
Abstract
Effects of blood glucose concentration on biochemical and neurologic outcome following lateral fluid percussion-induced traumatic injury of moderate severity (2.8 atm) in rats were studied using radioactive phosphorus (31P) magnetic resonance spectroscopy (MRS) and a battery of tests designed to evaluate posttraumatic neurologic motor function. Prior to injury, male Sprague-Dawley rats (n = 18) were randomly assigned to receive either dextrose, 2 ml 50% (wt/vol), zinc insulin (10 IU/kg) or no treatment, thus dividing the animals into hyperglycemic, hypoglycemic, and normoglycemic groups, respectively. Animals were then injured, monitored for 4 h by 31P MRS before being allowed to recover, and assessed for posttraumatic motor function. Following brain injury, there was no difference in brain intracellular pH between groups over the 4-h posttraumatic MRS monitoring period. Similarly, intracellular free magnesium, cytosolic phosphorylation potential, and neurologic outcome posttrauma were not significantly different between groups. We conclude that, unlike models of ischemia, blood glucose concentration may not be a significant factor affecting outcome in traumatic brain injury.
Keywords
It is now clearly accepted that blood glucose concentration has a major affect on outcome following brain ischemia (Myers and Yamaguchi, 1976). Hyperglycemia is deleterious to outcome, presumably by exacerbating tissue lactic acidosis (Plum, 1983; Siesjo, 1988) while moderate hypoglycemia results in less tissue acidosis and a smaller infarction area (Diemer and Siemkowicz, 1981). Based on these results, it has generally been assumed that a similar relationship would exist in traumatic brain injury. Indeed, experimental studies have shown that a transient increase in glycolytic rate does occur immediately following traumatic brain injury (Hayes et al., 1988; Hovda et al., 1992) and that, despite normal rates of oxygen consumption and blood flow, this increase in glycolytic rate does result in a slight and transient acidosis immediately following the traumatic event (Andersen and Marmarou, 1992). It is, therefore, possible that with increasing blood glucose concentration, elevated rates of cerebral glucose utilization would persist following trauma, with resultant exacerbation of acidosis and neurologic dysfunction. However, no studies have directly addressed the role that blood glucose concentration may play in the formation of lactic acid by the brain following traumatic injury or its subsequent affect on neurologic outcome.
Studies of experimental trauma using radioactive phosphorus (31P) magnetic resonance spectroscopy (MRS) have shown that brain tissue pH can be used to indirectly monitor brain lactic acidosis following traumatic brain injury (McIntosh et al., 1987). Furthermore, 31P MRS can also be used to determine free magnesium concentration and cytosolic phosphorylation potential, both of which have been correlated to neurologic outcome following brain trauma (Vink et al., 1988). In the present study, we used 31P MRS as well as a battery of tests designed to evaluate posttraumatic motor function in order to determine the effects of blood glucose concentration on biochemical and neurologic outcome following moderate traumatic brain injury in rats.
MATERIALS AND METHODS
Male Sprague–Dawley rats (n = 18; 250–300 g) were fed and watered ad libitum before being randomly assigned to receive either zinc insulin (hypoglycemia), dextrose (hyperglycemia), or no treatment (normoglycemia). Zinc insulin was administered by intraperitoneal injection (10 IU/kg) 3 h prior to injury so that the animals were moderately hypoglycemic by the time trauma was induced. Dextrose was administered as a 50% (wt/vol) solution into which a total of 2 mis was slowly infused intravenously over a 30 min interval immediately prior to induction of injury. Previous studies have shown that administration of dextrose by this regimen results in blood glucose concentrations >15 mM and brain glucose levels exceeding 30 μmol/g (Rehncrona et al., 1980). To confirm that blood glucose concentrations were in the appropriate range at the time of trauma, blood samples were obtained from the caudal artery immediately prior to injury and blood glucose concentration was determined using a Boehringer Mannheim glucometer (Boehringer Mannheim GmbH, Mannheim, Germany). Moderate hypoglycemia was defined as a blood glucose concentrations <2.0 mM, hyperglycemia as >15.0 mM, while the normoglycemic group recorded blood glucose concentrations between 4.0 and 10.0 mM. All blood glucose concentrations remained in their respective ranges throughout the MRS monitoring period with the exception of the hyperglycemic group, the levels of which fell to normoglycemic values between 3 and 4 h posttrauma.
Traumatic brain injury was induced in sodium pentobarbital anesthetized animals using a fluid percussion injury device, as previously described in detail elsewhere (McIntosh et al., 1989). This method is based on rapid injection of a saline pressure pulse epidurally into the closed cranial cavity of the rat causing transient tissue deformation and resultant injury. In the present study, injury was induced at 2.8 atm via a 4-mm craniotomy centered over the left parietal cortex. Prior to and for 4 h following injury, 3IP MRS spectra were obtained as previously described using a 7.0 T magnet interfaced with a Varian spectrometer console (Varian Associates, Palo Alto, CA, U.S.A.) (Emerson et al., 1993). Previous lateral fluid percussion injury studies have shown that injury is limited to the cortical and subcortical structures of the hemisphere receiving the impact (McIntosh et al., 1989). By using a 5 × 9 mm surface coil placed centrally around the trauma site and a 90° pulse width calibrated at 2 mm cortical depth, spectra were representative of the left hemisphere to a depth of 5 mm (Vink et al., 1987), thus encompassing the injury zone.
Intracellular pH was determined from the chemical shift of the inorganic phosphate peak (δPi) relative to phosphocreatine (PCr) in MRS spectra using the equation (Petroff et al., 1985)
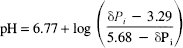
Free magnesium concentration was determined from the chemical shift difference between the α and β peaks of ATP (Gupta et al., 1978) using the equation (Malloy et al., 1986)
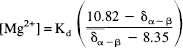
where δα–β is the chemical shift difference between the α and β peaks of ATP. The Kd for MgATP was assumed to be 50 μM at pH 7.2 and 0.15 M ionic strength and was corrected for pH as previously described (Bock et al., 1987). Cytosolic phosphorylation potential, ΔGp, was calculated as previously described in detail elsewhere (Vink et al., 1988) assuming preinjury values for phosphocreatine and ATP of 4.72 μmoles/g and 2.59 μmoles/g, respectively (Veech et al., 1979). Cytosolic ADP concentration was calculated from the creatine kinase equation after adjusting the equilibrium constant for pH and free magnesium concentration (Lawson and Veech, 1979; Vink et al., 1988). Brain water content was assumed to be 80% of total wet weight, with the intracellular compartment accounting for 78% of the total water (Siesjo, 1988).
Neurologic function was assessed at 1 and 2 weeks after injury using a battery of motor tests especially designed to detect abnormalities of motor function following lateral fluid percussion brain injury (McIntosh et al., 1989). Briefly, the tests used in the present study included (a) ability to maintain vertical, left horizontal, and right horizontal position on an inclined angleboard for 5 s, (b) left and right forelimb flexion upon suspension by the tail, and (c) resistance to both left and right lateral pulsion when attempting to roll the animal onto its back. All animals were graded on their performance in each task on a scale of 0–5, where 5 is normal and 0 is afunctional. A composite score ranging from 0 to 35 was obtained by combining the scores of all seven tests.
RESULTS
Prior to injury, brain intracellular pH in all animals was 7.09 ± 0.05 (mean ± SD), which is in excellent agreement with previously published results (McIntosh et al., 1987; Vink, 1993). After injury, there were no significant changes in brain intracellular pH irrespective of blood glucose concentration (Fig. 1). There was also no significant change in ATP concentrations following injury, consistent with previous results at this level of injury (McIntosh et al., 1987; Vink, 1993). Mean intracellular free magnesium concentration in brain was 0.62 ± 0.15 mM, which is similar to previously reported values (Emerson et al., 1993; Vink, 1993). Following moderate trauma, there was a significant decline in free magnesium concentration in all groups [p < 0.05, analysis of variance (ANOVA)] (Fig. 1), with no significant difference being detected between groups. Similarly, cytosolic phosphorylation potential declined from a mean preinjury ΔGp for all animals of 34.0 ± 6.5 mM–1 to ∼63% of preinjury values (p < 0.001, ANOVA), with no significant difference being detected between treatment groups (data not shown).
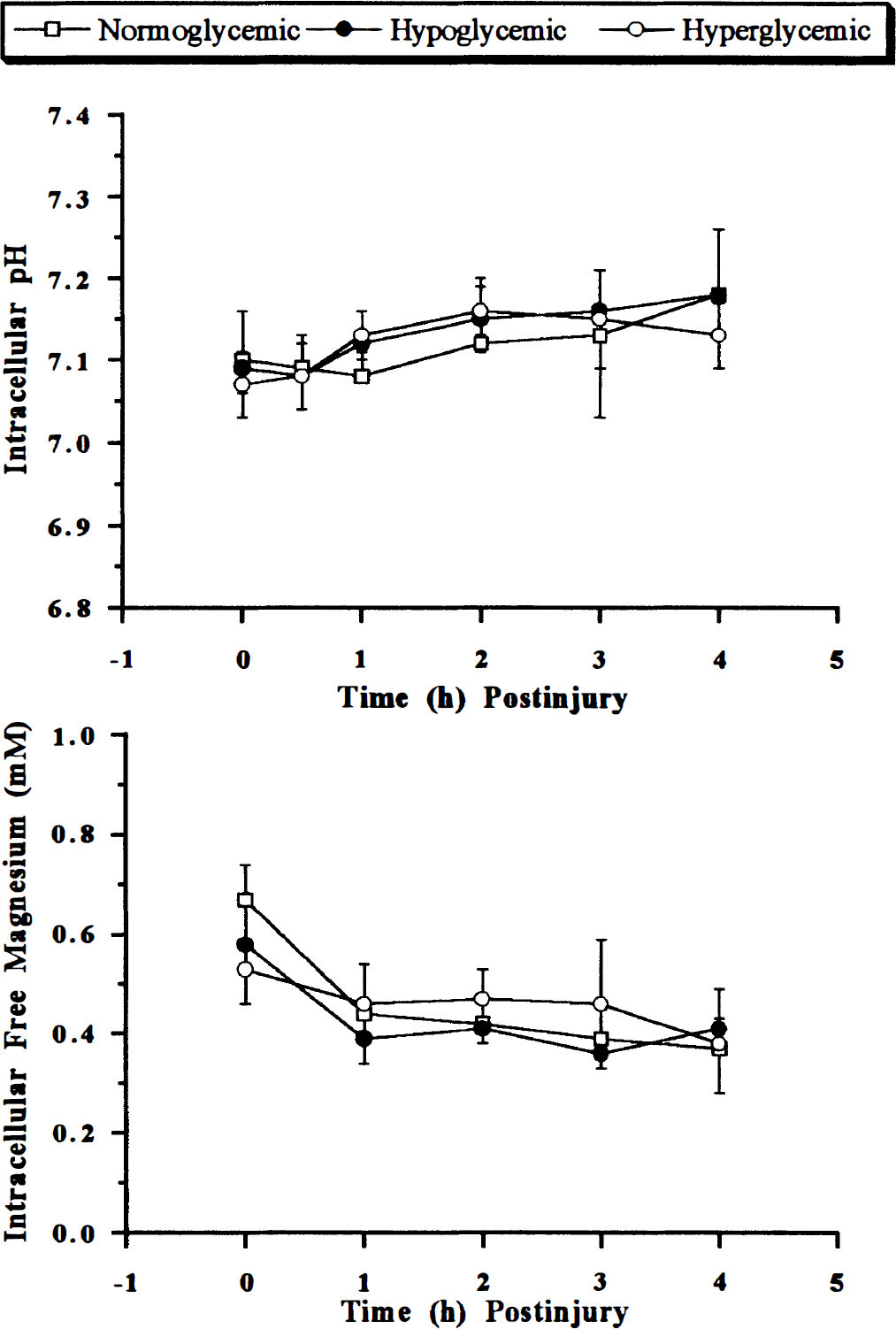
Changes in brain intracellular pH (top) and free magnesium concentration (bottom) following moderate fluid percussion-induced trauma in rats. Blood glucose concentrations were >15 mM in the hyperglycemic group, 4–10 mM in the normoglycemic group, and <2 mM in the moderate hypoglycemia group. All data are mean ± SD (n = 6).
Posttraumatic neurologic outcome in normoglycemic animals was consistent with moderate injury (McIntosh et al., 1989). Median score for individual tests was 4–4.5, while composite neuroscores were 28 and 31.5 in weeks 1 and 2, respectively. Outcomes in hyperglycemic and hypoglycemic animals were not significantly different from those of the normoglycemic animals (Fig. 2). Although there was a trend toward improved outcome in these groups at 1 week, this improvement was neither significant nor sustained.
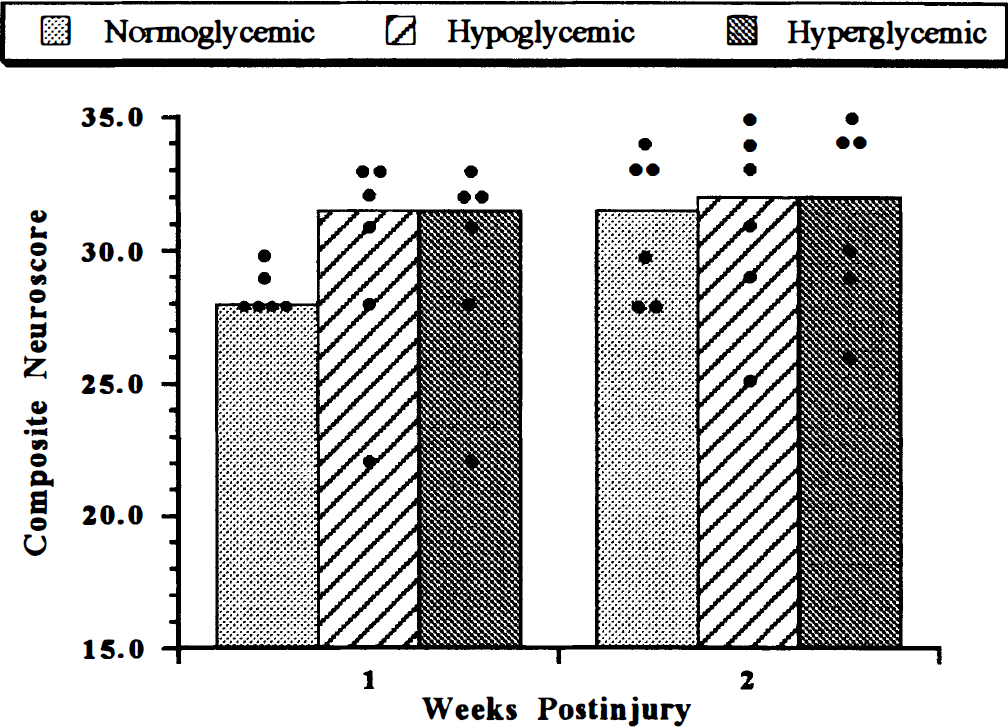
Composite neurologic outcome following fluid percussion-induced traumatic brain injury in rats. Dots represent scores of individual animals; bars represent the median score for each treatment group. No significant difference was noted at any time point between groups.
DISCUSSION
Previous studies have suggested that increased glycolytic rate following traumatic brain injury may result in formation of lactic acidosis with resultant exacerbation of injury (Andersen and Marmarou, 1992). The present studies demonstrated that irrespective of available glucose concentration in the blood, lactic acid production in moderately traumatized brain does not exceed either the acid extrusion capacity of the cells or the buffering capacity of the tissue. Two reasons may account for this observation. Firstly, cerebral blood flow following moderate trauma does not fall to levels wherein tissue oxygen delivery is compromised (Yamakami and McIntosh, 1989; Yuan et al., 1988). Secondly, studies with isolated mitochondria after traumatic brain injury have shown that the aerobic capacity of mitochondria are not affected (Vink et al., 1990). Moreover, in vivo studies of mitochondrial cytochrome aa3 have demonstrated that the terminal electron acceptor is not reduced after trauma but, rather, is oxidized (Duckrow et al., 1981). Therefore, it would appear that the tissue provides an adequate supply of oxygen to functional mitochondria, and a major switch to lactate production would be unlikely under these conditions. This idea is consistent with previous studies, which have shown that lactate formation is not a significant factor in mild-to-severe levels of brain injury (McIntosh et al., 1987).
Increased rate of glucose consumption following fluid percussion-induced traumatic brain injury is thought to be in response to the large ionic perturbations seen immediately after the concussive event (Hovda et al., 1992). Our present results confirm that there are significant ionic perturbations after trauma, at least with respect to free magnesium concentration. In the normoglycemic and hypoglycemic groups, the greatest degree of change in intracellular free magnesium concentration occurs during the first hour after trauma (34–35% decline). This coincides with the time course for the greatest increase in glucose consumption found by others (Andersen and Marmarou, 1992; Hayes et al., 1988; Hovda et al., 1992). However, in the hyperglycemic group, the decline in free magnesium concentration is only 11% in the first hour, followed by a further 14% decline 3–4 h posttrauma. Despite the decreased rate of magnesium decline in the hyperglycemic group over the first 3 h, these results still support the concept of increased glucose consumption being linked to early ionic changes. Glucose and magnesium are known to co-transport in a number of tissues (Henquin et al., 1983), and our results suggest that increased glucose consumption in brain after trauma may also result in magnesium co-transport. While blood glucose concentration remains in the hyperglycemic range, glucose/magnesium co-transport may attenuate the rate of decline of free magnesium. Since blood glucose falls to normal levels 3–4 h posttrauma, glucose/magnesium co-transport may then be insufficient to attenuate the magnesium decline. Interestingly, studies of cerebral ischemia have also shown higher levels of tissue magnesium concentration following an ischemic event under hyperglycemic conditions as compared to normoglycemic conditions (Warner et al., 1987).
Footnotes
Acknowledgment:
This study was supported, in part, by an Australian National Health and Medical Research Council grant (R.V.) and by a U.S.A. National Institutes of Heath grant, no. R01 NS26818 (T.K.M.).