
Abstract
Hyperglycemia worsens stroke, yet rigorous glycemic control does not improve neurologic outcome. An alternative is to target downstream molecular mediator(s) triggered by hyperglycemia but independent of prevailing glycemia. Soluble epoxide hydrolase (sEH) is a potential mediator of injury via its metabolism of neuroprotective epoxyeicosatrienoic acids (EETs). We tested whether hyperglycemia exacerbates cerebral injury by upregulating sEH and decreasing brain EET levels. Type 1 diabetes mellitus was modeled by streptozotocin (STZ;50 mg/kg per day intraperitoneally, 5 days) in male mice. At 4 weeks, STZ-treated and control mice underwent 45-minute middle cerebral artery occlusion (MCAO) with or without sEH blockade by trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid (t-AUCB;1 mg/kg intraperitoneally daily for 6 days before MCAO). The STZ-treated mice had increased sEH mRNA expression in cerebral vessels and decreased EET concentrations in brain. There was no difference in cortical perfusion between groups. The STZ-treated mice sustained larger brain infarct than controls. Pretreatment with t-AUCB eliminated the difference in infarct size and EETs concentration between STZ-treated mice and controls, without altering glycemia. We conclude that type 1 diabetes mellitus upregulates sEH mRNA and decreases concentrations of neuroprotective EETs within the brain, leading to worse stroke outcome. The data indicate that sEH antagonism may be beneficial in the setting of hyperglycemic stroke.
INTRODUCTION
Diabetes mellitus increases risk of stroke, and hyperglycemia exacerbates ischemic injury to brain.1–3 Coincidence of hyperglycemia at presentation for cerebral ischemia is associated with larger infarct size and worsened clinical outcome.3–6 Persistent hyperglycemia in the postischemic period is an independent predictor of worse neurologic outcome and 90-day mortality.7,8 Preexisting hyperglycemia also increases the ischemic brain infarct in numerous animal models, as recently reviewed. 9 Etiology of hyperglycemia, whether by insulin resistance, impaired insulin secretion, or exogenous glucose challenge, does not seem to be a deciding factor. The molecular mechanism by which hyperglycemia exacerbates ischemic injury to brain remains to be elucidated.
A 2001 study by van den Berghe et al 10 indicated that aggressive glycemic control with insulin in hyperglycemic critically ill surgical patients improved many clinical end points, including mortality. But two recent large, multicenter studies failed to show a beneficial effect of tight glycemic control. The ACCORD trial, enrolling 10,251 patients with type 2 diabetes mellitus, found that intensive glycemic control (target glycosylated Hb < 6.0) did not alter incidence of either fatal or nonfatal stroke, and increased overall mortality (relative risk 1.22) vs. less intensive therapy. 11 The NICE-SUGAR study, on 6,104 hyperglycemic critically ill patients, found that tight glycemic control (target < 180 mg/dL) also had no effect on neurologic outcome but increased mortality (relative risk 1.14) compared with standard care. 12 In both studies, tight glycemic control markedly increased the incidence of serious hypoglycemia.11,12 The GIST-UK study, 13 on 933 patients with acute stroke, showed that aggressive glycemic control did not improve mortality or 90-day neurologic disability, and patients with the largest poststroke decrease in blood glucose concentration experienced higher mortality. Together, the data indicate that rigorous glycemic control, either before or after cerebral ischemia, carries major risk accompanying a theoretical but unrealized protection against hyperglycemia-mediated ischemic brain injury.
An alternative approach to patients either acutely experiencing or at risk of hyperglycemic stroke is to interfere with downstream mediator(s) triggered by hyperglycemia but ultimately acting independent of the prevailing glycemic status. Soluble epoxide hydrolase (sEH), a product of gene EPHX2, is abundant in brain and is a potential mediator of ischemic injury via its clearance of protective epoxyeicosatrienoic acids (EETs).14,15 In the present study, we tested the hypothesis that hyperglycemia exacerbates cerebral ischemic injury by upregulating EPHX2 mRNA expression and by decreasing concentrations of vaso- and neuroprotective EETs in brain. We also tested whether specific sEH blockade with trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid (t-AUCB) can attenuate the worsened brain injury caused by hyperglycemia.
MATERIALS AND METHODS
Our study was conducted in accordance with National Institutes of Health guidelines for care and use of animals in research. All protocols were approved by the Institutional Animal Care and Use Committee of Oregon Health & Science University (Portland, OR).
Streptozotocin-Induced Type 1 Diabetes Model in Mice
Seven-week-old male C57BL/6 mice (Charles River, Hollister, CA, USA) were acclimatized to the animal facility and fasted for 4 hours before each injection. Mice received streptozotocin (STZ) 50 mg/kg (or saline vehicle in controls) intraperitoneally daily for 5 days. Glycemia in saphenous vein blood of nonfasted animals was measured weekly for 4 weeks by glucometer (Breeze 2; Bayer, Tarrytown, NY, USA) to confirm STZ-induced hyperglycemia. Insulin levels were measured by radioimmunoassay using a Sensitive Rat Insulin RIA Kit (Millipore, Billerica, MA, USA). Measurements were run in duplicate and performed according to the manufacturer's instructions.
Inhibition of Soluble Epoxide Hydrolase Activity
In vivo sEH activity was inhibited with t-AUCB, which was developed and generously provided by Dr Bruce Hammock, University of California, Davis, CA. 16 Mice received t-AUCB 1 mg/kg (or saline vehicle) intraperitoneally daily for the final 6 days before cerebral ischemia, with the final t-AUCB dose administered immediately after reperfusion.
Optical Microangiography
Optical microangiography (OMAG) was performed as previously described. 17 Blood perfusion was visualized and quantified based on endogenous light scattering from moving blood cells within the brain. A superluminescent diode with a central wavelength of 1,310 nm and a full-width, half-maximum bandwidth of 50 nm was used to illuminate the OMAG system. The spectral interferograms formed by the reference light and the light backscattered from the tissue sample were detected by a custom-built, high-resolution, and high-speed spectrometer. A final volume data cube of 1,000 × 500 × 512 (x – y – z) voxels was built, from which the three-dimensional OMAG structural and flow images were computed. The three-dimensional scan represented a physical volume with the following x – y – z dimensions: 2.5 × 2.5 × 2.0 mm3.
Middle Cerebral Artery Occlusion
Middle cerebral artery occlusion (MCAO) was performed at 4 weeks after completion of STZ treatment or vehicle for corresponding controls. Transient (45 minutes) focal cerebral ischemia was induced in overnight-fasted mice using an intraluminal MCAO technique as described previously. 18 Briefly, isoflurane-anesthetized mice were instrumented with a laser-Doppler probe and MCAO ischemia induced via insertion of a silicone-coated 6-0 nylon monofilament into the right internal carotid artery until MCA flow was <20% of baseline. The occluding filament was subsequently withdrawn for reperfusion. Mice were allowed to recover and observed for 1 day, after which they were anesthetized with isoflurane, and either killed by decapitation and brains removed for infarct size measurement, or perfused with heparinized ice-cold saline for cerebral vessel isolation, as described below. 18
Brain Infarct Size
Infarct size in cerebral cortex, caudate putamen, and total hemisphere was measured at 24 hours after MCAO in 2-mm thick coronal brain sections using 2,3,5-triphenyltetrazolium chloride staining and digital image analysis (SigmaScan Pro 5.0; Aspire Software, Ashburn, VA, USA) as previously described. 18 To account for edema, infarcted area was estimated by subtracting an uninfarcted region in ipsilateral hemisphere from contralateral hemisphere, and expressing infarct volume as a percentage of the contralateral hemisphere.
Isolation of Cerebral Vessels
Chilled brains were rapidly dissected, and pial vessels and large cerebral vessels were removed with forceps and placed on ice-cold phosphate-buffered saline (PBS) as previously described. 18 Remaining brain tissue was homogenized in ice-cold PBS and centrifuged at 2,000 g at 4°C for 5 minutes. Supernatant containing parenchymal brain tissue was discarded. The pellet containing vessels was resuspended in PBS, centrifuged at 2,000 g at 4°C for another 5 minutes, and the second pellet resuspended in PBS, layered over a 15% dextran density gradient (molecular weight 35,000 to 40,000 kDa), and centrifuged at 3,500 g at 4°C for 30 minutes. Supernatant was discarded, pellet resuspended in PBS, layered again over dextran gradient, and centrifuged at 3,500 g at 4°C for 30 minutes. The final pellet was filtered with ice-cold PBS over a 70-μm nylon mesh. Collected cerebral microvessels and capillaries were combined with the large vessels harvested earlier.
Real-Time Quantitative PCR
Levels of EPHX2 mRNA were determined by modification of Corenblum et al 19 In brief, RNA was isolated using a commercial kit (RNAqueous-Micro; Ambion, Austin, TX, USA), contaminant genomic DNA was removed by DNase treatment, and RNA reverse transcribed using a commercial high capacity cDNA archive kit (Applied Biosystems, Carlsbad, CA, USA). Resulting cDNA was amplified using TaqMan Universal PCR amplification in a commercial sequence detection system (ABI Prism 7000; Applied Biosystems). Quantitative PCR was performed in a 96-well plate using 50 μL total volume, in triplicate. The PCR was concurrently run on controls without template to assess DNA contamination and primer–dimer formation. Remaining RNA not reverse transcribed was included to control for genomic DNA amplification. 18S was measured as an internal control using an 18S rRNA control kit (FAM-TAMRA; Eurogentec, Seraing, Belgium). The EPHX2 primers used for cerebral vessels were designed using the specialized software (Primer Express, ABI; Applied Biosystems). Monitored sequences were forward 5′-GCTGGGAATCCCTCAAGCA-3′ and reverse 5′-AGAGAGCCATGTTCCACACCAT-3′. The EPHX2 primers used for brain were purchased as a Taqman Gene Expression Assay (Catalog # 4351372; Invitrogen, Grand Island, NY, USA). All final EPHX2 mRNA levels were normalized to 18S.
Western Blotting
Mice were perfused with ice-cold heparinized saline to remove blood and brains were collected. Brains were homogenized in lysis buffer containing sucrose (250 mmol/L), potassium chloride (60 mmol/L), tris-hydrochloride (15 mmol/L), sodium chloride (15 mmol/L), ethylenediaminetetraacetate (5 mmol/L), ethylene glycol tetraacetic acid (1 mmol/L), phenylmethane-sulfonylfluoride (0.5 mmol/L), dithiothreitol (10 mmol/L), 1 Complete Mini-EDTA free Protease Inhibitor Cocktail tablet (Roche Diagnostics, Indianapolis, IN, USA), and 10 μL/mL each of phosphatase inhibitor solution 1 and phosphatase inhibitor solution 2 (Sigma-Aldrich, St Louis, MO, USA). Lysates were then centrifuged at 2,000 g for 10 minutes at 4°C, super-natant collected and centrifuged at 17,000 g for 20 minutes at 4°C. Protein samples (40 μg) were separated by gel electrophoresis and then transferred onto polyvinylidene difluoride membranes. Blots were blocked in 5% dry milk, and incubated at 4°C overnight with a primary rabbit polyclonal antibody against murine sEH (1:500; Cayman Chemical, Ann Arbor, MI, USA). The signal was visualized using a horseradish peroxidase-linked rabbit secondary antibody (1:1,000; GE Healthcare, Piscataway, NJ, USA) followed by detection using Supersignal chemiluminescent reagents (Thermo Fisher Scientific, Waltham, MA, USA) with a FluorChem FC2 (Protein Simple, Santa Clara, CA, USA). Blots were stripped using Restore Western Blot Stripping Buffer (Thermo Fisher Scientific) and reprobed for β-actin (1:2,000; Sigma-Aldrich) and followed by horseradish peroxidase mouse secondary antibody (1:1,000; GE Healthcare) and reimaged. Densitometry was quantified using the AlphaView software (Protein Simple), and the sEH protein was normalized relative to β-actin.
Epoxyeicosatrienoic acids Concentration Measurement
Mice were perfused with ice-cold heparinized saline to remove blood and brains were collected. Epoxyeicosatrienoic acids quantification in brain was determined by liquid chromatography-tandem mass spectrometry as previously described. 20
Statistical Analysis
Data are expressed as mean ± s.e.m. Groups were compared by the t-test for two groups (EPHX2 mRNA), one-way ANOVA with the post hoc Newman-Keuls test for three or more groups (sEH protein, EETs concentration, OMAG, and single region infarct size), or two-way ANOVA with the post hoc Newman–Keuls test for multiple measures with multiple groups (blood glucose, laser-Doppler perfusion, and multiregional infarct size). Differences were considered as significant at P<0.05.
RESULTS
Soluble Epoxide Hydrolase Inhibition Does Not Alter Nonfasted Blood Glucose Concentration in Control or Streptozotocin-Treated Mice
Table 1 shows characteristics of study mice before MCAO, which did not differ among groups in age or temperature. Streptozotocin-treated mice have decreased body weight (P<0.01) and increased fasting blood glucose levels (P<0.001) compared with Control mice. Baseline plasma glucose concentration of mice before induction of STZ was comparable in all groups (Figure 1). Glycemia increased 1 week after beginning STZ treatment and persisted throughout the subsequent 4 weeks. In a subset of mice, the sEH inhibitor t-AUCB (1 mg/kg, intraperitoneally daily) or vehicle (saline) was administered for the final 6 days. At 4 weeks, nonfasting plasma glucose concentrations were elevated in STZ-treated mice compared with Control (P<0.001); an effect that was unaltered by t-AUCB treatment. Body weight was not altered by t-AUCB treatment (Table 1). Insulin levels were also not altered by t-AUCB treatment and were at the lowest level of detection in all STZ-treated mice regardless of vehicle or t-AUCB treatment (data not shown).
Characteristics of mice in all experimental groups before MCAO
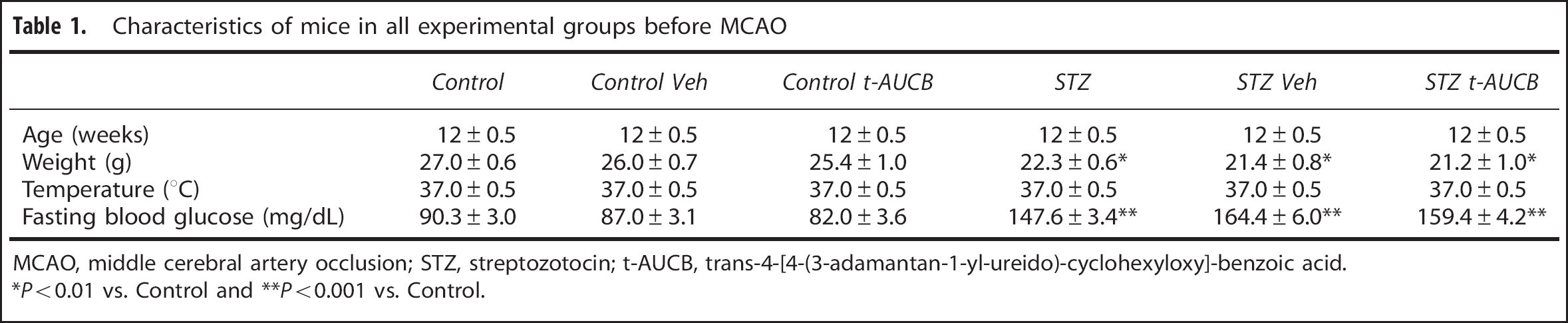
MCAO, middle cerebral artery occlusion; STZ, streptozotocin; t-AUCB, trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid.
P<0.01 vs. Control
P<0.001 vs. Control.
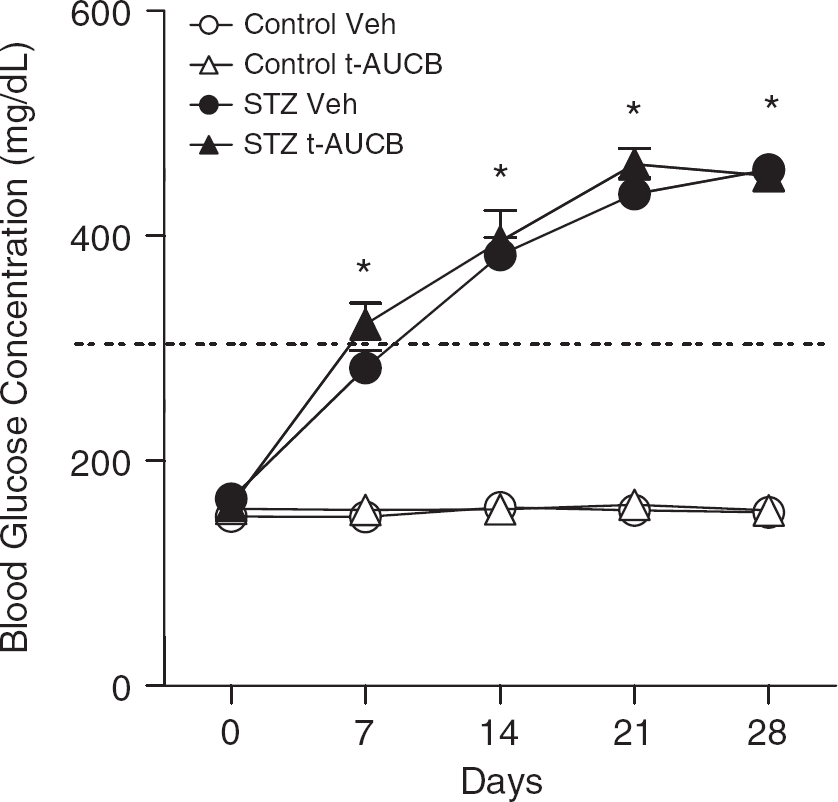
Soluble epoxide hydrolase (sEH) inhibition does not alter nonfasted blood glucose levels. Baseline, nonfasting blood glucose levels were within the normoglycemic range for both groups. Streptozotocin (STZ)-treated mice received STZ 50 mg/kg intraperitoneally daily for 5 days starting at day zero, and controls received vehicle only. Hyperglycemia (glucose >300 mg/dL, as indicated by dotted line) in STZ-treated mice began to occur at 7 days after final injection. sEH antagonist trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid (t-AUCB) (1 mg/kg; intraperitoneally) or vehicle (Veh) was administered daily over the final 6 days. *P<0.05 vs. time-matched Control Veh, n = 5 per group. White circles = Control Veh, white triangles = Control t-AUCB, black circles = STZ Veh, and black triangles = STZ t-AUCB.
Streptozotocin-Induced Type 1 Diabetes Increases Soluble Epoxide Hydrolase Expression in Cerebral Vessels, But Not in Brain
Figure 2A shows that, compared with controls at 4 weeks, STZ-treated mice exhibited a 1.8-fold upregulation of EPHX2 (gene encoding for sEH) mRNA in cerebral vessels (P<0.05 vs. Control). Figure 2B shows that EPHX2 mRNA expression in brain did not differ between Control and STZ-treated mice. Figure 3 shows that sEH protein expression in brain also did not differ between Control and STZ-treated mice, and that t-AUCB treatment had no effect on sEH protein expression in brain.
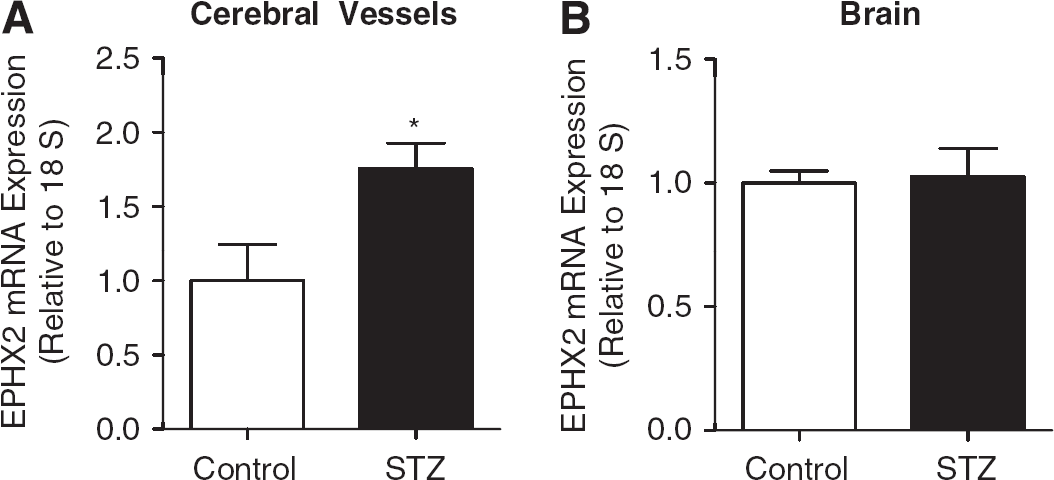
Streptozotocin (STZ) increases EPHX2 mRNA expression in cerebral vessels, but not in brain. STZ-treated mice received STZ 50 mg/kg intraperitoneally daily for 5 days starting at day zero, and controls received vehicle only. At 4 weeks, (
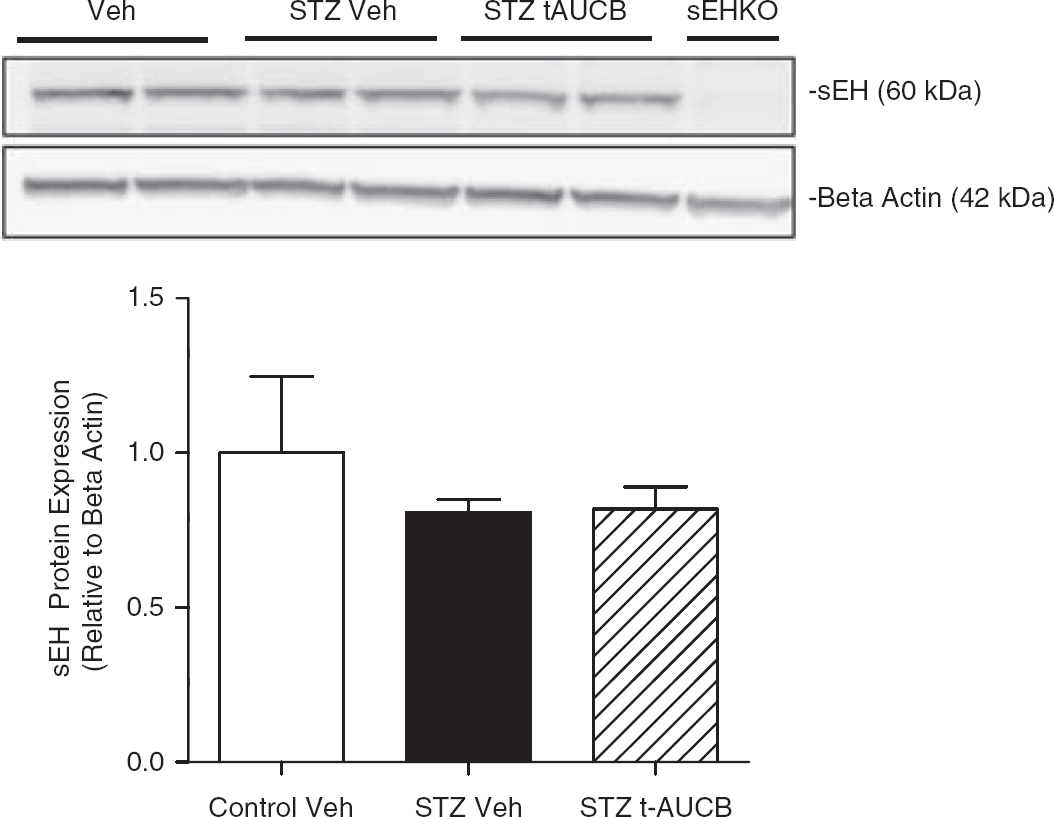
Neither streptozotocin (STZ) treatment nor soluble epoxide hydrolase (sHE) inhibition alters sEH protein expression in brain. STZ-treated mice received STZ 50 mg/kg intraperitoneally daily for 5 days. The sEH antagonist trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid (t-AUCB) (1 mg/kg intraperito-neally daily) or vehicle (Veh) was administered over the last 6 days before killing. At 4 weeks, brains were collected for measurement of sEH protein expression via western blotting. sEH protein expression was normalized to β-actin expression. Brain lysate from an sEH knockout mouse (sEHKO) is shown in the representative blot as a negative control. n = 4 to 5 per group. White bar = Control Veh, black bar = STZ Veh, and diagonally lined bar = STZ t-AUCB.
Soluble Epoxide Hydrolase Inhibition Prevents Streptozotocin-Induced Decreases in Epoxyeicosatrienoic acids Concentration in Brain
Although we did not detect changes in sEH expression in brain, we examined EETs concentration in whole brain as an indirect measure of sEH activity. Concentrations of 8,9-EETs, 11,12-EETs, and 14,15-EETs were measured in brain and added together for total EETs concentration. We were unable to measure 5,6-EETs in brain because of their well-known instability, with a half-life of only 8 minutes; 21 however, we were able to detect their metabolite 5,6-dihydroxyeicosatrienoic acids in all samples tested (data not shown). Figure 4 shows that, compared with controls at 4 weeks, STZ-treated mice had decreased (by 32%) concentration of total EETs (P<0.001 vs. Control vehicle) in brain. Furthermore, sEH inhibition was able to prevent the STZ-induced decrease in brain total EETs concentration (P<0.01 vs. STZ vehicle). Concentrations of all 8,9-EETs, 11,12-EETs, and 14,15-EETs were decreased in brains of STZ vehicle-treated mice (254±26.6, 132±13.0, and 169±10.5, respectively; mean ± s.e.m.) compared with Control vehicle-treated mice (382±16.3, 192±5.3, and 239±7.5, respectively; mean ± s.e.m., P<0.01). This decrease was prevented by t-AUCB treatment in STZ-treated mice for all three EET regioisomers (339±13.3, 183±7.2, and 223±11.6, respectively; mean ± s.e.m., P<0.01 vs. STZ vehicle).
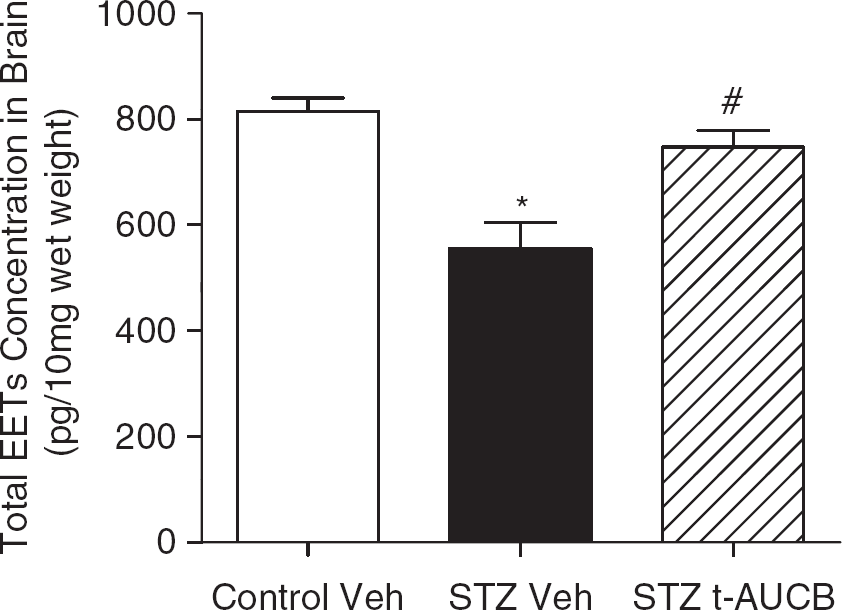
Soluble epoxide hydrolase (sEH) inhibition prevents streptozotocin (STZ)-induced decreases in epoxyeicosatrienoic acids (EETs) concentration in brain. STZ-treated mice received 50 mg/kg intraperitoneally daily for 5 days. The sEH antagonist trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid (t-AUCB) (1 mg/kg intraperitoneally daily) or vehicle (Veh) was administered over the last 6 days before killing. At 4 weeks, brains were collected for measurement of EETs concentration via liquid chromatographytandem mass spectrometry (LC-MS/MS). EETs concentration is shown as pg/10 mg of brain wet weight. *P<0.001 vs. Control Veh, #P<0.01 vs. STZ Veh, n = 4 to 5 per group. White bar = Control Veh, black bar = STZ Veh, and diagonally lined bar = STZ t-AUCB.
Neither StreptozotocinTreatment nor Soluble Epoxide Hydrolase Inhibition Alter Cerebral Perfusion at Baseline, During Ischemia, or During Early Reperfusion
Figure 5A shows that cortical perfusion, measured by optical microangiography, was not altered at baseline/before ischemia by STZ or t-AUCB treatment. Furthermore, Figure 5B shows that there was also no difference in relative changes in perfusion, as measured by laser-Doppler perfusion, of the MCA territory between STZ-treated and Control mice before, during, or shortly after MCAO.
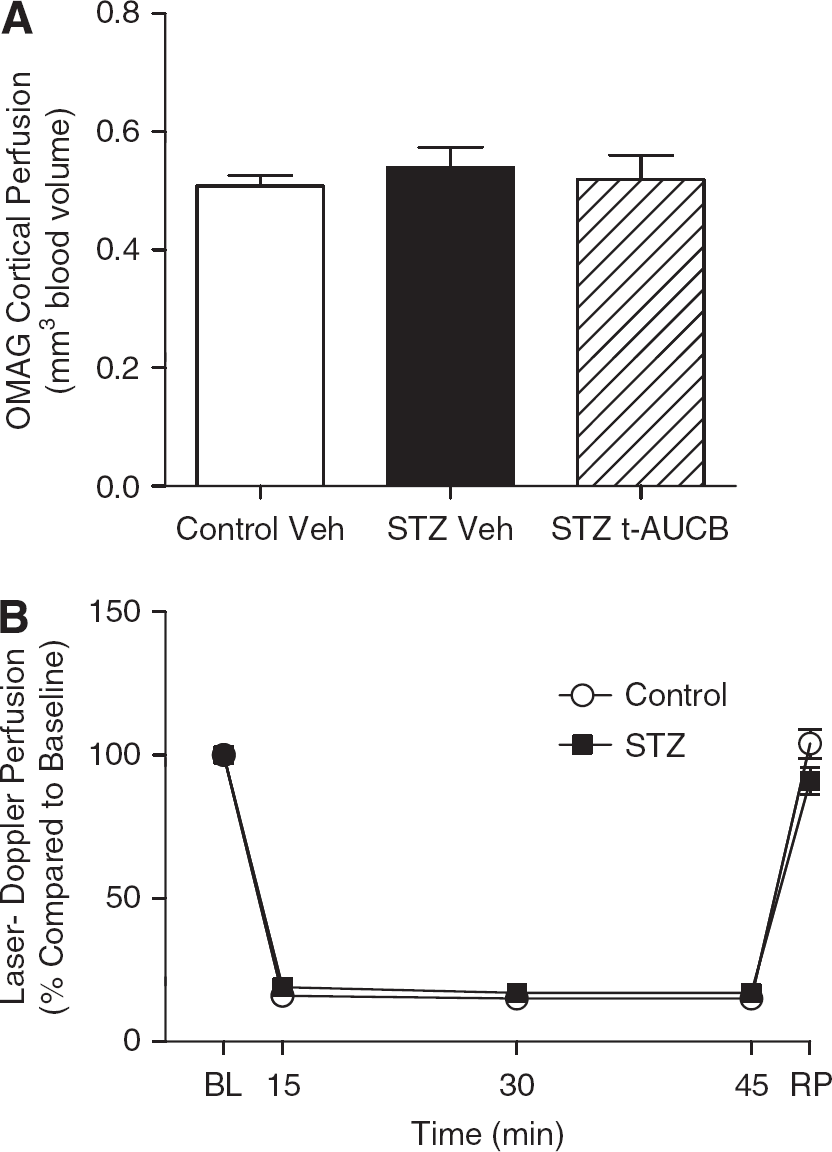
Neither streptozotocin (STZ) treatment nor soluble epoxide hydrolase (sEH) inhibition alters cerebral perfusion at baseline, during ischemia, or during early reperfusion. STZ-treated mice received STZ 50/kg mg intraperitoneally daily for 5 days. The sEH antagonist trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid (t-AUCB) (1 mg/kg intraperitoneally daily) or vehicle (Veh) was administered over the last 6 days before killing. (
Soluble Epoxide Hydrolase Inhibition Prevented Streptozotocin-Induced Increases in Infarct Size After Middle Cerebral Artery Occlusion
Despite the similar loss of perfusion during ischemia, Figure 6A shows that STZ-treated mice sustained a larger brain infarct size than Controls. Infarct size was increased by 54% in cerebral cortex (P<0.05), 47% in striatum (P<0.01), and 77% in whole hemisphere (P< 0.05) in the STZ-treated mice compared with Control mice. To test whether decreased EETs concentration in brains of STZ-treated mice might exaggerate vulnerability to stroke, we blocked sEH activity using the specific antagonist t-AUCB during the week before MCAO. Treatment with t-AUCB did not alter glycemic status of either STZ-treated or Control mice (Figure 1).
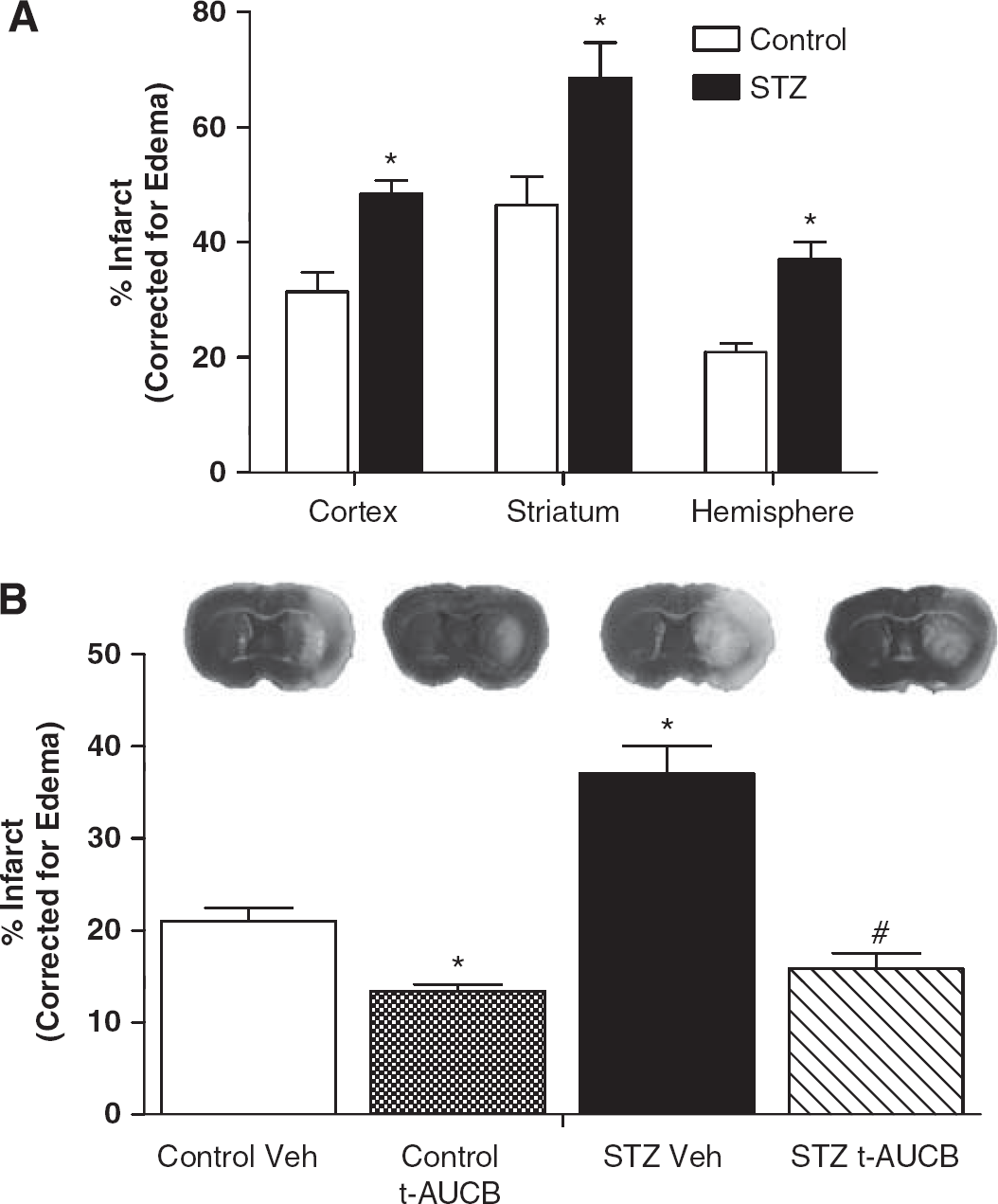
Streptozotocin (STZ)-treated mice had increased infarct size after middle cerebral artery occlusion (MCAO) that was prevented by soluble epoxide hydrolase (sEH) inhibition. STZ-treated mice received STZ 50 mg/kg intraperitoneally daily for 5 days. Animals were subjected to 45-minute MCAO at 4 weeks. Brains were harvested at 24 hours after MCAO, and infarct size was measured by 2,3,5-triphenyltetrazolium chloride (TTC) staining and corrected for edema as described in Materials and methods. (
Despite the lack of effect on prevailing glycemia, t-AUCB reduced MCAO-induced brain infarct size in both the STZ-treated (by 53%, P< 0.001 vs. STZ vehicle) and Control (by 45%, P<0.05 vs. Control vehicle) mice (Figure 6B). Further, t-AUCB abolished the difference in infarct size between STZ-treated and Control groups.
DISCUSSION
Our study shows that STZ-induced hyperglycemia increases vulnerability of multiple brain regions to transient focal ischemia, that STZ increases expression of EPHX2 (gene coding for sEH) in cerebral vessels and decreases the concentration of neuroprotective EETs in brain, that sEH blockade with t-AUCB improves infarct size and restores brain EETs concentrations without altering glycemic status or cerebral perfusion. Our findings support a novel role for sEH in mediating the deleterious effect of hyperglycemia on stroke, and indicate that sEH inhibition may serve as a potential prophylactic to counter stroke exacerbation in diabetes mellitus.
Clinical evidence indicates that diabetes is an independent risk factor that doubles the risk of stroke.22,23 Yet two recent large population studies show that aggressive attempt(s) to reduce hyperglycemia in outpatient diabetic patients fails to decrease risk or improve outcome of stroke.11,12 Once a stroke has begun, the coexistence of hyperglycemia at presentation, regardless of etiology, is associated with worse outcome.3–6 But initiation of tight glucose control for improving an evolving stroke has largely been futile, and possibly counterproductive, in terms of neurologic outcome, although the single large prospective clinical study to date had limitations. 13 We hypothesized that hyperglycemia may trigger a downstream exacerbating agent for stroke which, once activated, would act independently of the prevailing glycemic status.
Several lines of evidence point to sEH as a putative mediator to worsen stroke outcome in the setting of hyperglycemia. First, sEH is widely expressed in both human and mouse brain in numerous cell types, including neurons, astrocytes, oligodendrocytes, smooth muscle cells, and endothelial cells.15,24,25 Second, pharmacologic inhibition of sEH protects against injury in kidney, 26 heart,27,28 pancreas, 29 and brain.14,18,30,31 Third, deletion of EPHX2, the gene coding for sEH, protects against ischemic injury in kidney, 26 heart,27,28 pancreas, 29 and brain. 32 The effect of hyperglycemia on brain sEH expression had been unknown, but the current study shows that hyperglycemia at least of insulinopenic etiology upregulates expression of EPHX2, the gene that codes for sEH.
The mechanism by which sEH mediates ischemic brain injury is by metabolizing neuroprotective EETs, hydrolyzing them to form inert dihydroxyeicosatrienoic acids. 24 Epoxyeicosatrienoic acids are produced from arachidonic acid by cytochrome P450 epoxygenase and have been extensively reviewed.14,15 They protect against ischemic injury by multiple potential mechanisms, including inhibition of platelet adhesion, 33 antiinflammation, 34 reduction in oxygen-free radicals, 35 as well as antiapoptosis and activation of protective signal transduction.15,36 We have previously shown that sEH inhibition protects the brain from ischemic injury by a mechanism linked to preservation of EETs in nondiabetic mice. 18 In the current study, we found that brain EET concentrations were decreased by STZ-induced hyperglycemia and that this decrease in brain EETs was accompanied by an increase in infarct size after MCAO. Inhibition of sEH in hyperglycemic mice restored brain EET concentrations and reduced infarct size, suggesting that the sEH/EETs pathway has a detrimental role in the etiology of hyperglycemic stroke. In a previous study, 18 we have shown that another sEH inhibitor, AUDA-BE, was effective in reducing infarct when administered after stroked. In the current study, the sEH inhibitor was administered before stroke, which has limited value as a stroke therapy. However, our study was not designed to target sEH as a therapy for stroke, but rather, the study was designed to examine the role of sEH in mediating the effect of diabetes on stroke. Therefore, we inhibited sEH before stroke to determine its role in the exacerbation of stroke injury in diabetic brain; that is, if sEH inhibition is sufficient to reduce diabetic brain's increased vulnerability to ischemia.
Our finding suggests that STZ-induced hyperglycemia increases EPHX2 expression in cerebral vessels, but not in whole brain, and that cerebrovascular sEH may have a larger role in hyperglycemic stroke than other brain-derived sources of sEH. As previously mentioned, sEH is expressed in a variety of cells in the brain included cerebrovascular endothelium, vascular smooth muscle cells, neurons, oligodendrocytes, and astrocytes. 15 Our data in cerebral blood vessels are consistent with those of Thomas et al 37 in mouse liver. However, STZ has been reported to decrease sEH in mouse liver, 38 and to either decrease 38 or have no effect 39 on sEH in mouse kidney. Thus, the influence of diabetes and hyperglycemia on sEH expression appears to be tissue and even cell type specific.
An additional consideration is the effect of sEH blockade on glycemic status. We found that inhibition of sEH with t-AUCB had no effect on glycemic status or insulin levels in either STZ-treated or Control mice over a 6-day treatment course. Our finding in STZ-treated mice differs from that of Luo et al 29 who showed that sEH coding gene deletion or pharmacologic inhibition attenuated hyperglycemia in STZ-treated mice. Luo et al 29 found that sEH inhibition improved STZ hyperglycemia by limiting STZ-mediated damage to pancreatic β-cells, increasing insulin secretion. The lack of a glycemic effect in response to sEH blockade in our STZ-treated mice could be explained by more severe β-cell injury due to (1) more days of STZ injections (5 days vs. 3 days), (2) a different timing sequence (t-AUCB begun 3 weeks after STZ vs. immediately after STZ treatment), and (3) longer STZ incubation time (4 weeks vs. 3 days) compared with Luo et al. 29
The exacerbating effect of STZ on MCAO stroke injury occurred without apparent effect on immediate postreperfusion MCA blood flow. It is also possible that the protective effect of sEH blockade could be because of improved cerebral blood flow at later time points of reperfusion after MCAO, which could be a limitation of the present study as we only examined CBF for 5 minutes postreperfusion. However, sEH blockers, as a group, have weak vasodilating (antihypertensive) properties. 26 Furthermore, neither STZ treatment nor sEH inhibition altered cortical perfusion at baseline/before ischemia, making augmented cerebral tissue perfusion an unlikely scenario to eliminate the infarct size difference between STZ-treated mice and normoglycemic controls. As previously mentioned, EETs protect against ischemic injury by multiple potential mechanisms, including inhibition of platelet adhesion, 33 antiinflammation, 34 reduction in oxygen-free radicals, 35 reduction in apoptosis, and activation of protective signal transduction.15,36 In line with this, we have previously shown that brain EETs protect neurons against ischemia by mechanism(s) separate from cerebrovascular dilation in the absence of hyperglycemia. 40 Our current study supports a neuroprotective role of brain EETs that is likely independent of cerebral perfusion, this time in the context of hyperglycemia, as STZ decreased brain EET concentrations, while increasing infarct size without altering cerebral perfusion; effects that were reversed by sEH inhibition.
In summary, we found that (1) STZ-induced hyperglycemia exacerbates cerebral infarct size after MCAO in mice, (2) STZ hyperglycemia upregulates EPHX2 mRNA expression in cerebral vessels and decreases EETs concentration in brain, (3) sEH inhibition with t-AUCB increases brain EETs concentration, reduces brain infarct size after MCAO, and eliminates the difference in brain infarct size and brain EETs concentration between STZ-treated and Control mice, and (4) the neuroprotective effects of sEH blockade in STZ occurred without any effect on glycemic status or cerebral perfusion before, during, or immediately after MCAO. These findings indicate that sEH may have a role in the deleterious effect of hyperglycemia. More work is needed to determine whether the effect of sEH and its blockade extends to the setting of type 2 diabetes mellitus, a much more common hyperglycemic disease characterized by insulin resistance rather than absolute insulinopenia. Details of the mechanism(s) by which sEH influences hyperglycemic brain injury in stroke also remain(s) to be elucidated.
Footnotes
The authors declare no conflict of interest