PA secretion and t-PA mRNA are significantly up-regulated with serial passages in culture, while the urokinase type plasminogen activator gene that is normally not expressed in primary culture, becomes quite active at higher passage numbers (reviewed in Iba et al., 1991). These studies suggest that the expression of each of the two antigens may be regulated independently by various chemical or mechanical stimuli, and that under in vitro conditions their expression may be enhanced and/or modulated as a reaction to the tissue culture environment.
In vivo studies in non-human primates, in contrast to in vitro studies, suggest that TNF-α plays a pivotal role in the release of both t-PA and PAI-1 antigens during LPS (endotoxin) stimulation (Biemond et al., 1995). Release of both t-PA and PAI-1 by LPS in vivo has also been reported in rats (Emeis et al., 1995). In vivo studies in rats in contrast to in vivo studies in nonhuman primates indicated that neither TNF-α nor IL-1 are significantly involved in the induction of PAI-1 by LPS (Emeis et al., 1995). Among other differences, expression of t-PA in cerebral microvessels in nonhuman primates in vivo has been found to be limited to some precapillary arterioles and postcapillary venules (Levin and del Zoppo, 1994), while in rats and guinea-pigs, t-PA antigen is expressed in cerebral capillaries in vivo (Zlokovic et al., 1995b). It has also been shown recently that regulation of t-PA and PAI-1 in rat aorta in response to LPS differs from that observed in mouse aorta and rat carotid artery (Padro et al., 1994). Thus, taken together, these studies caution against extrapolation of results and conclusions from one tissue or species subjected to specific types of stimuli to another, as well as from the in vitro setting to an in vivo situation.
The present study demonstrates that chronic nicotine administration depletes brain capillary t-PA antigen in vivo, while the relative abundance of PAI-1 antigen and PAI-1-t-PA complexes is unaffected by this treatment. These results suggest that nicotine could significantly reduce “local” fibrinolytic potential in brain microcirculation by completely depleting the free pool of microvascular t-PA. The only t-PA that was found in brain capillaries of nicotine-treated rats remained bound to PAI-1, and therefore, is likely to be functionally inactive. The unchanged level of cerbromicrovascular PAI-1 antigen suggests that at this stage of chronic nicotine treatment (14 days), PAI-1 most likely is not involved in reducing the level of t-PA antigen. The absence of increased amounts of PAI-1-t-PA complexes in the microvasculature of nicotine-treated rats supports this hypothesis.
In vitro studies in bovine aortic endothelial cells have shown that acute nicotine exposure is associated with direct pharmacological stimulation of t-PA secretion (Kuo et al., 1989). It has been also shown that acute smoking transiently elevates plasma t-PA antigen, either via the direct effect of nicotine, elevated vasopressin level, and/or elevated catecholamines (as a secondary nicotine effect), all of which release endothelial t-PA (Allen et al., 1985). In contrast, chronic exposure to tobacco smoke and nicotine diminishes fibrinolytic activity, reduces levels of t-PA antigen in plasma and peripheral vascular vessel walls, and decreases t-PA release following stimulation with vasopressin (Allen et al., 1985; Kjaelgaard and Larsson, 1986; Haire et al., 1989; Ozdemir et al., 1992).
The mechanism of t-PA antigen depletion in brain microvessels by nicotine is presently not understood. The relative abundance of t-PA mRNA versus β-actin mRNA in cerebral microvessels of nicotine-treated rats remains unchanged in comparison to that in control vehicle-treated rats. Although absolute changes in t-PA mRNA expression were not determined in the present study, it is likely that alterations in transcriptional activity or mRNA stability would be reflected by differences in band intensity following semiquantitative RT-PCR. Thus, the results suggest that the effect of nicotine is mediated at the posttranscriptional level. There are many possible explanations for the dissociation between t-PA mRNA and protein, including posttranslational modifications that result in altered t-PA turnover, and release, secretion, or antibody recognition. Variations in protein levels in the face of unchanging mRNA levels are well documented (e.g., Hengst and Reed, 1996). A discrepancy between t-PA mRNA and t-PA antigen changes has also been reported for the adventitia of rat aorta, in which an increase in t-PA mRNA after LPS treatment was unaccompanied by a change in the level of t-PA antigen (Padro et al., 1994).
In vitro studies with systemic endothelial cells have demonstrated that environmental stress may influence t-PA gene expression independently of PAI-1 gene expression (Diamond et al., 1989; Iba et al., 1991). It has been suggested that morphological changes in endothelial cells (i.e., cytoskeletal rearrangement, increase in monolayer permeability, cellular shape change) in response to stress (i.e., shear, stretch, generation of oxygen radicals, hypertonicity) correlate well with stress-stimulated t-PA mRNA production (reviewed in Levin et al., 1993). Ultrastructural analysis of brain microvasculature in the present nicotine model in rodents failed to demonstrate any morphological changes in brain endothelial cells in vivo, as reported for guinea-pigs (Barron et al., 1992) and 14-day treated rats (data not shown). This is in contrast to giant cell formation of aortic endothelial cells in vitro in response to nicotine (Talloss and Booyse, 1978), ruffled membranes, and extensive cellular vacuolation of rabbit aortic endothelium following oral consumption of nicotine (Booyse et al., 1981), and mitochondrial swelling in the aortic endothelium in mouse in vivo (Zimmerman and McGeachie, 1987). Lack of ultrastructural alterations in brain endothelial cells in guinea pigs and rats may reflect either subthreshold exposure to nicotine, different response of these cells to the toxin, and/or species differences.
The present study demonstrates a 21 % increase in the injury volume of the gray matter corrected for edema in nicotine-treated versus control rats. This increase was caused by expansion of injury into the cortical regions; increase in the injury size in the pallium was 36%. No significant change in cortical CBF was observed in nicotine-treated rats prior to MCA occlusion. However, whether the reactivity of vessels to CO2 during reperfusion is altered by nicotine treatment—and, thus, the status of vascular nitric oxide synthase that normally may be protective after MCA occlusion in mice (Huang et al., 1994)—are presently unknown. Vasospastic effects of nicotine patches followed by stroke have been reported in chronic smokers (Jackson, 1993). Our earlier work showed that chronic nicotine treatment in rats specifically down-regulates the α2 subunit of the sodium pump in cerebral microvessels and neuron-associated glia (Zlokovic et al., 1993; Wang et al., 1994). It is possible that because of impaired NA,K-ATPase function after MCA occlusion (Yang et al., 1992), nicotine-treated rats may become more prone to develop postischemic cellular brain edema and microvascular luminal narrowing that could impair CBF during reperfusion.
In experimental models of thromboembolic stroke (Gross et al., 1995) and MCA occlusion (Nakajima et al., 1993), thrombolytic therapy with exogenous t-PA administered within the therapeutic window reduced the size of infarction and edema, and partially restored CBF (Gross et al., 1995). Using a model of reversible MCA occlusion, the present study demonstrated that depletion of endogenous brain capillary t-PA antigen in nicotine-treated rats correlates well with diminished restoration of CBF during reperfusion, poor neurologic outcome, and larger volumes of brain infarction and edema in comparison to control rats. During reperfusion, nicotine-treated rats exhibited a trend toward impaired CBF restoration in the periphery of the ischemic region, perhaps due to normal endogenous microvascular thrombolysis, although other multiple possible effects of nicotine on neurons and the microvasculature may also be involved. This might be similar to the recently reported association between depleted cerebromicrovascular t-PA antigen and focal ischemic brain injury in a diabetic model in rats (Kittaka et al., 1996).
It is not certain whether nicotine effects in habitual smokers are comparable to results of this study. Intermittent bolus-like dosing of nicotine from cigarettes could have different effects from those produced by the continually released transdermal nicotine of patches and/or the continuous chronic s.c. administration of nicotine via minipumps, as in this study. However, an association between depletion of t-PA antigen from coronary vessels and subsequent development of coronary artery disease in patients has been reported (Lasarsere et al., 1995), and it is possible that depletion of the free t-PA antigen pool within the brain microcirculation may compromise brain fibrinolytic capacity in smokers, predisposing to larger and more disabling strokes. On the other hand, it might be that direct acute effects of nicotine (i.e., within several hours) on t-PA mRNA and antigen expression in brain endothelial cells in vitro may be different from chronic in vivo effects as in this study (i.e., over a 2-week exposure).
It is well established that free radical production plays a role in brain injury in reperfusion models (Siesjo, 1992). It has been shown that acute exposure to nicotine increases formation of reactive oxygen species in epithelial tissues (Wetsher et al., 1995a; Wetsher et al., 1995b), but whether chronic nicotine administration results in free radical production in brain in vivo has not been investigated. Thus, the possibility that the effect of nicotine on brain injury is due to both vascular and neuronal effects should be considered, and, in particular, in relation to vascular changes in t-PA expression, as suggested by the present study, and the possible role of t-PA in neuronal degeneration induced by excitotoxins (Tsirka et al., 1995), such as nicotine.
Footnotes
Acknowledgment:
This work was supported by NIH grant NS31945 and the Cigarette and Tobacco Surtax Fund of the State of California through the Tobacco-Related Disease Program grant 2RT0071. We wish to thank Dr. Mark Fisher for his helpful discussion.