
Abstract
We tested the hypothesis that the neuropathologic outcome following recovery from incomplete ischemia is similar in normoglycemia and diabetes. Incomplete global ischemia was induced for 20 min in two groups of dogs: (a) normoglycemic, nondiabetic controls (n = 11) and (b) chronic (3 months), diabetic hyperglycemic subjects (n = 12). Animals were allowed to recover from surgery for 7 days after which they were perfusion-fixed for neuropathology. On paraffin processed tissue stained with hematoxylin and eosin (H&E), ischemic neurons were counted and the per cent of cell damage determined. All control animals survived for 7 days postischemia. Four of 12 diabetic animals survived for 7 days, with the remaining eight diabetic dogs dying within the first 3 days. On day 7, the percentage of neurons showing ischemic cell change in the four diabetic survivors and the 11 nondiabetic controls was similar in the cerebellum, CA1, superior temporal gyrus, and caudate. However, diabetic dogs that did not survive the 7-day recovery period showed cerebellar swelling, reduced Purkinje cell densities, and herniation. During the 3 months prior to ischemia, morning (10.7 ± 4.4 versus 11.2 ± 5.2 mM) and afternoon (8.8 ± 5.0 versus 9.4 ± 5.3 mM) blood glucose levels in the four surviving and eight nonsurviving diabetic animals, respectively, were similar. However, preischemic blood glucose was significantly elevated in animals that did not survive (7.8 ± 2.8 versus 15.8 ± 7.3 mM in survivors and nonsurvivors, respectively). This study shows that diabetic animals surviving 7 days postischemia and nondiabetic controls have similar neuropathology. However, diabetic animals in which glucose control deteriorated during the 24-h prior to ischemia did not survive, possibly due to severe hindbrain edema. These results show that in diabetes, blood glucose control immediately prior to incomplete global brain ischemia is an important determinant of morbidity and neuropathology.
Diabetes is associated with an increased incidence of cardiovascular morbidity (Garcia et al., 1974) and a poor neurologic outcome following stroke (Pulsinelli et al., 1983). In addition, hyperglycemia, with or without the presence of diabetes, is associated with a poor neurologic outcome (Pulsinelli et al., 1983). Autopsy studies have shown that diabetics have an increased incidence of stroke (Alex et al., 1962). However, it is unclear what influence chronic hyperglycemia has on stroke outcome given the vascular pathology that occurs with this disease. In addition, the effect of acute preischemic hyperglycemia is known to be detrimental, while that of chronic preischemic hyperglycemia is not (Pulsinelli et al., 1982).
Following global brain ischemia, whether complete or incomplete, animal experiments have consistently shown that acute, moderate hyperglycemia (200–300 mg/dl) existing prior to a severe ischemic or hypoxic event worsens neurological damage (Pulsinelli et al., 1982), impairs recovery of high-energy phosphates, and causes greater delayed hypoperfusion (Welsh et al., 1980; Kågström et al., 1983). A proposed mechanism for hyperglycemia-enhanced ischemic brain damage is that hyperglycemia increases intracellular acidosis during ischemia (Hurn et al., 1991). Therefore, it was predicted that any preischemic hyperglycemic state, whether acute or chronic, would compromise neurologic outcome. However, our laboratory has previously shown that following global incomplete ischemia, chronically diabetic, hyperglycemic dogs have a similar acute recovery of brain pH as do nondiabetic, normoglycemic animals, despite attaining an end-ischemic brain pH comparable to that of nondiabetic dogs made acutely hyperglycemic (Sieber et al., 1994). Thus, diabetic, chronic hyperglycemia may cause adaptations in the brain that hasten the acute recovery of pH following ischemia and result in better maintenance of energy metabolism, evoked potentials, and tissue water content compared to nondiabetic, acute hyperglycemia. However, it is unclear if the acute postischemic brain ATP and pH responses of diabetic animals influence morbidity and delayed neuropathologic outcome. To determine how diabetic, chronic hyperglycemia influences neuropathology and neurologic outcome, we tested the hypothesis that the neuropathologic outcome following recovery from incomplete ischemia is similar in normoglycemia and diabetes.
METHODS
Diabetic model
To produce diabetes, total pancreatectomy under halothane anesthesia was performed on conditioned, pure bred, male beagle dogs as described previously (Sieber et al., 1993a,b). Surgery was followed by a 3-month period of blood glucose management with subcuticular injections of ultralente insulin. Blood samples were drawn from a foreleg vein at 8–9 a.m. (overnight fasting) and at 3 p.m. (after main feeding) for daily analysis of blood glucose. The dose of insulin was adjusted individually to maintain blood glucose between 10 and 17 mM throughout the day and averaged 1.4 U/kg/day (Sieber et al., 1993a,b). On the day of the ischemic protocol, the usual morning insulin dose was withheld. This allowed us to control for the effects of insulin during ischemia. A group of nondiabetic controls (n = 11) underwent sham operation involving abdominal incision and closure. Following sham operation, the nondiabetic controls survived for 3 months.
Ischemic protocol
After placing a peripheral intravenous catheter, all dogs were anesthetized with fentanyl (50 μg/kg, i.v.) and pentobarbital (10 mg/kg, i.v.) followed by a continuous pentobarbital infusion (3 mg/kg/h, i.V.). Pancuronium bromide (0.1 mg/kg, i.v.) was injected for muscle paralysis. The trachea was intubated and the lungs mechanically ventilated. A femoral artery was cannulated for blood pressure measurement and blood sampling. The left temporalis muscle over the lambdoidal suture was retracted from the skull. After placing a small burr hole, a thermistor was inserted between bone and dura to monitor epidural temperature. After placing a second small burr hole, a silastic ventricular drain catheter with multiple side ports was inserted into the lateral ventricle for measuring intracranial pressure (ICP) and infusing artificial cerebrospinal fluid (CSF) (Hum et al., 1991; Sieber et al., 1994; Palmon et al., 1995). Arterial line placement, intraventricular catheter placement, and production of ischemia were all performed using strict sterile precautions. The body of the dog was wrapped in a plastic bag and placed on a blanket perfused with recirculating warm water. Heat lamps were used, if necessary, to maintain epidural temperature at 37–38°C. End-tidal CO2 was monitored and ventilation adjusted to maintain Paco2 at 35–40 mm Hg. Mean arterial blood pressure (MABP) and ICP were continuously recorded. Inspired O2 was at ˜30% to maintain oxyhemoglobin saturation. Supplemental oxygen was administered until ˜30 min postischemia (when the animal was extubated). Although prolonged postischemic hyperoxia may exacerbate neurologic dysfunction, 30 min of postischemic hyperoxia does not influence neurologic outcome (Mickel et al., 1987).
Incomplete ischemia was produced in two groups of dogs—nondiabetic (n = 11) and diabetic (n = 12)—by infusion of sterile artificial CSF into the lateral ventricle to produce an ICP 10 mm Hg below that of the animal's MABP. The artificial CSF was prewarmed and maintained at a temperature of 38°C. A moderate pressor response resulted, but ICP was easily manipulated to help keep cerebral perfusion pressure constant at 10 mm Hg for 20 min. Cerebral blood flow (CBF) during ischemia ranged from 2 to 11 ml min−1 100 g−1 (Sieber et al., 1994). This was sufficient to reduce cerebral O2 uptake, flatten the somatosensory evoked potentials, and reduce phosphocreatine and ATP levels (Sieber et al., 1994; Palmon et al., 1995). To start reperfusion, the CSF pressure reservoir was disconnected and ICP allowed to normalize.
MABP and ICP were measured continuously with Statham pressure transducers (Quest Medical, Allen, TX, U.S.A.). Arterial Po2, Pco2, and pH were measured with Radiometer BMS3 electrodes and analyzer (Radiometer, Nevada, Denmark). Arterial oxygen content and saturation, and hemoglobin concentration were determined with a Hemoximeter 3 (Radiometer). Arterial glucose concentration was measured with a Yellow Springs glucose/lactate analyzer (Yellow Springs, Yellow Springs, OH, U.S.A.). All analyzers were calibrated routinely throughout each experiment as per our previous work (Hurn et al., 1991). Following the ischemic episode, all catheters were removed, bleeding controlled, and skin incisions sutured prior to awakening the animal from anesthesia.
In animals surviving ≥24 h postischemia, a neurological deficit scoring system for dogs was used, as previously described (Sieber et al., 1995). The neurologic deficit evaluation provided a single score that may range from a low of 20, which was the best possible, to a high of 62. Neurologic scoring was performed daily during the postischemia recovery period by a blinded observer.
In both groups, daily blood glucose was measured in the a.m. and p.m. following ischemia. In the diabetic group, postischemic insulin administration was adjusted to a blood glucose of ˜10 mM for the duration of the reperfusion period.
At 7 days postischemia, dogs were anesthetized with pentobarbital, anticoagulated with heparin, and vasodilated with sodium nitrate, after which their brains were perfused with phosphate-buffered saline followed by 4% paraformaldehyde. After removal, each brain was divided midsagittally and the hemispheres were cut systematically into 1-cm thick coronal slabs from anterior to posterior. From the left hemisphere, the mid-hippocampus and surrounding temporal neocortex, frontal cortex, caudate (head), and cerebellum were sampled consistently for paraffin histology and hematoxylin and eosin (H&E) staining. Our preliminary studies confirm that in this model of ischemia, neuronal damage was similar bilaterally. Four areas were evaluated quantitatively for ischemic injury: the hippocampus (CA1), the crown of the superior temporal gyrus, the sulcus of the superior temporal gyrus, and the anterior cerebellar lobule at the midline. We selected these areas since we had showed previously that these regions were consistently damaged in this dog model of incomplete cerebral ischemia (Sieber et al., 1995). Purkinje cells in the cerebellum were counted in four fields at 200× in the rostral cerebellar folia (anterior/rostral vermis) against the pons and above the fourth ventricle. The left hippocampal CA1 region was counted at the mid-hippocampal level in six fields at 600×. Pyramidal neurons in layer III of the superior temporal gyrus were counted in six fields at 600× at the crown (moving towards the sulcus) and in six fields at the sulcus. In sections stained for H&E, the number of neurons were counted and the % of neurons with ischemic damage was determined using the criterion of microvacuolar change within the cell body, peripheralization of chromatin, perinuclear eosinophilia, or perikaryal shrinkage with eosinophilic cytoplasm. Ischemic neuronal damage was determined by a blinded investigator (L.M.), who evaluated the four regions of interest separately.
For animals not surviving 7 days postischemia, the animals either were perfused intra-aortically with 4% paraformaldehyde when spontaneous ventilations deteriorated (n = 4) or the brains were removed postmortem and immersion-fixed in 4% paraformaldehyde (n = 4). In immersion-fixed brains, the postmortem interval delay was 6 (n = 3) and 16 (n = 1) h. Paraffin-processed brain samples and H&E slides from each of the above-mentioned regions were prepared and the number of neurons/mm2 was calculated for each region as an index of postischemic brain swelling.
In 7-day survivors, the % neuronal damage in each region was averaged for each animal and groups were compared on a regional basis using a t-test. The number of neurons/mm2 was calculated for each region in all animals. Comparisons of the number of neurons/mm2 were made between survivors and nonsurvivors, and between diabetic and nondiabetic animals using a t-test. Neurologic deficit evaluation scores were obtained for each postischemic day and compared by the Wilcoxon rank-sum test for two groups.
RESULTS
Twenty-three dogs underwent 20 min of global incomplete ischemia. Arterial blood gas and hemoglobin (Hb) values immediately preischemia were not different between groups [pH, 7.42 ± 0.06; Pco2, 31 ± 6 mm Hg; Po2, 243 ± 171 mm Hg; Hb, 14.0 ± 1.8 g/dl; and pH, 7.46 ± 0.05; Pco2, 31 ± 4 mm Hg; Po2, 246 ± 147 mm Hg; Hb, 13.4 ± 2.3 g/dl in control and diabetic groups, respectively (mean ± SD)]. Immediately preischemia, mean blood glucose was elevated >threefold in the diabetic group (4.0 ± 0.5 mM versus 12.1 ± 7.6 mM in control and diabetic groups, respectively). Epidural temperature during ischemia was 37.8 ± 0.6°C and not different between groups. No difference between groups was observed in MABP or ICP pre-, during, and up to 15 min postischemia.
All animals in the control group survived for 7 days postischemia. However, in the diabetic group, only four of 12 animals survived for 7 days postischemia. Seven diabetic animals died within the first 24 h postischemia with one dog surviving for 3 days postischemia. During the 3 months prior to ischemia, morning and afternoon blood glucose in the four surviving and eight nonsurviving diabetic animals was similar. Both diabetic survivors and nonsurvivors received the same dose of insulin during their 3-month period of diabetes (Ultralente, 14 ± 2 U/day). The range of daily insulin doses given was 12–22 and 8–24 U/day in surviving and nonsurviving diabetic animals, respectively. However, preischemic blood glucose was elevated in those animals that did not survive (Table 1). In nonsurviving diabetic dogs, preischemic blood glucose was ˜four- and twofold higher than in control and surviving diabetic dogs, respectively.
Blood glucose levels in diabetic animals 3-month convalesence
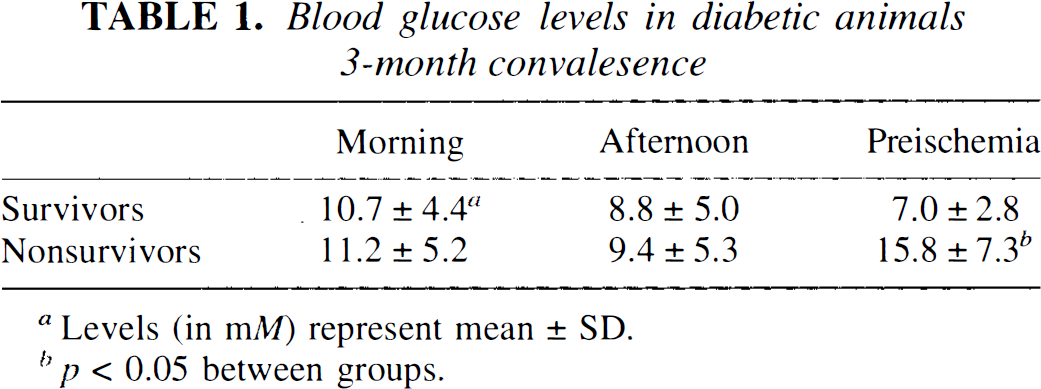
Levels(in mM)represent mean±SD.
p < 0.05between groups.
In animals surviving 7 days postischemia, no focal neurologic deficits or overt seizures were observed during the 7-day recovery period. Neurologic scores were similar in surviving diabetic and nondiabetic animals. The neurologic score on postischemic days 1, 2, and 3 (24 ± 8, 22 ± 4, and 22 ± 3, respectively) was increased from the scores obtained on postischemic days 5, 6, and 7 (21 ± 2, 21 ± 1, and 20 ± 1, respectively). Nonsurviving diabetic animals were generally unresponsive to external stimuli and, thus, were not evaluated neurologically.
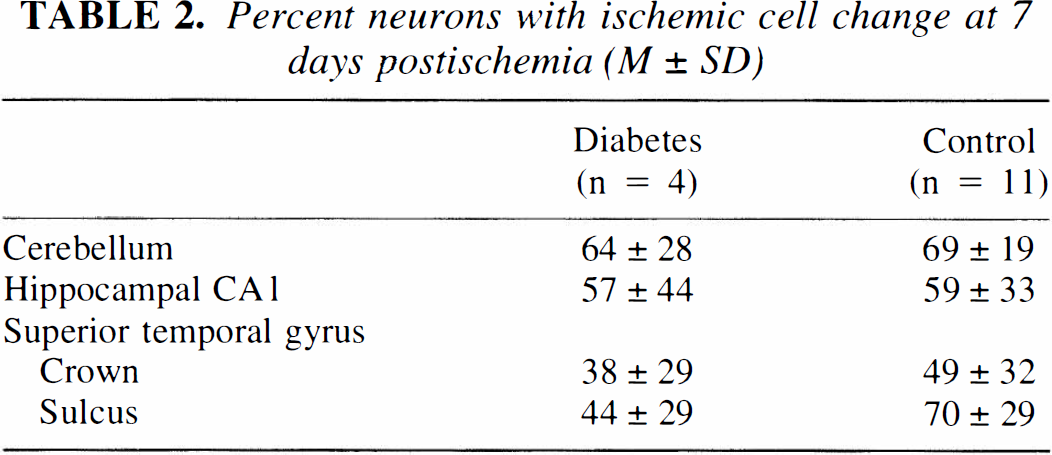
In the dogs surviving for 7 days, neurons in the neocortex, hippocampus, caudate, and cerebellar cortex showed ischemic cell change, as shown previously in this model (Sieber et al., 1995). Microscopically, neurons in the forebrain and cerebellum were shrunken and highly eosinophilic with pyknosis of the nucleus (Fig. 1). The severity of postischemic neuropathology was determined as a function of remaining neurons that showed ischemic cell injury. At 7 days postischemia, the % of neurons displaying evidence of ischemic cell change was similar between groups (Table 2). Diabetic dogs showed no evidence of cerebral microangiopathy.
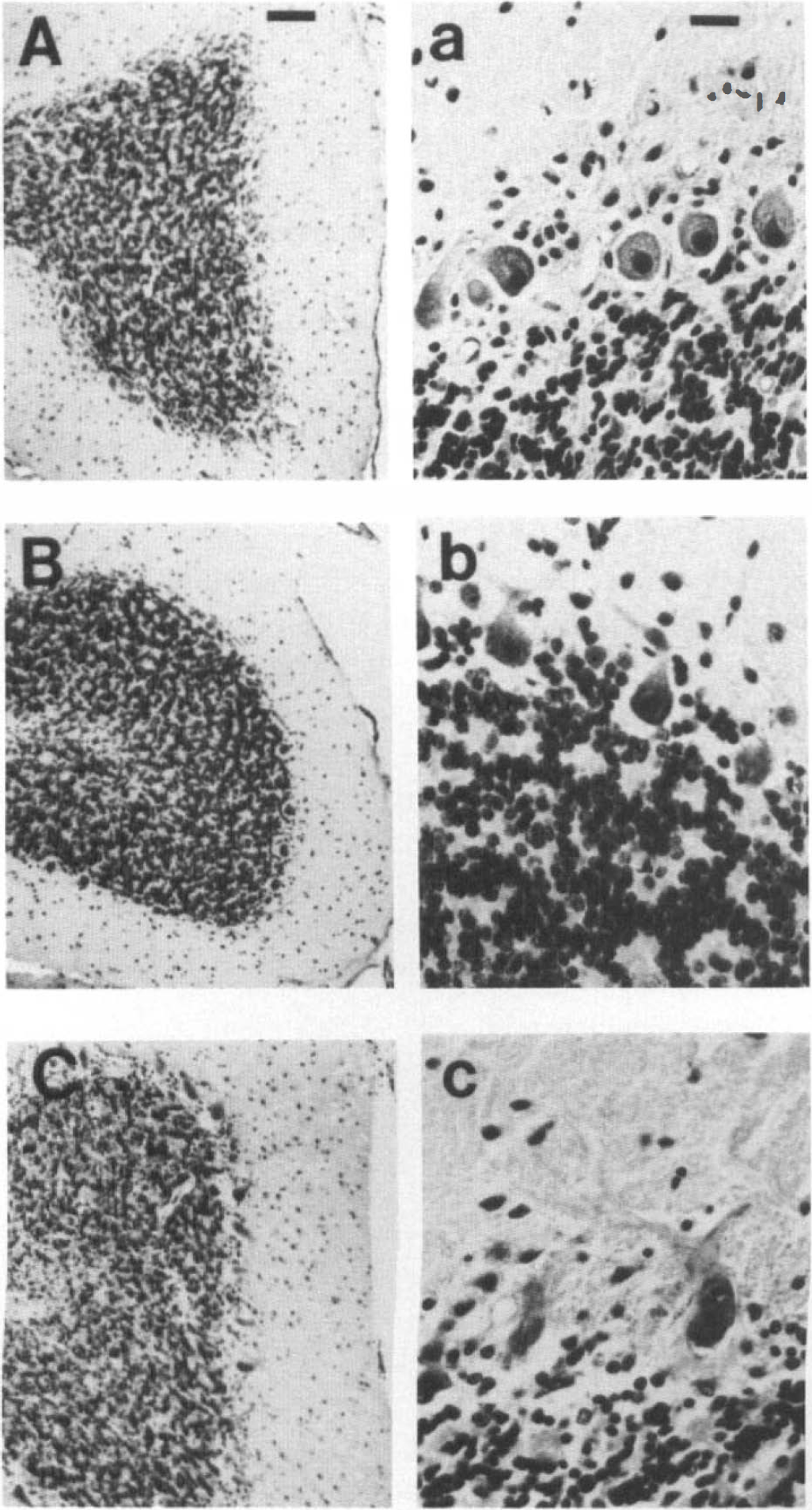
Postischemic cerebellar damage in nondiabetic controls
Percent neurons with ischemic cell change at 7 days postischemia (M ± SD)
Postmortem, brains from the eight diabetic nonsurvivors showed evidence of brain edema. On gross examination, the posterior cerebellar lobule was consistently herniated partially through the foramen magnum. However, the cerebral hemispheres were symmetrical and, on brain cutting, the ventricular system was not distorted. These findings were consistent with the presence of hindbrain herniation in the absence of forebrain herniation. Microscopically, large perineuronal spaces surrounded most injured neurons, probably representing swollen astrocytic processes. These microscopic observations are consistent with previous neuropathologic descriptions of hyperglycemic ischemia (Pulsinelli et al., 1982). In diabetic nonsurvivors, the number of neurons/mm2 was significantly decreased in the cerebellum (but not in forebrain regions) from that of ischemic controls (Table 3). This finding is consistent with the occurrence of cerebellar swelling (Fig. 1C,c). In nonsurviving diabetic animals, neuropathology was similar in immersion-fixed and perfused brains, indicating that immersion fixation produced little artifact. Dark neurons were observed infrequently in immersion-fixed brains.
Regional Neuronal Densities (cells/mm2) Postischemia
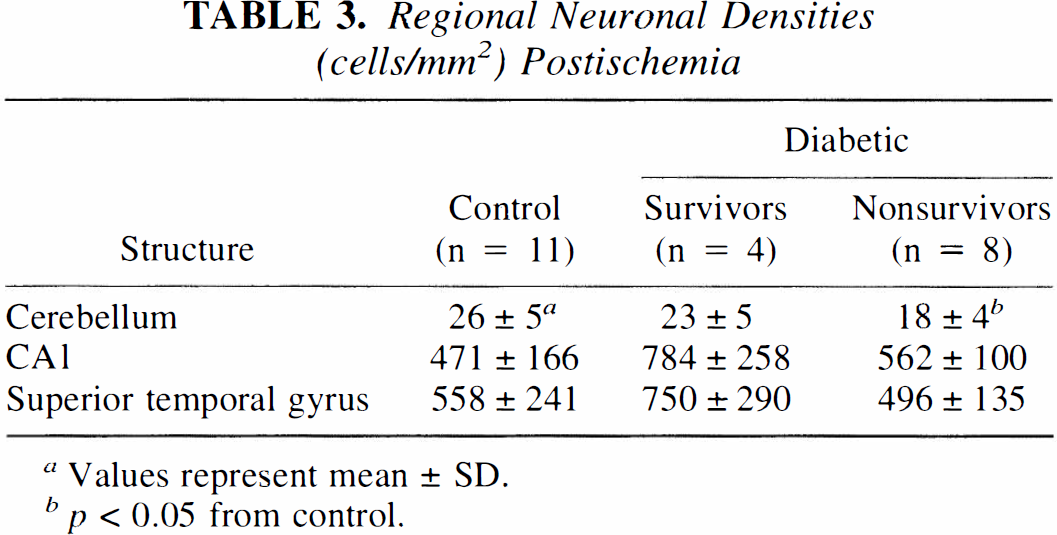
Values represent mean ± SD.
p < 0.05 from control.
DISCUSSION
This study shows that diabetic animals without microangiopathy surviving 7 days postischemia have a similar neurologic and neuropathologic outcome as non-diabetic controls despite chronic hyperglycemia. However, diabetic animals in which glucose control deteriorated during the 24 h prior to ischemia did not survive. These results show that in diabetes, insulin-based blood glucose control immediately prior to incomplete global brain ischemia is an important determinant of morbidity and neuropathologic outcome.
The association between diabetes and poor neurologic/neuropathologic outcome following stroke and cardiac arrest has long been recognized (Garcia et al., 1974; Pulsinelli et al., 1993; McCall, 1992; Biller and Love, 1993). Studies suggest that the glucose level may be an important determinant of outcome, although the role of vascular disease cannot be ruled out. The pancreatectomized dog model of diabetes allows the study of the effects of chronic low dose insulin management of blood glucose on the ischemic response of the diabetic brain without the confounding influence of diabetic microangiopathy. The pancreatectomized dog model of diabetes develops progressive diabetic end organ vascular disease. In other words, the spectrum of the effects of diabetes can be examined from acute hyperglycemia (acute glucose bolus and infusion), to chronic hyperglycemia without histologically-defined vascular disease, to diabetes with microangiopathy (histologically reproducible retinopathy and glomerulopathy, which is similar to humans and first occurs at 24–30 months in diabetic dogs) (Engerman et al., 1977). The current study reports the brain ischemic response in diabetic animals without histologically defined microangiopathy.
The interpretation of our results provides insight into the factor(s) that dictate the responses of the diabetic brain to ischemia. Because evidence suggests that insulin ameliorates brain ischemic damage, especially after prolonged periods of recovery (Voll and Auer, 1991; Strong et al., 1990; Izumi et al., 1992), a possible interpretation for differences in postischemic morbidity of diabetic dogs would be insulin-mediated neuroprotection. However, insulin doses were similar in diabetic survivors and nonsurvivors. In addition, insulin was withheld in all diabetic animals on the day of the ischemic insult. It will be difficult to eliminate insulin effects in the reperfusion period in the diabetic group since without insulin, the diabetic animals would die. However, these conditions are similar to those in the clinic, where insulin is not withheld from diabetic persons following stroke.
Diabetes alters brain responses to focal ischemia. With focal ischemia in animals, diabetes causes an increase in infarct volume and decreases selective neuronal injury in the peri-infarct region compared to normoglycemia (Nedergaard et al., 1988; Nedergaard and Diemer, 1987). In addition, with transient focal ischemia, diabetes may convert selective neuronal injury to infarct (Nedergaard, 1987). In humans, increased glucose levels on hospital admission are associated with increased mortality from stroke in nondiabetic, but not in diabetic, patients (Woo et al., 1990; Jørgensen et al., 1994). These reports have been interpreted as showing that the association between hyperglycemia and stroke outcome may actually reflect stroke severity rather than the adverse effects of glucose in the clinical situation. However, the effects of diabetes on global ischemia are unclear.
Acute hyperglycemia augments ischemic brain damage in animal models of global ischemia (Pulsinelli et al., 1982; Sieber and Traystman, 1992). Our findings in the cerebellum of nonsurviving animals are consistent with the neuropathology in this region following acute hyperglycemic ischemia (Pulsinelli et al., 1982). The swelling of cerebellum and herniation of hindbrain, but not forebrain, in our dog model of global incomplete ischemia may be related to the tendency for a greater reduction of CBF in hindbrain as compared to forebrain regions (Duckrow et al., 1987). However, it is unlikely that diabetes worsens regional neuropathologic damage following global brain ischemia as the neuropathologic damage in surviving diabetic and nondiabetic normoglycemic animals was similar. This suggests that following 3 months of diabetes, delayed ischemic brain damage is unlikely to be influenced significantly by diabetes-induced adaptations within the brain (Sieber et al., 1994). In nondiabetic animals, global ischemic brain damage begins to be enhanced at a threshold plasma glucose of 10–13 mM (Li et al., 1994). We could not demonstrate a clear-cut threshold value for blood glucose at which diabetic dogs die following ischemia. At present, we are uncertain as to why blood glucose levels were significantly higher immediately preischemia in nonsurviving versus surviving diabetic dogs. Age, gender, care and handling, as well as insulin treatments, were similar for all animals. Perhaps surviving ectopic islets of Langerhans are responsible for the resistance to hyperglycemia in some dogs. During total pancreatectomy, visual affirmation was used to determine that the pancreas had been completely removed. However, postoperatively, we did not document insulin levels or perform tolbutamide stress tests. We believe that the current study suggests that diabetes has less of an impact on neurologic and neuropathologic outcome than does the preischemic blood glucose level following global cerebral ischemia.
In summary, this study examines the effects of chronic hyperglycemia on neuropathology and neurologic outcome following global brain ischemia. Results suggest that any adaptations occurring in the diabetic brain as a result of chronic hyperglycemia do not significantly influence neurologic outcome. It appears that the preischemic blood glucose level is an important determinant of morbidity and neuropathologic damage following global brain ischemia, regardless of the presence of diabetes. This emphasizes the importance of preischemic insulin-based blood glucose control in determining neurologic outcome following brain ischemic insults.
Footnotes
Abbreviations used
Acknowledgment:
The authors would like to thank Rosie Cousins, Dawn Spicer, and Ann Price for their technical assistance, and Lee Palmer for her expert preparation of this manuscript. This study was supported, in part, by grants from the United States Public Health Service National Institutes of Health (NS 01380, NS 20020, and AG 07914).