
Abstract
We examined time-dependent changes in protein synthesis and in the immunoreactivities of representative contraction-related structural proteins in smooth muscle cells of canine basilar arteries after experimental subarachnoid hemorrhage (SAH). Protein synthesis was assessed by the percentage of polyribosome-forming ribosomes to total ribosomes (aggregation rate), a morphological index of the activity of protein synthesis. The aggregation rates in prostaglandin F2α- (PGF2α) and 12-O-tetradecanoyl-phorbol-13-acetate (TPA)-induced contracted basilar arteries were 70.0 ± 7.0% and 71.4 ± 8.7%, respectively, quite similar to the value in normal basilar artery (73.0 ± 8.0%). In the single-SAH group with little delayed histological changes in the basilar arteries, the aggregation rate was significantly decreased to 30.5 ± 6.4% by 24 h after the SAH, and recovered to 52.3 ± 9.0% and 70.2 ± 7.6% at 7 and 14 days postSAH, respectively, when the vasospasm was moderately and completely ameliorated. In contrast, in the double-SAH group in which the basilar arteries developed delayed smooth muscle cell death and long-lasting arterial contraction, a significant decrease in the aggregation rate (25.0 ± 5.0% on day 4) persisted for 14 days. The in vitro incorporation of [3H]-leucine in the basilar arterial cells was also significantly suppressed 4 and 7 days after the initial SAH (1.2 ± 0.4 and 1.4 ± 0.3 × 103 dpm/mg protein) in the double-SAH group, as opposed to no significant decrease in the basilar artery at 7 days postSAH in the single-SAH group (1.9 ± 0.6 × 103 dpm/mg protein). The immunoreactivity of α-smooth muscle actin, a contractile protein, demonstrated by immunohistochemistry and immunoblots, was not altered for up to 14 days even in the double-SAH group, but that of calponin and of h-caldesmon, contraction-inhibiting proteins, was markedly reduced 4–14 days after the initial SAH. Persistent impairment of protein synthesis and relative reduction of immunoreactivities of the contraction-inhibiting proteins were observed in arteries with severe vasospasm and loss of smooth muscle cells, as noted in the double-SAH subjects. These abnormalities may cooperate to cause cerebral arterial narrowing accompanied by degeneration of smooth muscle cells after SAH.
Smooth muscle cells in cerebral arteries affected by vasospasm following subarachnoid hemorrhage (SAH) due to rupture of an aneurysm develop sustained contraction and delayed degeneration represented by so-called myonecrosis (Alksne and Greenhoot, 1974; Fein et al., 1974; Mayberg et al., 1978). The pathogenesis of prolonged smooth muscle contraction has not been clearly demonstrated, because of the limited participation of known biochemical pathways in inducing physiological smooth muscle contraction, such as those mediated by calmodulin (Adelstein et al., 1980; Murphy et al., 1983; Small and Sobieazek, 1977) and protein kinase C (PKC) (Nishizuka, 1986; Rasmussen et al., 1987), in the pathogenesis of cerebral vasospasm (Sakaki et al., 1989; Sako et al., 1993). The mechanism of delayed death of smooth muscle cells in cerebral arteries with vasospasm also remains unknown. Among the essential factors for maintenance of cellular viability are high-energy phosphates, which have been reported to decrease from 24 h to 14 days in cerebral arterial smooth muscle cells following SAH (Nozaki et al., 1990; Kim et al., 1992; Yoshimoto et al., 1993). Continuous protein synthesis is also a crucial factor for cell survival and maintenance of intracellular structure. However, there have been no reports of changes in protein synthetic activity in cerebral arterial smooth muscle cells following SAH.
In the present study, we evaluated chronological changes in protein synthesis in smooth muscle cells of vasospastic basilar arteries in single- and double-SAH models in dogs with smooth muscle cells developing little or massive degeneration, respectively. Evaluation was performed by observing ultrastructural changes in the polyribosomes, that catalyze protein synthesis for intracellular use and by in vitro assays of radiolabeled amino acid incorporation. We also examined immunoreactivities of several contraction-related structural proteins in smooth muscle cells following SAH, using immunohistological and immunoblotting techniques, to explore the possibility that smooth muscle contraction is caused by the derangement of the contraction-related structural proteins themselves, since, if protein synthesis for intracellular use were impaired, the quality or quantity of the proteins might be altered.
MATERIALS AND METHODS
Animal preparation
Experiments were conducted in compliance with the guidelines for animal experimentation at Ehime University School of Medicine. Adult mongrel dogs of both sexes, weighing 7–13 kg each, were anesthetized with an intramuscular injection of ketamine hydrochloride (5 mg/kg) and intubated following an intravenous injection of sodium thiamylal (10 mg/kg) and atropine sulfate (0.5 mg/kg). Catheterization of the right femoral artery was performed for continuous monitoring of systemic blood pressure and pulse rate. Arterial blood gases and body temperature were monitored and maintained within the physiological range using artificial ventilation and a heating blanket.
Eighty-four dogs were divided into four groups: 21 in the normal control group, nine with drug-induced vasoconstriction, 21 with single-SAH-induced vasospasm, and 33 with double-SAH-induced vasospasm. For the drug-induced vasoconstriction study, 10−4 M prostaglandin F2α (PGF2α) (dissolved in saline, was applied topically to the basilar artery by transclivai exposure under an operating microscope) or 6.0 ng/kg of 12-O-tetradecanoylphorbol-13-acetate (TPA) (dissolved in 1 ml of 1% dimethyl sulfoxide) was injected intracisternally through a puncture of the cisterna magna with a 22-gauge needle without drawing cerebrospinal fluid (CSF), under sterile conditions. This concentration of dimethyl sulfoxide had no effect on canine basilar arterial diameter. For the single-SAH-induced vasospasm, 0.8 ml/kg of autologous arterial blood obtained from the femoral arterial catheter was slowly injected intracisternally in the same manner, and for the double-SAH-induced vasospasm, intracisternal injection of arterial blood was repeated after 48 h (Varsos et al, 1983).
Assessment of basilar arterial diameter
Vertebrobasilar angiography was performed before and after intracisternal injection of autologous arterial blood in 20 other single- and double-SAH dogs. Then, 5 ml of lopamidol (Iopamiron) (Japan Schering Co., Tokyo, Japan) was injected via a femoral catheter placed in the left vertebral artery. All angiograms were taken under fixed magnification. The diameter of the basilar artery was measured with an MICD imaging analyzer (Imaging Research, St. Catherine, Ontario, Canada) and contraction was assessed by determining the mean percentage of the pretreatment diameter at a point 10 mm proximal from the basilar head.
Histological examination
Light microscopic examination of the basilar artery was performed 14 days after the initial SAH in the single- and double-SAH groups (in four dogs of each group). Five cross-coronal sections of 20 μm, cut perpendicularly to the long axis of the basilar artery, were made per each animal. For assessment of delayed smooth muscle cell death, the number of smooth muscle cell layers was counted on five arbitrary crosslines from the intima to the adventitia, i.e., per whole tunica media, taking into consideration the difference of diameter due to the remaining vasospasm between the single- and double-SAH groups.
Morphological study of ribosomes
Morphological studies were performed in the drug-induced vasoconstriction group 15 min after topical application of PGF2α and 4 h after the intracisternal injection of TPA, at which time elicitation of the maximal basilar arterial contraction responding to the drugs was reported. (Kohno et al., 1991; Sako et al., 1993). In the single-SAH group, studies were performed 1 and 24 h, and 7 and 14 days after the arterial blood injection, and in the double-SAH group, 4, 7, and 14 days after the first arterial blood injection.
Under the same anesthesia mentioned earler, animals were killed by intracardiac perfusion fixation using 1.0 L of ice-cold saline followed by 1.0 L of 3% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) at a constant pressure of 120 mm Hg during exsanguination via a femoral venous catheter. Basilar arteries were removed, postfixed with 2% OsO4 for 2 h at 4°C, stained with uranyl acetate, and dehydrated through a graded ethanol series. Specimens were embedded in Quetol 812 (Nishin E. M. Co., Tokyo, Japan), and 70 nm sections were cut with an ultramicrotome and stained again with uranyl acetate and lead citrate. They were observed under a Hitachi H-800 transmission electron microscope (Tokyo, Japan). From each basilar artery, 10 microphotographs of smooth muscle cells not showing severe degenerative changes were chosen randomly. Using the MICD imaging analyzer, the ribosome aggregation rate in a smooth muscle cell was calculated as the percentage of ribosomes forming polyribosomes to total ribosome numbers, ribosomes in aggregates of four or more were regarded as a polyribosome.
Amino acid incorporation in basilar artery
The validity of morphological assessment of protein synthesis was ascertained with an in vitro assay of amino acid incorporation in normal basilar artery, the basilar arteries 7 days in the single-SAH model, and 4 and 7 days after the first arterial blood injection in the double-SAH group. Animals were killed by intracardiac perfusion with 1.0 L of ice-cold saline, and 10–20 mg of basilar artery was removed and homogenized in a glass homogenizer in 2.0 ml of 25 mM Tris-HCl buffer (pH 7.4) containing 100 mM KCl, 10 mM MgCl2, 0.1 mM ethylendiamine tetraacetic acid (EDTA), and 2.5 mM dithiothreitol. The homogenate was centrifuged for 30 min at 1,500 g, and the supernatant filtered in Sephadex G-25 columns (PD-10: Pharmacia LKB, Uppsala, Sweden) for removal of free amino acids and stored at −70°C. Protein content of the supernatant was measured with a Bio-Rad protein assay kit (Bio-Rad, Richmond, VA, U.S.A.). The supernatant was mixed with 50 μl of a reagent consisting of 60 mM KCl, 80 mM Tris-HCl (pH 7.4), 4 mM dithiothreitol, 2 mM ATP, 0.4 mM GTP, 30 mM phosphocreatine, 5 μCi [3H] leucine (Amersham, Arlington Heights, IL, U.S.A.), 120 μM each of the other 19 amino acids, and 200 μg/ml creatine kinase at 0°C, and incubated at 37°C for 60 min. The reaction was terminated by the addition of 0.5 ml of cold 10% trichloroacetic acid (TCA). The precipitate was washed three times with cold 5% TCA and the last washing was conducted under heating at 90°C for 15 min. The sample was extracted with ethanol-ether (1:1, vol/vol) then again with ether. The dried sample was dissolved in 1.0 N sodium hydroxide and the radioactivity incorporated into the protein was measured in an ACS II scintillant (Amersham, Arlington Heights, IL, U.S.A.) with a liquid scintillation counter. The amount of incorporated [3H]leucine was expressed as dpm/mg protein.
Immunohistochemistry of contraction-related proteins
Immunohistochemical studies of the contraction-related proteins, calponin, h-caldesmon, and α-smooth muscle actin, in basilar arteries were performed non-treated in the control group; 4 h after the TPA injection in the drug-induced vasoconstriction group; 24 h after the SAH in the single-SAH group; and 4, 7, and 14 days after the initial SAH in the double-SAH group. Under anesthesia, animals were killed by intracardiac perfusion fixation with 1.0 L of saline and 1.0 L of 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.2) as in the above procedure. The basilar artery was removed, postfixed in the same buffer at 4°C for 4 h, and immersed in 0.1 M phosphate buffered saline (PBS) (pH 7.2) containing 30% sucrose. Frozen sections (7 μm thick) were cut with a cryostat and processed for immunohistochemical examination of calponin, h-caldesmon, and α-smooth muscle actin with monoclonal antibodies against each protein—clones hCP, C21 and 1A4, respectively (Sigma Chemical Co., St. Louis, MO, U.S.A.). Sections were treated for 24 h with biotinylated anti-mouse IgG followed by peroxidase-conjugated streptavidin and subjected to a modified version of the cobalt-glucose-oxidase-diaminobenzene (DAB) intensification method (Sakanaka et al., 1987).
Immunoblotting of contraction-related proteins
Unfixed basilar arteries obtained at the same period as those for immunohistochemistry were homogenized in 50 mM imidazole-HCl buffer (pH 6.9) containing 300 mM KCl, 5 mM ethyleneglycol-bis (β-aminoefhylefher) N, N′-tetraacetic acid (EGTA), 1 mM dithiothreitol, 0.5 mM phenylmefhylsulfonyl fluoride, 10 μg/ml leupeptin, and 1 mM sodium tetrathionate in a glass homogenizer. Proteins in the homogenate were separated by SDS-polyacrylamide slab gel electrophoresis (12.5% wt/vol for calponin and α-smooth muscle actin and 7% wt/vol for h-caldesmon) with the buffer system of Laemmli (1970) and transferred to a polyvinylidine difluoride (PVDF) membrane (Millipore, Bedford, MA, U.S.A.) The membrane was processed for immunoblotting examination with the same antibodies and DAB method as in the immunohistochemistry study. Densitometrie quantification of each specific band was performed with the MICD image analyzer, and the quantity after SAH was expressed as the percentage of that in the normal basilar artery.
Statistical analysis
All values are given as means ± SD. Statistical comparison was made by a two-factor analysis of variance (ANOVA) (Scheffé‘s test for multiple comparisons).
RESULTS
Changes in basilar arterial diameter
In the single-SAH group, the basilar artery contracted to 74 ± 6 and 71 ± 5% of the pretreatment diameter 24 and 48 h after the SAH induction, respectively, and its diameter had almost completely returned to the pretreatment values 7 and 14 days after the SAH, respectively. In contrast, in the double-SAH group, basilar arterial narrowing was more severe (to ˜60% of pretreatment diameter) and more persistent than in the single-SAH group after the second arterial blood injection; a 40% decrease in the diameter persisted 4–14 days after the initial SAH (Fig. 1).
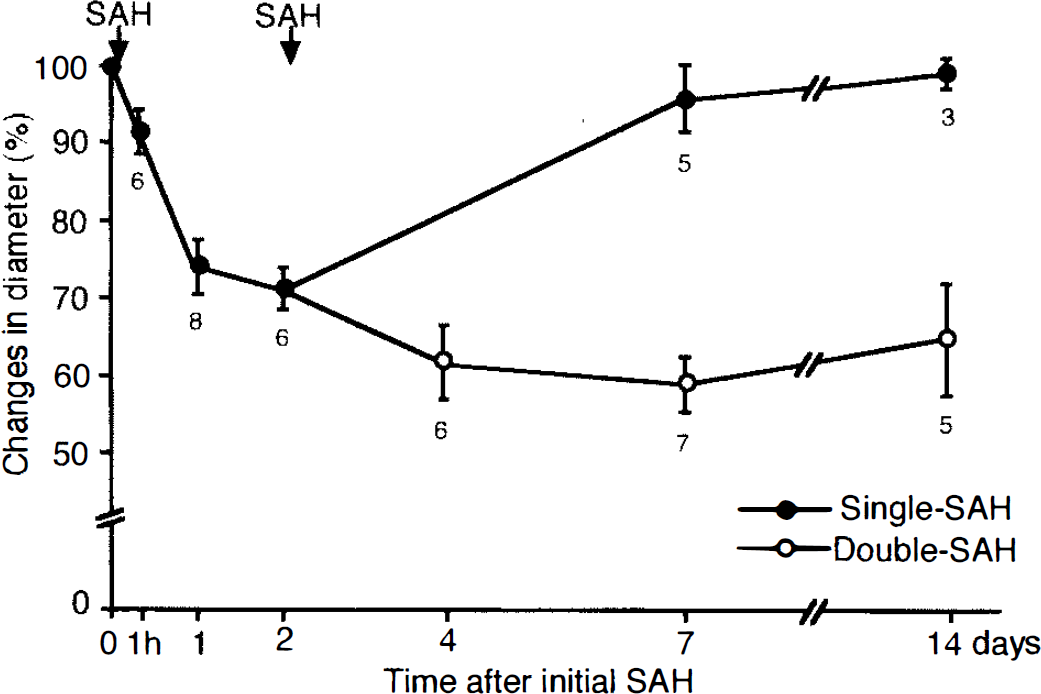
Changes in diameter of the basilar artery after SAH (control diameter was taken as 100%). Each value represents the mean ± SD. In the single-SAH group, the diameter of the basilar artery was significantly decreased 24 h after the initial SAH and showed gradual recovery up to day 7. In the double-SAH group, significant basilar arterial narrowing persisted 4–14 days after the initial SAH.
Histology of basilar artery
In the normal basilar artery, the number of smooth muscle cells was 10.8 ± 1.9 layers/tunica media (n = 4) on the crossline of the coronal section. The number of surviving smooth muscle cells was the same (9.7 ± 1.3; n = 4) 14 days after SAH in the single-SAH group, and smooth muscle cells showed almost no degenerative changes. In the double-SAH group, the number was significantly decreased to 7.6 ± 1.6 layers/tunica media (n = 4) 14 days after the initial SAH, and several surviving smooth muscle cells also featured a loss of nuclei and appearance of vacuoles.
Changes in ribosome aggregation rate
Electron microscopy of smooth muscle cells in a normal basilar artery showed that almost all ribosomes that were located predominantly around the perikarya were in aggregates of four or more (Fig. 2A), and the aggregation rate of ribosomes was 73.0 ± 8.0%. In smooth muscle cells of the basilar artery 15 min after topical application of PGF2α and 4 h after the intracisternal injection of TPA, when the basilar arteries were maximally contracted to ˜70 and 60% of the pretreatment diameter, respectively, the aggregation rate was 70.0 ± 7.0% and 71.4 ± 8.7%, respectively, the same as in the normal artery. In the single-SAH group, 1 h after the SAH, the aggregation rate was 62.5 ± 6.0%; 24 h after SAH, it was significantly decreased to 30.5 ± 6.4%; 7 days later, it had increased to 52.3 ± 9.0% (72% of control value) and returned to 70.2 ± 7.6%, the same as the control value, by 14 days (Fig. 3). In the double-SAH group, the aggregation rate was significantly decreased to 25.0 ± 5.0% 4 days after the initial SAH (Fig. 2B), and it remained significantly low at 14 days, 38.0 ± 10.5% at 7 days and 42.2 ± 12.3% at 14 days after initial SAH, even in the surviving smooth muscle cells with almost normal appearance (Fig. 3).
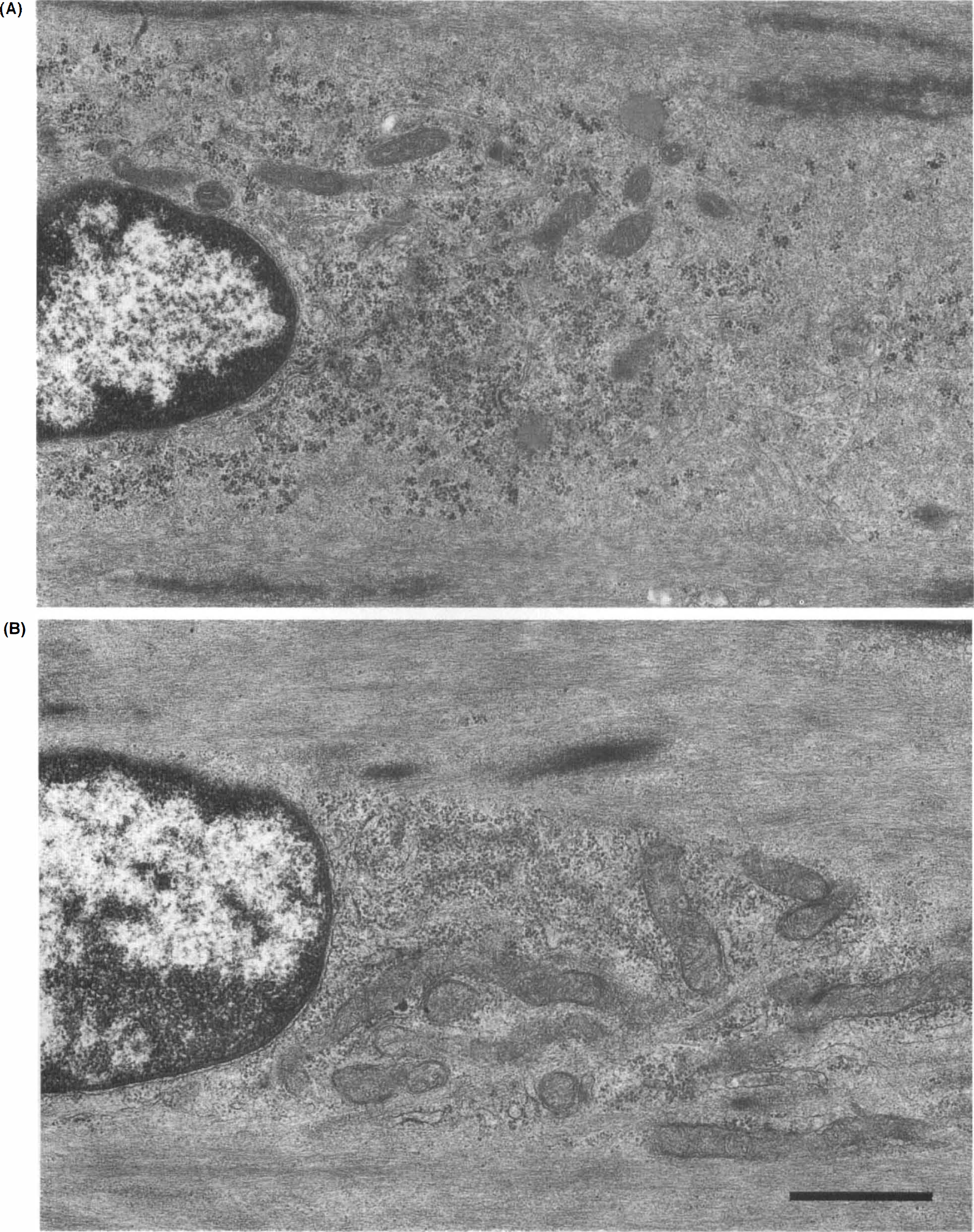
Electron micrographs showing smooth muscle cells of basilar arteries.
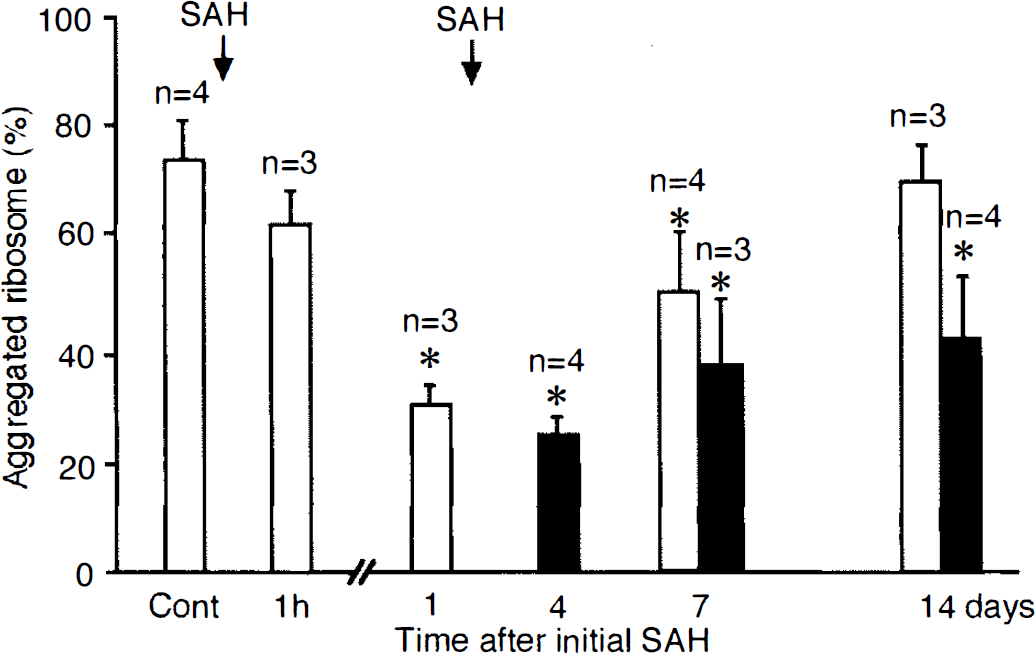
Changes in rate of aggregation of ribosomes in the single- and double-SAH groups. Open and closed bars indicate the single- and double-SAH groups, respectively, and each value represents the mean ± SD. In the single-SAH group, the ribosome aggregation rate was significantly decreased 24 h after the initial SAH and tended to recover 14 days after the initial SAH. In the double-SAH group, a significant reduction in the aggregation rate remained at 14 days after the initial SAH (*, p < 0.05).
Amino acid incorporation to proteins in basilar artery
In vitro amino acid incorporation into proteins was 2.2 ± 0.6 × 103 dpm/mg protein in the normal basilar artery (n = 7). It was slightly decreased to 1.9 ± 0.6 × 103 dpm/mg protein (n = 3) in the basilar artery at 7 days postSAH in the single-SAH group. In the double-SAH group, it was significantly suppressed to 1.2 ± 0.4 and 1.4 ± 0.3 × 103 dpm/mg protein (each n = 4) in the basilar arteries at 4 and 7 days after the initial SAH, respectively.
Changes in immunoreactivities of contraction-related proteins
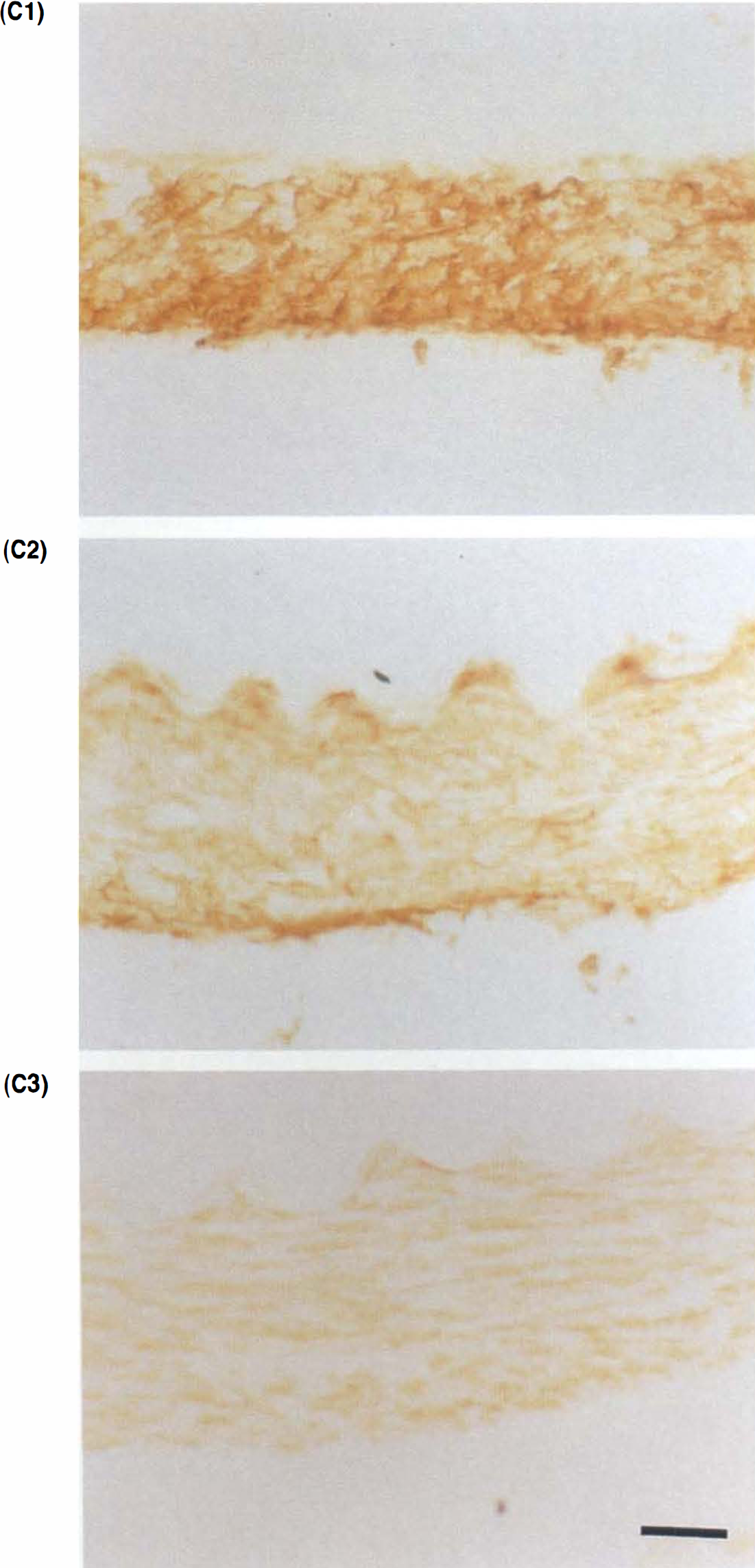
Figures 4 and 5 show the immunohistochemistry and immunoblotting for α-smooth muscle actin, calponin, and h-caldesmon. Immunoreactivity was seen predominantly in the tunica media of the basilar arteries (Fig. 4). In immunoblotting studies, the immunoreactive bands indicating α-smooth muscle actin, calponin, and h-caldesmon were specifically recognized at 40, 32, and 120 kDa, respectively (Fig. 5A–C). Changes in the densitometric quantities of immunoreactive bands are shown in Table 1. In the TPA-induced contracted basilar artery, each immunoreactivity was observed to be unaltered in the immunohistochemical examination (Fig. 4A–C). In the double-SAH group, both immunoreactivity in the histochemical examination and intensity of the band for α-smooth muscle actin were preserved for 14 days after the initial SAH (Figs. 4A, 5A, Table 1); in contrast, immunoreactivity and intensity for calponin and h-caldesmon were obviously decreased at 4 to 14 days after the initial SAH without any cross reactivities to degraded filaments (Figs. 4B,C, 5B,C, Table 1). The relative imbalance in the immunoreactivities of these structural proteins became obvious 4 days after the SAH, prior to the usual histological evidence of degeneration.
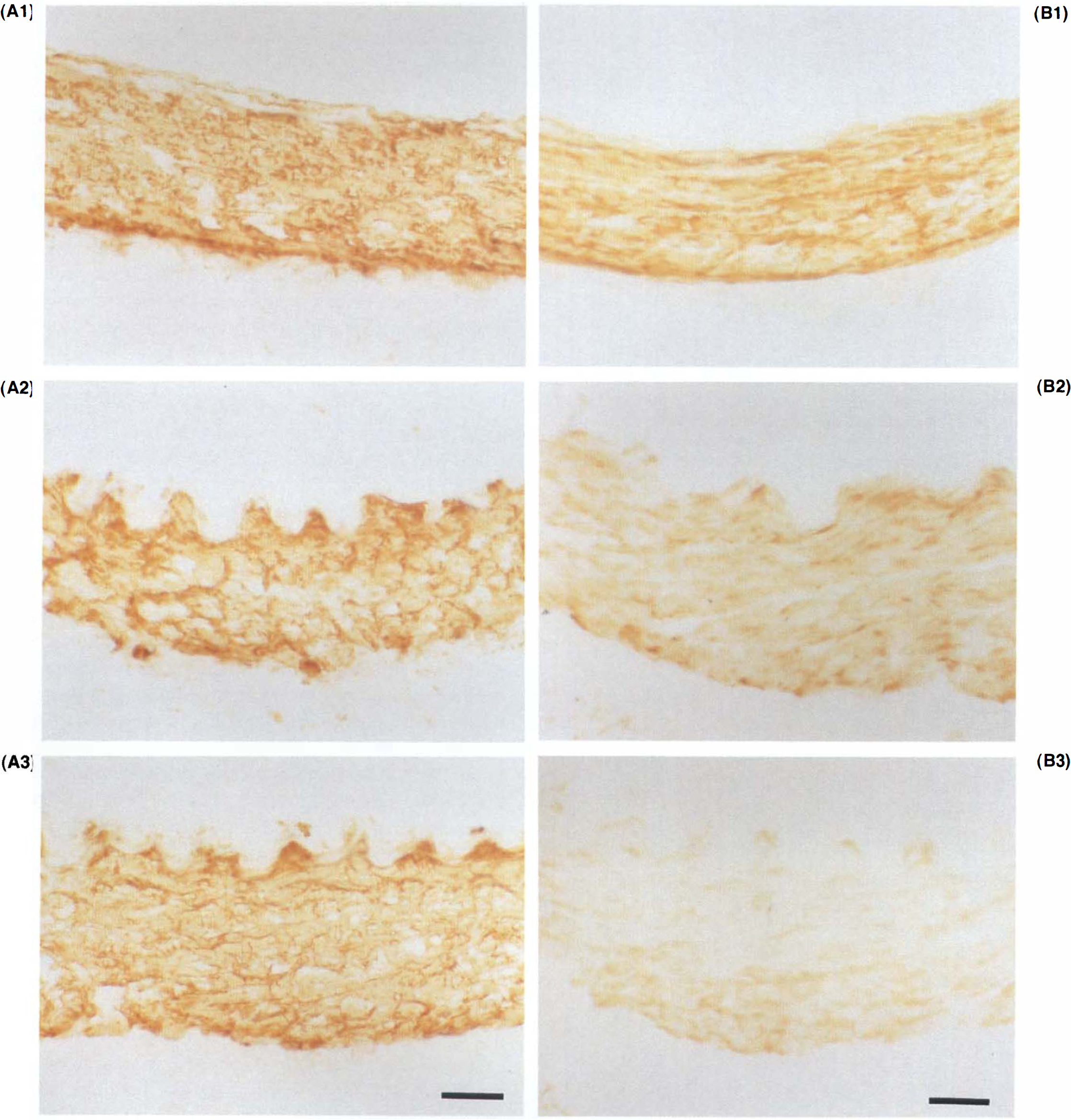
Photographs of immunohistochemical changes in α-smooth muscle actin, calponin, and h-caldesmon in normal basilar arteries (
See legend on page 1340.
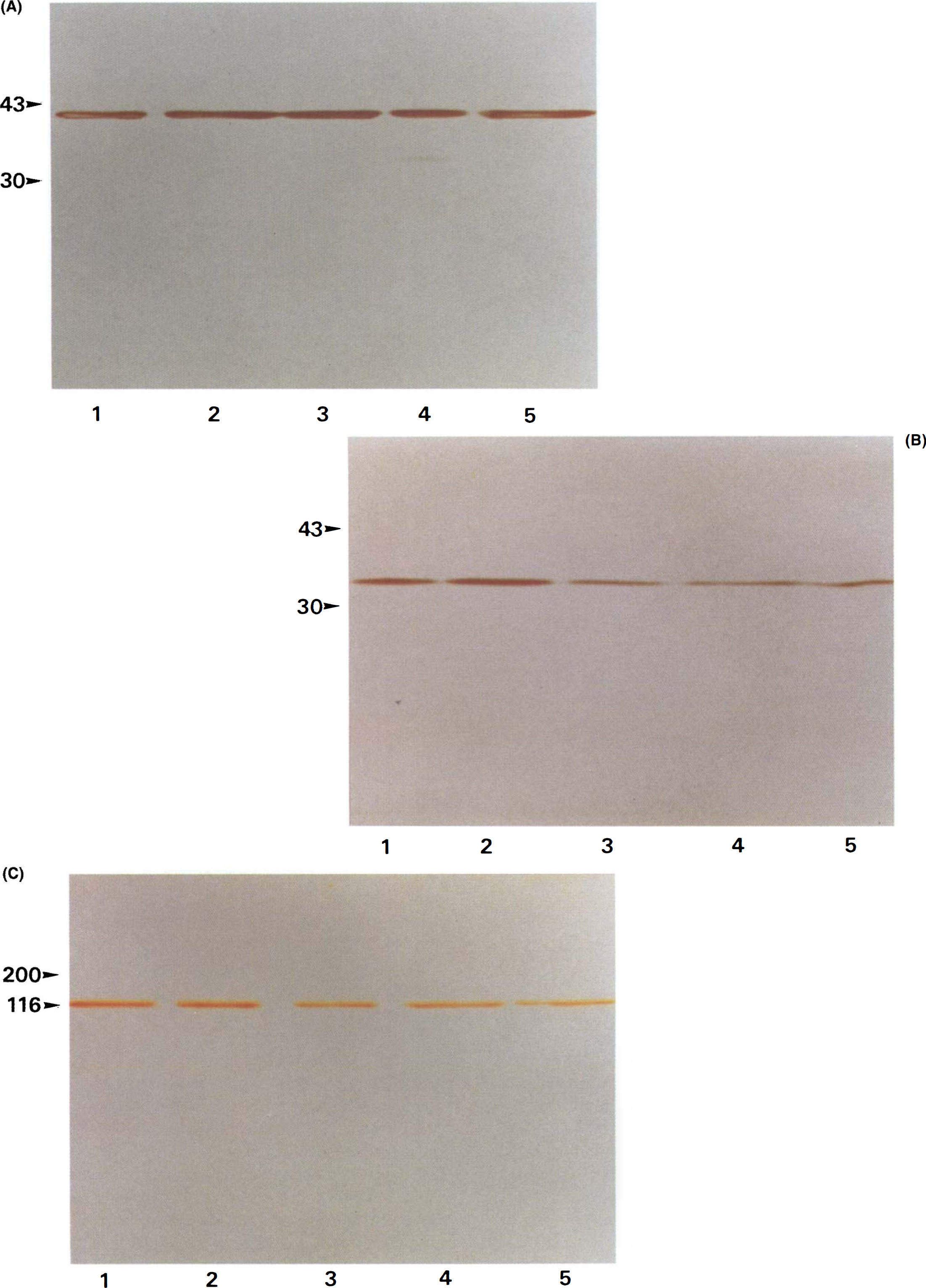
Photographs showing immunoblots of 40 kDa α-smooth muscle actin (
Changes in densitometric quantities of immunoreactive bands to α-smooth muscle actin, calponin, and h-caldesmon in the basilar artery after SAH
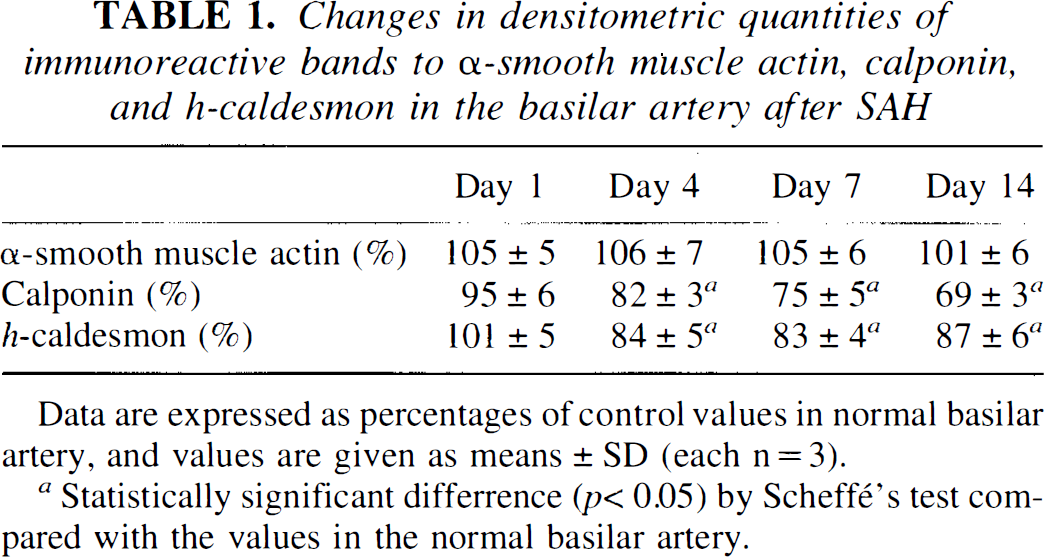
Data are expressed as percentages of control values in normal basilar artery, and values are given as means ± SD (each n = 3).
Statistically significant differrence (p < 0.05) by Scheffé‘s test com pared with the values in the normal basilar artery.
DISCUSSION
The etiology of the cerebral vasospasm following SAH due to a ruptured intracranial aneurysm remains unknown, and peculiar abnormalities appear in the smooth muscle cells of contracted arteries: a sustained contraction, which is originally the most representative function of the smooth muscle cells, and histological evidence of the development of marked degeneration, which must result in dysfunction of smooth muscle cells (Alksne and Greenhoot, 1974; Fein et al, 1974; Mayberg et al., 1978; Findlay et al., 1989). The possibility that excessive and long-lasting functional smooth muscle contraction may be an important element of vasospasm has led to studies on the alteration of the physiological regulatory pathways (e.g., those controlled by calmodulin or PKC, in an attempt to clarify their participation in the pathogenesis of cerebral vasospasm) in smooth muscle cell contraction and relaxation; however, no convincing explanations of the two peculiar phenomena, i.e., sustained contraction and histological degeneration, in cerebral vasospasm have been obtained (Sakaki et al., 1989; Sako et al., 1993). We conducted the present study with the hypothesis that progressive smooth muscle degeneration may be more important in the pathogenesis, not only of the delayed occurrence of smooth muscle cell death, but also of smooth muscle contraction. We first examined the serial changes in basilar arterial smooth muscle cells in the activity of protein synthesis, which represents the intracellular metabolism crucial for cell survival as well as energy supply, using the single-SAH model, in which milder vasospasm develops with short duration and little histological degeneration in the basilar artery, and the double-SAH model, in which long-lasting and more severe vasospasm develops, with massive degeneration and subsequent occurrence of smooth muscle cell death.
Protein synthesis in smooth muscle cells was evaluated by the morphological changes of the polyribosomes, since the translation of mRNA into peptide chains is catalyzed simultaneously by four–six ribosomes, appearing in rosette-like formations called polyribosomes or aggregated ribosomes (Rich, 1963), and protein synthetic activity can be morphologically estimated by the percentage of polyribosomes to total ribosomes. The application of PGF2α and TPA to the basilar artery to induce a physiological contraction through Ca2+-calmodulin and PKC pathways did not change the percentage of aggregated ribosomes. The finding showed that the physiologically functional contraction was not related to the rate of aggregation of ribosomes, as determined by electron microscopic examination. In the single-SAH group, the aggregation rate was significantly decreased 1 day after SAH and returned to normal 14 days after SAH, when the vasospasm was completely ameliorated and almost all smooth muscle cells survived, suggesting that the smooth muscle cells once damaged by the SAH insult still possessed the capacity to resume protein synthesis. In contrast, in the double-SAH group, much more disaggregation of polyribosomes was observed after the second SAH insult, and persisted longer, and the aggregation rate was still significantly below normal even in the surviving smooth muscle cells 14 days after the initial SAH. Thus, most of the smooth muscle cells that were damaged by the double SAH insults developed severe vasospasm and failed to recover from the impairment of protein synthesis.
An in vivo autoradiography for the assessment of protein synthesis in cerebral arterial smooth muscle cells of dogs is difficult since the animals are too big and the specimens of the basilar artery to be examined are too small. We conducted an in vitro radiolabeled amino acid incorporation assay to confirm the validity of the results obtained from morphological examination and found that incorporation was significantly reduced at 4 and 7 days after the initial SAH in the double-SAH group, whereas it was not significantly suppressed in the basilar artery at 7 days in the single-SAH group. Depletion of high-energy phosphates was not responsible for this supression, since they were replenished exogenously in the medium. Suppression of protein synthetic activity may be a phenomenon accompanied by the process of smooth muscle cell death, since it is sometimes observed in slowly progressing cell death, such as apoptosis (Cidlowski, 1982; Cohen, 1993). However, the persistent suppression of protein synthesis may be one of the causes of delayed smooth muscle degeneration and cell death, since the application of protein synthesis inhibitors induced delayed-type cell death in various organs (Chow et al., 1995; Martin et al., 1990).
Although it was not clear in the present study whether suppression of protein synthesis was a cause or not, it is important that the process leading toward smooth muscle cell death may be initiated in the quite early period after SAH insult before ordinary morphological smooth muscle cell degeneration becomes evident. One candidate for the mechanism that inhibits protein synthesis is intracellular Ca2+ overload, which has been observed just after SAH insult (Kohno et al., 1991). In neurons that are vulnerable to minimal transient ischemic injury, translation of mRNA into the peptide chain was impaired, with disaggregation of ribosomes due to the inactivation of initiation factor 2 brought about by the excessive overload of intracellular Ca2+ (Duncan and Hershey, 1984; Furuta et al., 1993; Nakagomi et al., 1993; Nowak et al., 1985; Ogura et al., 1988), and the intracellular Ca2+ level occasionally plays a pivotal role in the induction of apoptosis (Kizaki et al., 1989; Yasutomi et al., 1992).
In addition, Rosenblum et al. (1995) reported that protein synthesis was essential for the recovery of the endothelium-dependent arterial dilatation, even from the minimal injury of the cerebral artery with no histological degeneration. It was also reported that endothelium-dependent arterial dilatation was severely impaired in the vasospastic artery after SAH (Kim et al., 1988; Kanamaru et al., 1989). Therefore, in the present single-SAH group, the cerebral arterial contraction might have been relieved according to the resumption of endothelium-dependent arterial dilatation as the protein synthesis was restored, and the long-lasting reduction of protein synthesis in the severe vasospasm in the double-SAH group might have elevated the arterial susceptibility to contraction for a long duration by the failure to recover the potential for endothelium-dependent arterial dilatation.
Using immunohistochemistry and immunoblotting, we examined the changes in the immunoreactivities of some contractile and contraction-inhibiting structural proteins in the smooth muscle cells of basilar arteries in the double-SAH group. There have been reports that levels of l-caldesmon, an immature type of caldesmon, and myosin light chains were reduced in the smooth muscle cells, although tropomyosin was not significantly affected, and that the immunoreactivity of h-caldesmon, a mature type, was not reduced in the contracted smooth muscle cells of experimental SAH rabbits with minor histological degeneration (Takenaka et al., 1993). There have also been conflicting reports that degradation of actin occurs (Mayberg et al., 1990) or does not occur (Macdonald et al, 1992) in spastic arteries. In the present study, immunoreactivities of representative contraction-inhibiting proteins, i.e., calponin and h-caldesmon, were significantly more decreased after day 4 preceding the apparent degeneration of smooth muscle cells, than was that of the representative contractile protein, α-smooth muscle actin. PKC-dependent smooth muscle contraction has been reported to be induced via withdrawal of the inhibition of actomyosin ATPase by calponin and caldesmon through their phosphorylation (Naka et al., 1990; Tanaka et al., 1990). Therefore, even in the physiological PKC-dependent smooth muscle contraction, their immunoreactivities may be altered; we previously examined the immunoreactivities of these proteins by immunohistochemical examination in the PKC-dependent contracted basilar artery induced by TPA application (Sako et al., 1993). It was then suggested that phosphorylation of calponin and h-caldesmon by PKC did not significantly influence immunoreactivities. It was reported that calpain, a protease, was activated in the basilar artery after SAH (Minami et al., 1992), and catalyzed the proteolysis of calponin and caldesmon (Lee et al., 1993; Tsunekawa et al, 1989). The prolonged reduction in the immunoreactivities of both proteins observed in the present study might imply their quantitative changes, which could be induced with ease under the condition of suppression of the generation of proteins, in addition to degradation by calpain. PKC has been shown to be activated from days 4 to 7 in the basilar arteries of the same double-SAH model (Matsui et al., 1991; Sako et al, 1993). If there had been quantitative reductions or degradation of contraction-inhibiting proteins relative to the contractile proteins after SAH, the efficacy of the interaction between actin and myosin could be enhanced with activation of PKC. We hypothesized that the imbalance of contraction-related proteins could explain the smooth muscle contraction accompanying smooth muscle cell degeneration in cerebral vasospasm after SAH, if the reduction of immunoreactivities had been caused by their quantitative alteration. However, the validity of our hypothesis should be further examined using an appropriate quantitative measurement of contraction-related structural proteins, since reduction of immunoreactivity basically indicates qualitative changes of the protein, including the physiological conformational changes, rather than quantitative changes.
In conclusion, we observed an early-occurring and prolonged impairment of protein synthesis in smooth muscle cells of severely vasospastic cerebral arteries after SAH insult accompanying subsequent reduction of immunoreactivities predominantly in contraction-inhibitory structural proteins. The mechanism underlying suppression of protein synthesis and imbalance of immunoreactivities between contraction-related proteins must be examined further.
Footnotes
Abbreviations used
Acknowledgment:
We are grateful to Mr. Masachika Syudo of the Central Research Laboratory, and Mr. Kansei Wada and Mr. Kazushige Ohno of the Laboratory Animal Center at Ehime University School fo Medicine for technical assistance, and Mr. Daniel Mrozek for language support.