
Abstract
Calpain, a neutral protease activated by calcium, may promote microtubular proteolysis in ischemic brain. We tested this hypothesis in an animal model of focal cerebral ischemia without reperfusion. The earliest sign of tissue injury was observed after no more than 15 min of ischemia, with coiling of apical dendrites immunolabeled to show microtubule-associated protein 2 (MAP2). After 6 h of ischemia, MAP2 immunoreactivity was markedly diminished in the infarct zone. Quantitative Western analysis demonstrated that MAP2 was almost unmeasurable after 24 h of ischemia. An increase in calpain activity, shown by an antibody recognizing calpain-cleaved spectrin fragments, paralleled the loss of MAP2 immunostaining. Double-labeled immunofluorescent studies showed that intraneuronal calpain activity preceded evidence of MAP2 proteolysis. Perikaryal immunolabeling of τ protein became increasingly prominent between 1 and 6 h in neurons located within the transition zone between ischemic and unaffected tissue. Western blot experiments confirmed that dephosphorylation of τ protein occurred during 24 h of ischemia, but was not associated with significant loss of τ antigen. We conclude that focal cerebral ischemia is associated with early microtubular proteolysis caused by calpain.
Cytoskeletal degradation, represented by the apparent loss of micro tubule-associated, pro teins (MAPs) shown by immunochemical methods, is a sensitive indicator of neuronal injury in ischemic brain. Now documented in several models of global and focal ischemia (Inuzuka et al, 1990; Yanagihara et al, 1990; Yoshimi et al., 1991), it is known to follow different patterns according to choice of animal, type of vascular occlusion, and region of brain selected for study. A growing body of evidence suggests that cytoskeletal changes observed in ischemic neurons may result from microtubular proteolysis caused by calpain, a nonlysosomal, calcium-activated neutral protease (see review in Saido et al., 1994). Calpain is distributed ubiquitously in all mammalian cells as pro-calpain, an inactive precursor composed of a 75- to 80-kDa catalytic subunit and a 30-kDa regulatory fraction (Melloni and Pontremoli, 1989). The two isozymic forms of calpain in cerebral tissue differ in the composition of the catalytic subunit and in the cell of origin. Calpain I (μ-calpain) is localized to neurons and may be activated in the presence of micromolar concentrations of calcium. Calpain II (m-calpain) is present in glial cells and in axonal tracts, but must be exposed to millimolar concentrations of calcium to be activated. The intracellular concentration of calcium in ischemic neurons will reach the micromolar range required for activation of calpain I, due to the release of ion from stores within cellular organelles or enhancement of calcium conductance from the extracellular space (Siesjö, 1988). Calcium-mediated activation of calpain begins with the translocation of pro-calpain to a target substance in the cell membrane or cytoskeleton, followed by autolytic transformation of the precursor to the active calpain moiety. Once activated, calpain may cause proteolysis of neurofilaments, MAPs, and other cytoskeletal proteins before returning as a soluble, inactive form to the cytoplasm (Suzuki and Ohno, 1990).
Roberts-lewis et al. (1994) have developed a family of antibodies that recognize calpain-cleaved spectrin in gerbil brain. Immunostaining with one of these antibodies, Ab-37, became intensified in CA1 pyramidal neurons after 5 min of bilateral common carotid artery (CCA) occlusion. Immunoreactivity persisted from 24 to 48 h after reversal of occlusion and correlated with the well-recognized vulnerability of CA1 neurons to cellular death associated with reperfusion following global ischemia (Kirino, 1982; Pulsinelli et al., 1982; Smith et al, 1984). Immunostaining was also observed in the neocortex, striatum, and thalamus as early as 30 min after reversal of carotid occlusion, but had begun to fade after 24 h. These results confirm that persistence of Ab-37 immunoreactivity is associated with calpain-induced cytoskeletal proteolysis and neuronal degeneration in global cerebral ischemia. We hypothesized that a similar relationship between calpain activity and neuronal death may apply during maturation of a cerebral infarct. We tested this hypothesis by using Ab-37 as a functional assay of calpain activity in a well-established model of focal brain infarction in the rat. We performed immunocytochemical studies, double-labeled immunofluorescent imaging, and Western blot analysis to demonstrate the effect of calpain on MAPs in ischemic brain tissue.
Materials and Methods
Focal cerebral ischemia
All experimental methods were approved by the Institutional Animal Care and Use Committee of the University of Kentucky. Male spontaneously hypertensive rats (SHRs; Harlan Sprague-Dawley; Indianapolis, IN, U.S.A.) of 250–350 g body weight were used for each experiment described in this report. CCA-middle cerebral artery (MCA) occlusion was performed by a modification of the technique described by Brint et al. (1988). All rats were fasted overnight prior to surgical preparation. Each animal was anesthetized via intraperitoneal injection with 0.5 mg/g chloral hydrate for isolation of the MCA and CCA. One femoral artery was cannulated for blood sampling and measurement of MABP. Arterial blood was sampled 10 min before CCA–MCA occlusion in each animal prepared for immunocytochemical studies (group A; see below) and used to determine pH, Pao2, Paco2, hematocrit (Hct), and glucose. Rectal and temporalis muscle temperatures were maintained at 36–37°C by external warming. The left CCA was isolated through an anterior incision in the neck. For identification of the ipsilateral MCA, a second incision was made between the lateral canthus of the left eye and the corresponding external auditory canal to bare the underlying skull. The MCA was exposed through a 2-mm burrhole drilled 2–3 mm rostral to the fusion of the zygomatic arch and the squamosal bone under direct visualization with a Zeiss operating microscope. The dura was opened with a sharp needle, and an alloy wire (0.1 mm diameter) was inserted beneath the MCA just superior to the inferior cortical vein. The MCA was elevated from the cortical surface and cauterized by applying electrical current to the wire. The CCA was tied off with 4–0 silk and both incisions were closed. The animal was then returned to its cage and given free access to water and rat chow.
Thirty-two rats (group A) were killed and prepared for immunocytochemical studies after permanent CCA–MCA occlusion lasting for 15 min or 1, 6, 24, or 72 h (n = 5 or 6 at each time point). Group A also included five sham-ischemic animals subjected to nonocclusive manipulation of the left CCA and craniotomy performed without opening the dura before being killed for immunocytochemical studies 24 h later. An additional 16 animals (group B) underwent permanent CCA–MCA occlusion for 15 min or 1, 6, or 24 h (n = 4 at each time point), before being killed for sampling of normal and ischemic brain tissue used in Western blot analysis.
Primary antibodies
The primary antibodies used for immunocytochemical studies and Western blot analysis included anti-MAP2 (clone AP20, Boehringer–Mannheim Corporation, Indianapolis, IN, U.S.A.), Ab-37 (gift from Dr. Robert Siman of Cephalon, Incorporated, West Chester, PA, U.S.A.), and anti-T protein (τ-1, Boehringer–Mannheim Corporation). The AP20 monoclonal antibody recognizes all isoforms of dendrite-specific MAP2. Ab-37 is a polyclonal antibody that identifies a proteolytic fragment of spectrin produced by activated calpain (Roberts-Lewis et al., 1994). The anti-τ monoclonal antibody recognizes a phosphorylation-sensitive, nonphosphorylated epitope usually confined to axons (Binder et al., 1985). However, enzymatic dephosphorylation of tissue sections taken from adult rat brain will enhance τ-1 immunoreactivity in somatodendritic structures (Papasozomenos and Binder, 1987). Our Western blot studies were designed to include a second anti-τ monoclonal antibody, 8C11 (Athena Neurosciences, South San Francisco, CA, U.S.A.), that identifies a phosphorylation-independent epitope. Whereas τ-1 will recognize only nonphosphorylated protein, 8C11 will immunolabel all isoforms of τ. Optimal dilutions of all primary antibodies were verified in preliminary experiments and in recent publications from our laboratory (Geddes et al., 1994a,b; Schwab et al., 1994). Production, characterization, and specificity have been described for each antibody [AP20 (Binder et al., 1986); Ab-37 (Roberts-Lewis et al., 1994; Geddes et al., 1995); τ-1 (Binder et al., 1985; Papasozomenos and Binder, 1987; Szendrei et al., 1993); 8C11 (Vigo-Pelfrey et al., 1995)]. In the present study, controls for the specificity of the immunostaining included testing of primary antibodies in Western blots containing purified target antigen and omission of the same antibodies from immunocytochemical preparations (Schwab et al., 1994).
Immunocytochemistry and double-labeled immunofluorescence
After the designated period of CCA–MCA occlusion, each rat in group A was reanesthetized with an intraperitoneal injection of 0.5 mg/g chloral hydrate and perfused transcardially with 50 ml of heparinized normal saline, followed by 200 ml cold 4% paraformaldehyde in phosphate buffered saline (pH 7.4). The brain was removed and stored in the same fixative for 24 h, before being cryoprotected in 30% sucrose and frozen in powdered dry ice. The frozen brain was stored at −70°C before being cut into 30-μm coronal sections with a Leitz freezing sledge microtome. Three changes of Tris-buffered saline (TBS; 50 mM, pH 7.5) were used to rinse free-floating tissue sections. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide followed by transfer of the sections into TBS containing 0.1% Triton X-100. All tissue sections designated for immunolabeling of MAP2 or τ protein were then incubated with 1.5% normal horse serum, while those chosen for Ab-37 immunostaining were exposed to 1.5% normal goat serum. Sections prepared for immunolabeling of MAP2 or τ protein were incubated overnight in a solution of primary antibody (AP20 1:10,000; τ-1 1:20,000) maintained at room temperature. Other sections used to demonstrate calpain-mediated spectrin proteolysis were incubated with Ab-37 (1:10,000) under the same conditions. Biotinylated secondary antibodies were used for identification of AP20 and τ-1 (anti-mouse preadsorbed against rat serum, Vector Laboratories, Burlingame, CA, U.S.A.) or Ab-37 (anti-rabbit, Zymed Laboratories, Inc., South San Francisco, CA, U.S.A.). With colorimetric detection by the avidin-biotin technique (Elite ABC kit, Vector Laboratories). Diaminobenzidine was chosen as the peroxidase substrate. Cresyl violet was used to stain corresponding tissue sections.
Differences in the intensity of immunostaining between normal and ischemic tissue were quantified with a computer-assisted image analysis system, consisting of a Macintosh IIci computer (Apple Computer, Cupertino, CA, U.S.A.), a Quick Capture frame grabber card (Data Translations, Marlboro, MA, U.S.A.), an Hitachi CCD camera mounted on an Olympus BH2 microscope, and NIH Image Analysis software, v. 1.55. The system was calibrated against a Kodak Optical Density Standard (Eastman Kodak, Rochester, NY, U.S.A.). Quantification of immunostaining was conducted on brain sections obtained from the group A rats and exposed to the AP20, Ab-37, or τ-1 antibodies. The region of altered immunoreactivity within ischemic tissue was outlined in each coronal section and the optical density was determined in comparison to the equivalent area in the opposite hemisphere. Two tissue sections were examined for each animal and averaged to yield a final result.
After completion of the immunocytochemical studies, selected specimens were prepared for double-labeled immunofluorescence to determine if there was a temporal or spatial correlation between the cytoskeletal changes and the activation of calpain. Tissue sections were blocked in 1.5% horse and 1.5% goat serum before being incubated with both the mouse monoclonal AP20 (1:2,000) and the rabbit polyclonal Ab-37 (1:7,500) overnight at room temperature. The sections were washed three times in TBS and incubated for 1 h with a fluorescein isothiocyanate (FITC)-conjugated horse anti-mouse secondary antibody (Vector). To maximize the specificity of the Ab-37 antibody, the same sections were incubated for 1 h each with a biotinylated goat anti-rabbit secondary antibody and Texas Red avidin D (both Vector products). The sections were mounted on glass slides, cover-slipped with VectaShield to retard quenching of the fluorescent signal, and viewed with the Olympus BH2 microscope equipped for epifluorescence and filtered to differentiate the FITC and Texas Red flurophores.
Western blot analysis
Each animal in group B was reanesthetized with an intraperitoneal injection of 0.5 mg/g chloral hydrate, as described in the preceding paragraphs. It was then perfused transcardially with 30 ml of 15% India ink (v/v) diluted in normal saline. The brain was removed from the skull and chilled with a fluorinated hydrocarbon spray (Cryokwik, International Equipment Corporation, Needham Heights, MA, U.S.A.), before being cut into 2-mm coronal sections. Examination of the brain sections revealed that normally perfused tissue had been stained by the ink, whereas unperfused cortex remained pale. The core of unperfused tissue in the left hemisphere was resected with a scalpel and a comparable volume was removed from the same location in the right hemisphere. The samples were packaged individually and snap-frozen in liquid nitrogen. Each hemispheric sample was homogenized (20% w/v) in Tris-saline containing leupeptin (10 μ/M), EDTA-Na2+ (1 μM), pepstatin A (1 μM), and AEBSF (0.25 μM). The homogenates were centrifuged at 100,000 g for 45 min at 4°C. Preliminary studies showed that MAP2 and τ protein immunoreactivity was confined almost exclusively to the supernatant fractions, which were chosen for all quantitative procedures. The protein levels in the supernatants were determined by the micro-BCA method (Pierce Chemical Company, Rockford, IL, U.S.A.) and equivalent aliquots (each containing 20 μg protein) were separated by SDS-polyacrylamide gel electrophoresis. Isolation of MAP2 was conducted with 6.5% gels, but τ protein fractions were separated on preformed 10–20% linear gradient gels (Millipore, Bedford, MA, U.S.A.). A standard supernatant prepared from normal rat cortex was applied to each gel to ensure reproducibility and allow comparison of assays. The proteins were transferred electrophoretically onto polyvinylidene fluoride membranes (Millipore) and immunoreacted with the antibodies of interest (AP20 1:1,000; τ-1 1:2,000; 8C11 1:10,000). Formal quantification of immunolabeled proteins was accomplished by using the peroxidase substrate SG (Vector Laboratories) for colorimetric detection by a red laser densitometer (LKB Ultroscan XL, Pharmacia, Piscataway, NJ, U.S.A.). The relative quantities of proteins in the ischemic cortex were expressed as percentages of those in matching tissue obtained from the unaffected right hemisphere.
Statistical analysis
The physiologic data obtained from group A animals were expressed as means ± SD. Mean whole blood glucose levels, Hct, and arterial blood pH, Paco2, and Pao2 were compared between subgroups identified by duration of CCA–MCA occlusion, using Kruskal-Wallis one-way analysis of variance (ANOVA) on ranks. The mean optical density measurements made on unaffected and ischemic tissue immunolabeled with the different primary antibodies were tested by the same method, followed by post hoc analysis with paired t tests. Densitometrie readings of Western blots performed to determine the concentration of MAP2 and τ protein in the ischemic and unaffected cortex were compared by the same approach undertaken for the optical densities calculated from tissue sections, except that post hoc analysis was conducted with the Student-Newman-Keuls method for nonparametric multiple comparisons.
RESULTS
Physiologic data obtained from the arterial blood samples in group A confirmed that all animals were uniformly stable prior to CCA–MCA occlusion. There was no significant difference in any of the physiologic variables compared between subgroups defined by the duration of ischemia. Aggregate data compiled from arterial blood gas samples taken from all of the subgroups showed Pao2 70 ± 9 mm Hg, Paco2 57 ± 6 mm Hg, and pH 7.26 ± 0.09. The hypercapnea and mild respiratory acidosis resulted from suppression of ventilatory rate in animals anesthetized with chloral hydrate. The whole blood glucose level taken from the same aggregate data set was 4.8 ± 0.6 mmol/L and Hct was 52 ± 2%. Mean arterial blood pressure was 91 ± 17 mm Hg across all subgroups.
Immunocytochemistry of MAP2
Figure 1 shows representative 30-μm coronal sections of brain taken from the right cerebral hemisphere serving as an unaffected control and the left hemisphere demonstrating ischemic tissue sampled at 15 min or 1, 6, 24, or 72 h after initiation of CCA–MCA occlusion. Each section was immunostained with AP20 identifying all isoforms of MAP2, τ-1 recognizing a nonphosphorylated epitope of τ protein, or Ab-37 labeling calpain-cleaved spectrin by-products. The pattern of immunoreactivity observed with the AP20 antibody was identical in unaffected hemispheres and in sham-ischemic controls. The antibody labeled MAP2 fibers in healthy dendrites (Fig. 2A), as described in previous reports (Bernhardt and Matus, 1984; DeCamilli et al., 1984). Immunoreactivity was relatively consistent across cortical regions but was much fainter in the striatum. At higher magnification, a rich plexus of fiber staining was evident in the molecular layer of cerebral cortex. Layers 3 and 4 of the cortex featured prominent immunolabeling of straight apical dendrites and fainter background staining of dendritic branches. In layer 5, neuronal perikarya were only faintly immunostained (Fig. 3A).
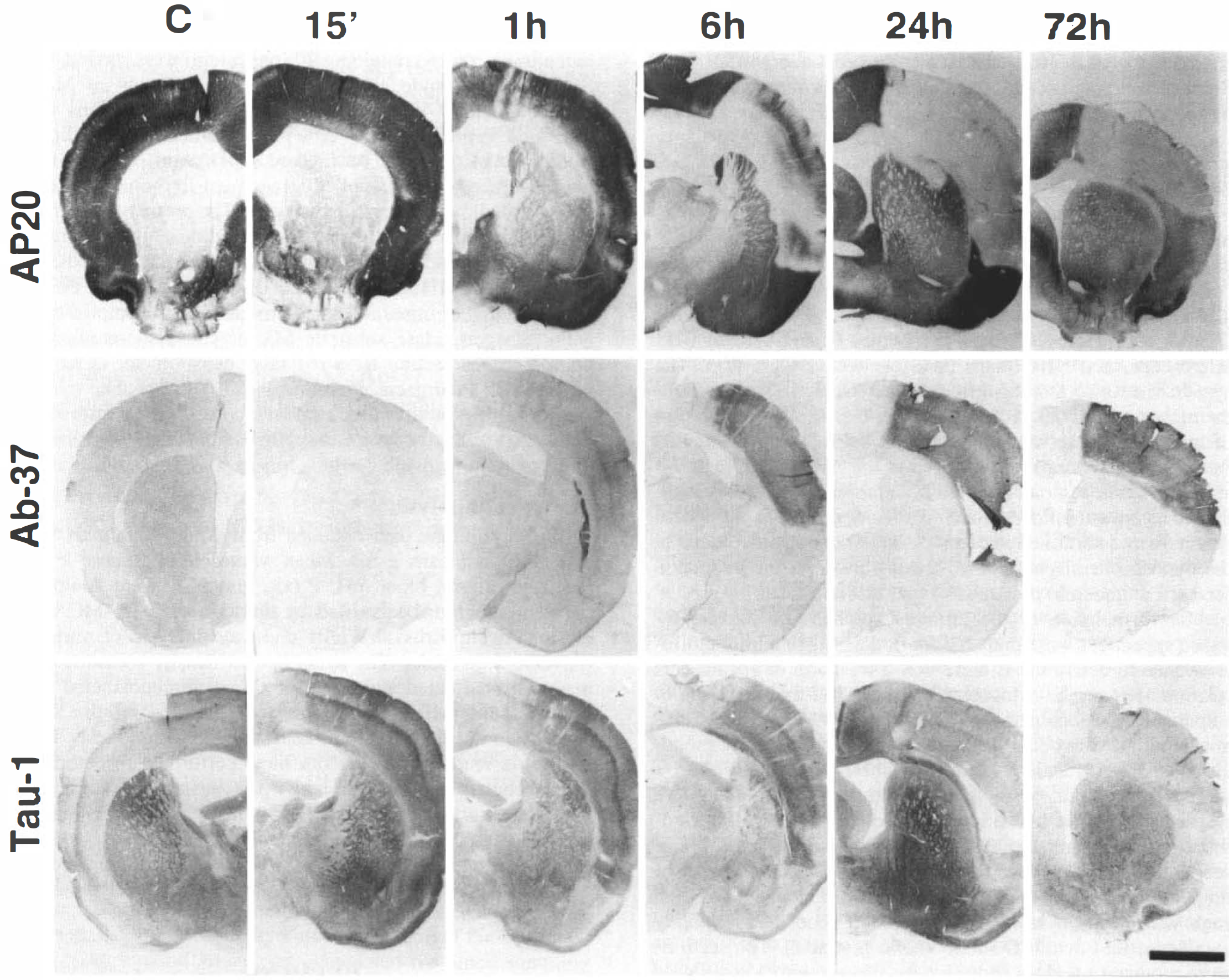
Representative 30 μm coronal sections of rat brain taken from the unaffected right hemisphere (C) or the ischemic left hemisphere between 15 min and 72 h after initiation of CCA–MCA occlusion. All photographs were taken at 1× magnification. The loss of MAP2 immunostaining by the AP20 antibody is gradual, beginning in isolated patches of ischemic tissue between 15 min and 1 h and progressing to complete absence of signal within the infarct zone after 24 h. Corresponding inversely to the loss of MAP2 signal is a gradual increase in the intensity of immunostaining with the Ab-37 antibody, representing accumulation of spectrin breakdown products resulting from calpain-mediated proteolysis. Tau-1 immunostaining of nonphosphorylated τ protein becomes intensified within the infarct zone as early as 15 min after CCA–MCA occlusion, but appears diminished in the adjacent transition area representing the ischemic penumbra. This apparent concentration of τ-1 immunostaining persists for 6 h, but decreases gradually between 24 and 72 h. The scale bar represents 2 mm.
Within 15 min of CCA–MCA occlusion, the dendritic processes of some neurons within the infarct had become coiled (Fig. 2B). Fine dendritic processes appeared less intense in ischemic tissue, such that the surviving apical dendrites stood out against an unstained background. After 1 h of CCA–MCA occlusion, MAP2 immunoreactivity appeared patchy within the infarct zone and could not be detected in the dorsolateral aspect of the ipsilateral striatum (see AP20 immunolabeling in Fig. 1). The loss of MAP2 immunolabeling was most prominent in layers 5 and 6 of ischemic cortex, as well as in the outer molecular layer. Toward the boundary of the infarct adjacent to the transition zone representing the ischemic penumbra, MAP2 immunostaining was diminished across all cortical layers. Coiled dendrites were abundant in the regions of decreased MAP2 immunostaining (Fig. 2C). Increased perikaryal MAP2 immunoreactivity was also evident (Fig. 3B), particularly in the transition area. After 6 h of ischemia, MAP2 immunolabeling was markedly diminished in the infarct zone (see AP20 immunolabeling in Fig. 1). While some apical dendritic fragments were visible, neuronal perikarya and the background neuropil were not immunostained. The infarct zone remained devoid of MAP2 immunostaining after 24 and 72 h of ischemia. There was an abrupt transition between the infarct and adjacent normal tissue, with an occasional neuron in the transition zone displaying enhanced perikaryal MAP2 immunoreactivity.
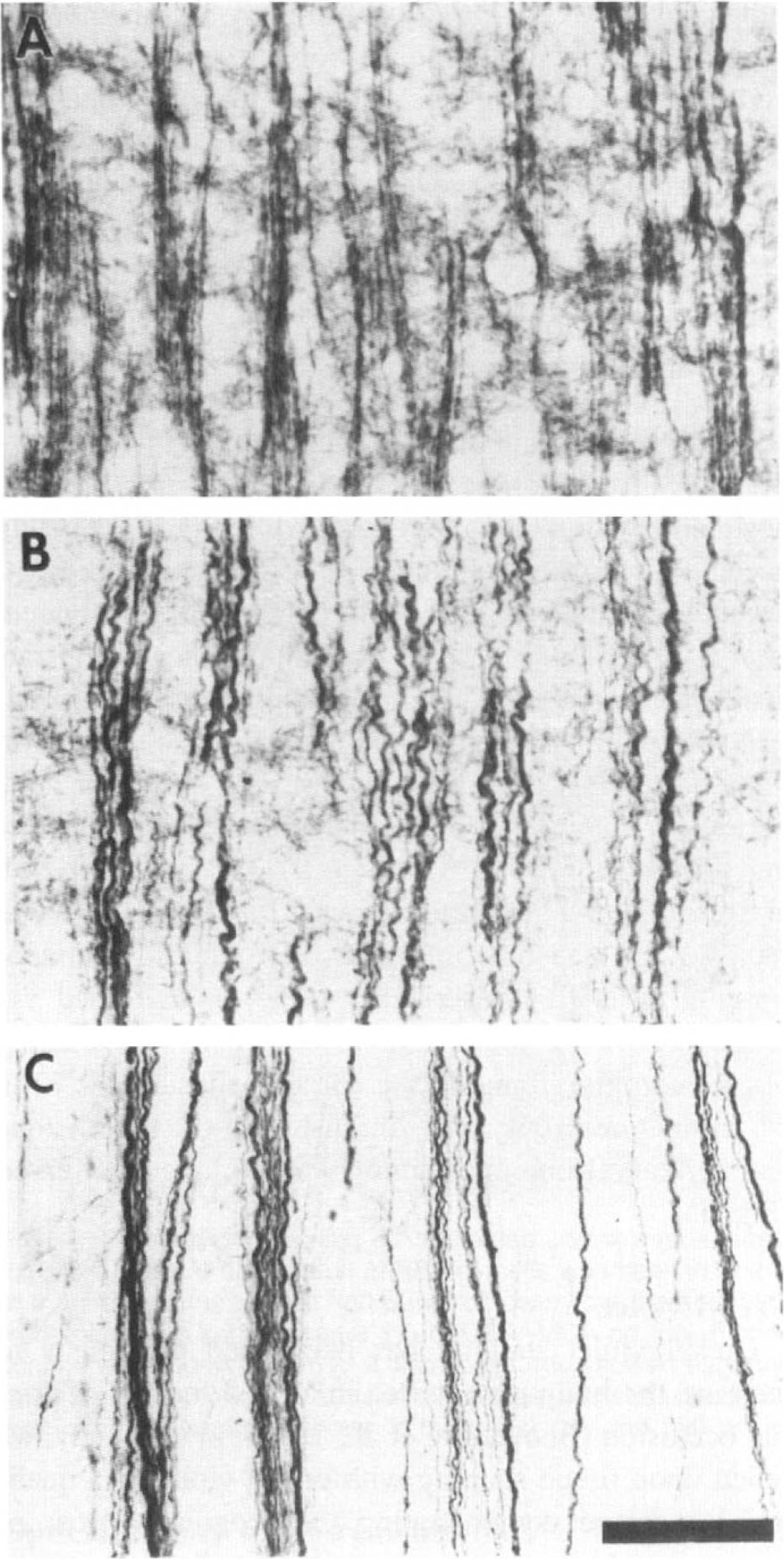
Representative photomicrographs (×40) of alterations in dendritic MAP2 immunoreactivity during focal ischemia. In the unaffected right hemisphere
Immunocytochemistry of calpain-cleaved spectrin fragments
As shown in Fig. 1, no specific Ab-37 immunoreactivity was observed in normal, nonischemic tissue. Scattered neuronal perikarya were visible in unaffected cortex of ischemic animals in both hemispheres of sham-ischemic rats, but the staining intensity was barely above background. After 15 min of CCA–MCA occlusion, isolated neurons within the infarct zone displayed enhanced perikaryal immunostaining (Fig. 3C). After 1 h of ischemia, neuronal perikarya were labeled more intensely, with immunoreactivity also evident in apical dendrites (Fig. 3D). The staining intensity of background neuropil was increased throughout the infarct zone. Figure 1 shows that intense Ab-37 immunolabeling was observed in a discrete region of the dorsolateral striatum ipsilateral to the cortical infarct in two of five rats, corresponding to the loss of MAP2 immunoreactivity seen in the same region. At 6 h following arterial occlusion, intense Ab-37 immunolabeling was evident throughout the infarct in all animals studied. Neuronal perikarya were no longer stained, but dendrites remained immunopositive. Of the five rats examined at this time point, one showed perikarya immunostaining of isolated cells in the striatum contralateral to the infarction, a second animal displayed perikaryal immunolabeling in cells of the ipsilateral reticular thalamic nucleus, and a third exhibited a large patch of immunostaining in the ipsilateral striatum. After 24 h of ischemia, Ab-37 immunostaining of the infarct remained intense and well demarcated in four of the five animals examined. The remaining animal showed no difference in Ab-37 immunoreactivity of ischemic tissue compared to its unaffected cortex or sham-ischemic controls. After 72 h of CCA–MCA occlusion, Ab-37 immunoreactivity persisted in the infarct zone and was also evident in the ipsilateral external capsule (3/6 rats), the lower ipsilateral cortex (1/6), and the striatum (2/6).
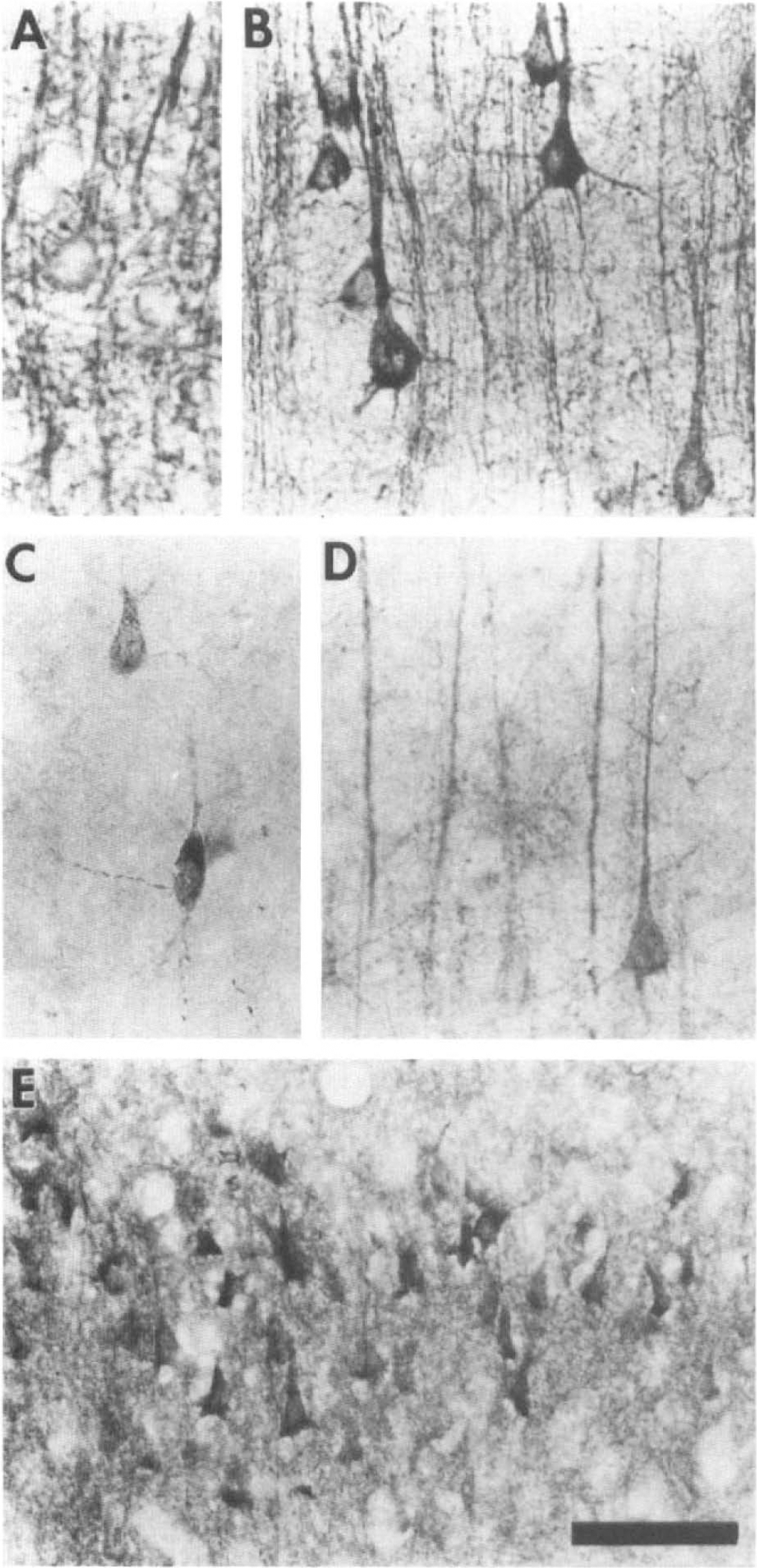
Representative photomicrographs (×40) demonstrating increased perikaryal immunostaining of cytoskeletal proteins during focal ischemia. In the unaffected hemisphere
Immunocytochemistry of τ protein
Immunostaining with τ-1 in normal cortex was confined primarily to axons, as reported originally by Binder et al. (1985). In the cortex and striatum of sham-ischemic controls and in the unaffected hemisphere of animals subjected to CCA–MCA occlusion, τ-1 immunoreactivity was present in the neuropil whereas neuronal perikarya remained unlabeled. There were regional differences in τ-1 immunostaining of normal cortex. The frontoparietal region displayed prominent τ-1 immunostaining in cortical layers 3 and 4. However, this pattern of immunoreactivity was not seen in primary olfactory cortex, which had a clear zone in layer 2 and prominent labeling of the outer molecular layer. As is demonstrated in Fig. 1, alterations in the normal pattern of τ-1 immunoreactivity were noticeable within 15 min of CCA–MCA occlusion. There was enhancement of τ-1 immunostaining within the infarct zone, while immunolabeling became diminished within the transition area representing the ischemic penumbra. This pattern was also evident after 1 and 6 h of arterial occlusion, when some rats displayed increased τ-1 immunostaining in the dorsolateral striatum ipsilateral to the infarct (Fig. 1). Perikaryal immunostaining of neurons within the transition zone between ischemic, and unaffected tissue became increasingly prominent between 1 and 6 h (Fig. 3E). After 24 h of ischemia, τ-1 immunoreactivity in the neuropil was reduced and had been replaced by enhanced immunostaining in neuronal perikarya (Fig. 1). Increased τ-1 immunoreactivity was also evident in cells, presumably oligodendroglia, found in the corpus callosum. At 72 h, τ-1 immunoreactivity was diminished in the infarct zone and perikaryal immunostaining was no longer observed (Fig. 1).
Cresyl violet
Cresyl violet staining has been used previously to measure the brain infarct area in animal models of arterial occlusion (Benveniste et al, 1991). In this study, we relied upon tissue staining with cresyl violet as a qualitative measure demonstrating the progressive loss of neuronal architecture over time. We sought to determine the correlation between the early changes in the distribution of τ protein, MAP2, or calpain-cleaved spectrin and the alteration of Nissl substance stained by the cresyl violet. Figure 4 shows that only healthy cells with good laminar detail were observed in the unaffected cortex (C), as expected. There was no significant difference in cresyl violet staining of cells in the unaffected cortex and those in the infarct zone after 15 min of CCA–MCA occlusion. After 1 h of ischemia, there was minimal loss of staining but no pyknosis or neuronolysis. After 6 h of arterial occlusion, inspection of ischemic cortex showed shrinkage of neuronal volume with an inflammatory infiltrate in one animal and loss of approximately 30% of cells in another. By 24 h, the infarct zone had developed islands of necrosis with loss of 50–70% of neurons in all animals. Complete loss of cells occurred after 72 h, and the infarct zone appeared uniformly necrotic.
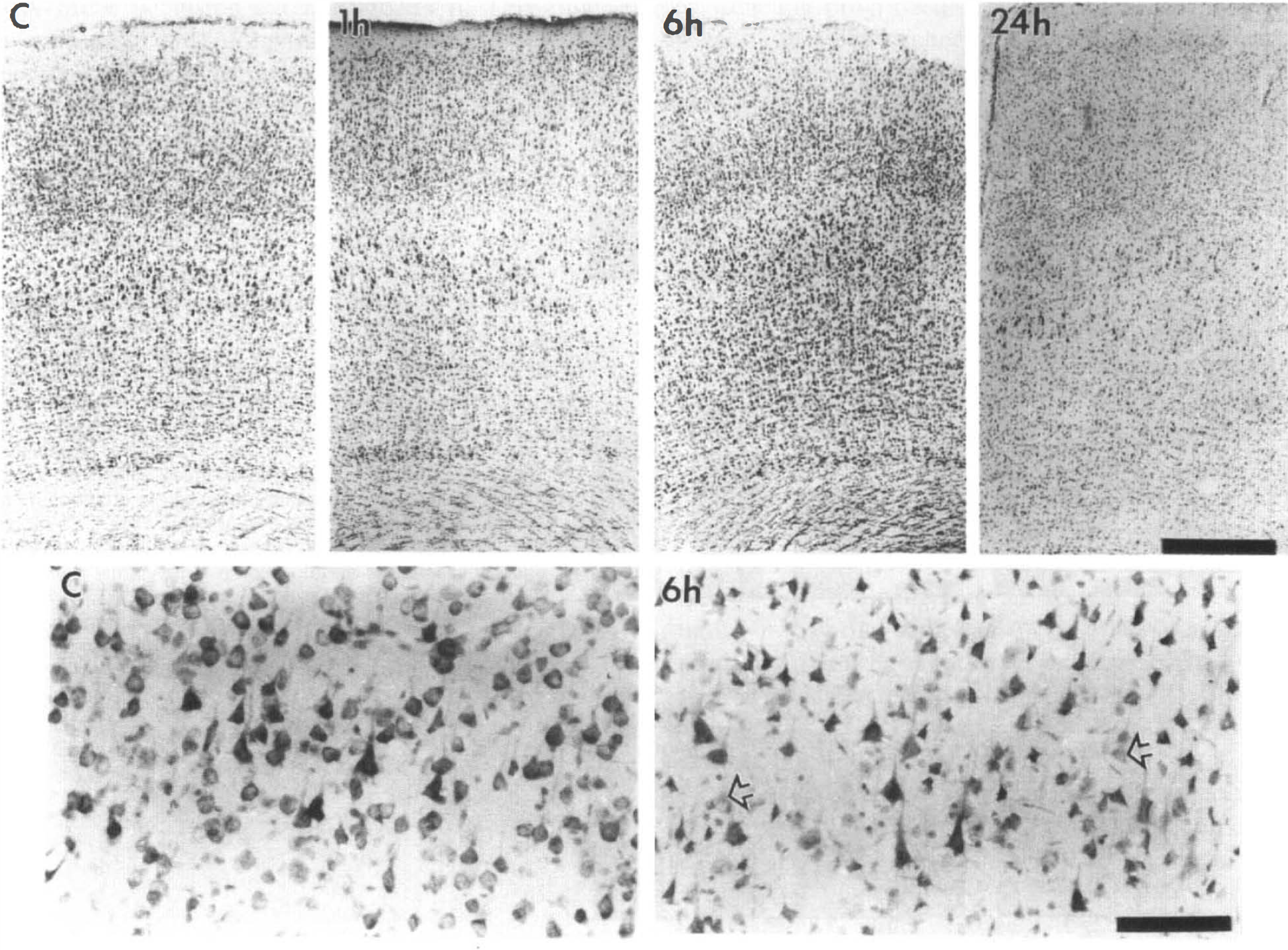
Cresyl violet staining of unaffected cortex and ischemic tissue sampled at 1, 6, or 24 h (upper row: ×4; lower row: ×20). The expected laminar distribution of healthy cells was seen in unaffected cortex (C), shown in the upper row. The higher magnification view of the same sample (C in first panel of lower row) revealed the neuronal density and appearance found customarily in normal cortex. Ischemic tissue sampled after 1 h of CCA–MCA occlusion showed no pyknosis or neuronolysis, but widespread shrinkage of neuronal volume was observed at 6 h. Isolated neurons showed degenerating nuclei in tissue sampled after 6 h (arrows in second panel of lower row). By 24 h, the infarct zone in all animals showed islands of necrosis with loss of 50–70% of neurons. The scale bars represent 0.5 mm in the photographs shown in the upper row and 100 pm in the higher magnification views in the lower row.
Optical density measurements
Figure 5 shows optical densities measured from normal and ischemic cortex obtained from group A animals and processed for immunolabeling of MAP2, calpain-cleaved spectrin, and τ protein. The upper panel in Fig. 5 reveals data acquired from tissue immunolabeled with the AP20 antibody used to identify MAP2. Comparison of group means using the Kruskal-Wallis ANOVA revealed that MAP2 immunoreactivity differed significantly in the infarct zone over varying durations of ischemia (p ≤ 0.002). Post hoc analysis by paired t testing confirmed that ischemic tissue was significantly less immunoreactive than unaffected cortex after 1 (p ≤ 0.05), 6 (p ≤ 0.0001), 24 (p ≤ 0.0001), and 72 (p ≤ 0.02) h of CCA–MCA occlusion.
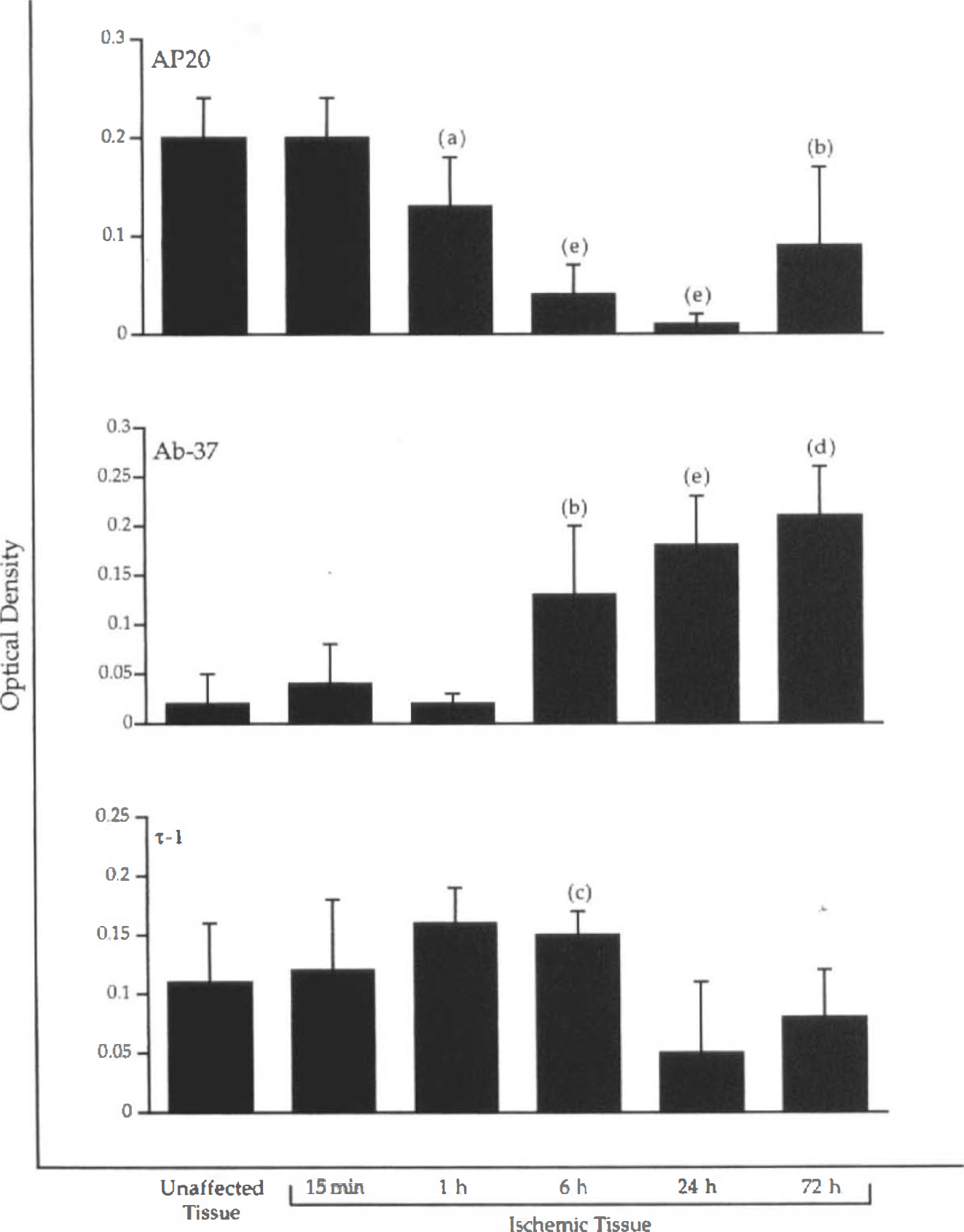
Histogram of mean optical densities (ODs) ± SD measured in immunocytochemical specimens prepared from animals subjected to varying durations of ischemia (n = 5 or 6 for each sampling time). An overall mean OD measured from unaffected cortex in all animals (n = 32) is shown for interpretive convenience. For AP20 immunolabeling, the mean OD in ischemic tissue was significantly lower than that measured in unaffected cortex taken from the same animals after 1, 6, 24, and 72 h of CCA–MCA occlusion. For Ab-37, mean OD in ischemic cortex differed significantly from unaffected tissue at 6, 24, and 72 h. For the τ-1 antibody, only the relative increase in immunoreactivity measured in ischemic tissue at 6 h was significantly different from unaffected cortex, (a) ≤ 0.05, (b) ≤ 0.02, (c) ≤ 0.02, (d) p ≤ 0.001, and (e) p ≤ 0.0001.
The middle panel of Fig. 5 shows mean optical densities measured from ischemic cortex immunolabeled with Ab-37 to identify calpain-cleaved spectrin. Kruskal–Wallis ANOVA again revealed a highly significant difference between optical density means in groups sampled at varying durations of ischemia (p ≤ 2.0 × 10−10). Post hoc analysis showed that immunoreactivity of calpain-cleaved spectrin rose significantly in ischemic cortex at 6 (p ≤ 0.02), 24 (p ≤ 0.0001), and 72 (p ≤ 0.001) h. The lower panel of Fig. 5 shows mean optical densities measured from unaffected and ischemic cortex immunolabeled with τ-1. Kruskal-Wallis ANOVA revealed a significant difference between optical density means calculated for ischemic tissue in groups sampled at varying times (p ≤ 0.002). Post hoc analysis demonstrated that the increased τ-1 immunoreactivity observed in the infarct zone at 6 h differed significantly from the baseline signal in the unaffected cortex (p ≤ 0.002).
Quantitative Western blot studies
We performed quantitative Western blot analysis to determine if the qualitative differences in immunoreactivity between normal and ischemic cortex were associated with actual changes in the concentration of τ or MAP2 protein. Figure 6 shows a photograph of a representative Western blot incubated with the AP20 antibody recognizing MAP2 in unaffected and ischemic cortex. A single immunoreactive band at 220 kDa was used for quantitation of changes in MAP2 protein concentrations. Sampling of tissue during maturation of the infarct demonstrated a progressive loss of MAP2 as the duration of CCA–MCA occlusion extended to 24 h. This time-dependent loss of MAP2 protein correlated with the disappearance of MAP2 immunoreactivity observed in the infarct zone after 6 h, as shown in Fig. 1.
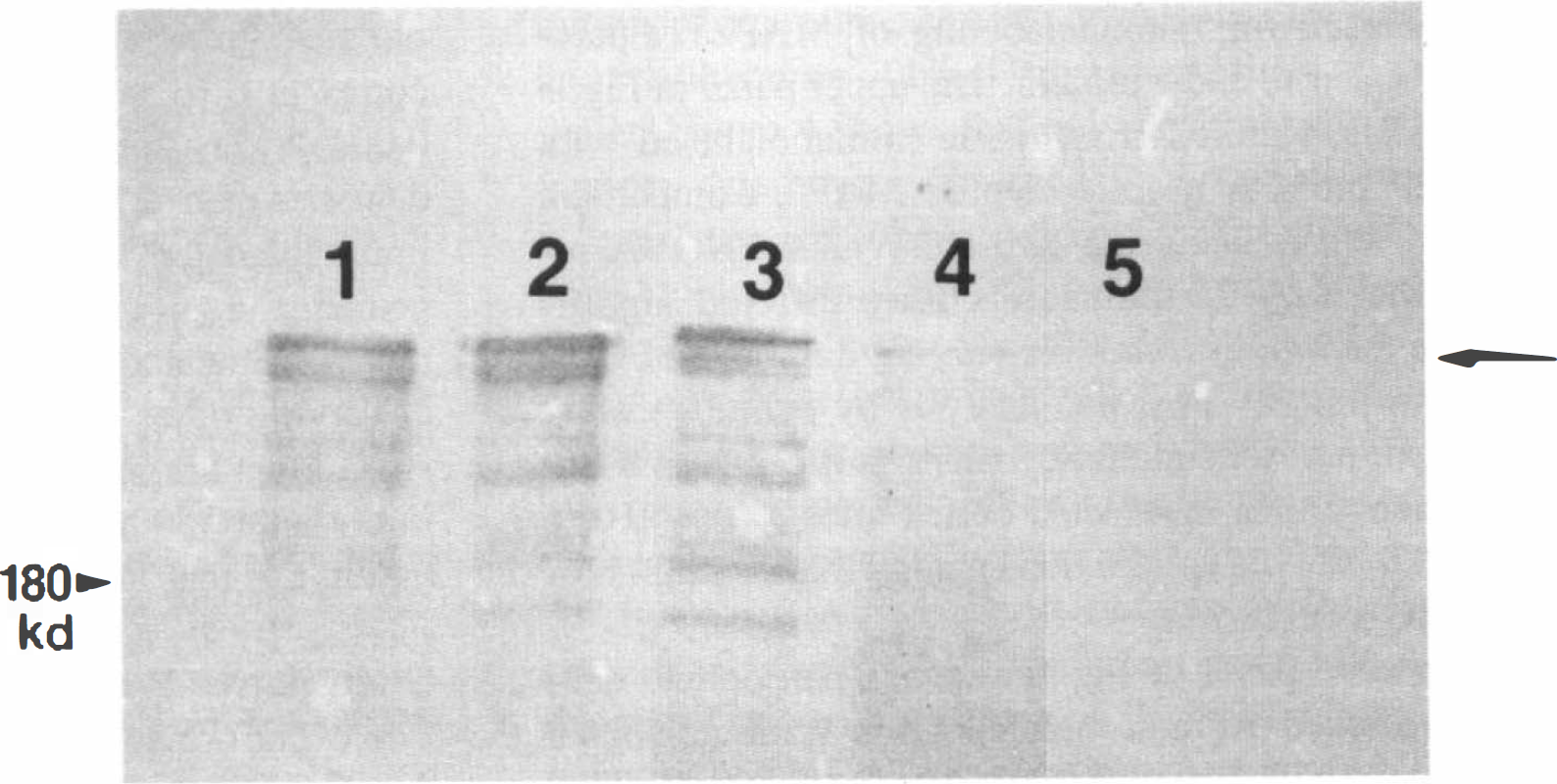
Representative Western analysis of MAP2 in supernatant prepared by centrifugation of homogenized cortical tissue. Lane 1 shows the MAP2 content in cortical tissue removed from the unaffected hemisphere of an ischemic rat after 15 min of CCA–MCA occlusion. Lane 2 contains MAP2 from ischemic cortical tissue in the same animal. The remaining lanes show MAP2 in ischemic cortex sampled after 1 (lane 3), 6 (lane 4), or 24 (lane 5) h of arterial occlusion. Note the progressive loss of MAP2 protein from ischemic tissue over this time course. A single molecular weight marker of 180 kDa is shown on the left margin. The arrow on the right indicates the 220-kDa band used for quantification of MAP2 by laser densitometry.
Figure 7 contains two photographic images of the same samples displayed in Figure 6, but now immunoreacted with τ-1 (panel A) or 8C11 (panel B). Im-munolabeling with either antibody reveals that a well-demarcated pair of bands exceeding 48.5 kDa was observed in unaffected cortex. This doublet lost immunoreactivity and converted into a single band as the duration of CCA–MCA occlusion extended to 24 h. Correspondingly, a single band of lower molecular weight observed in unaffected cortex became more pronounced as ischemia progressed. This reciprocal phenomenon suggests that dephosphorylation of τ protein occurs as cortical tissue becomes more ischemic, correlating with our observation of a similar process in postmortem brain (Schwab et al, 1994).
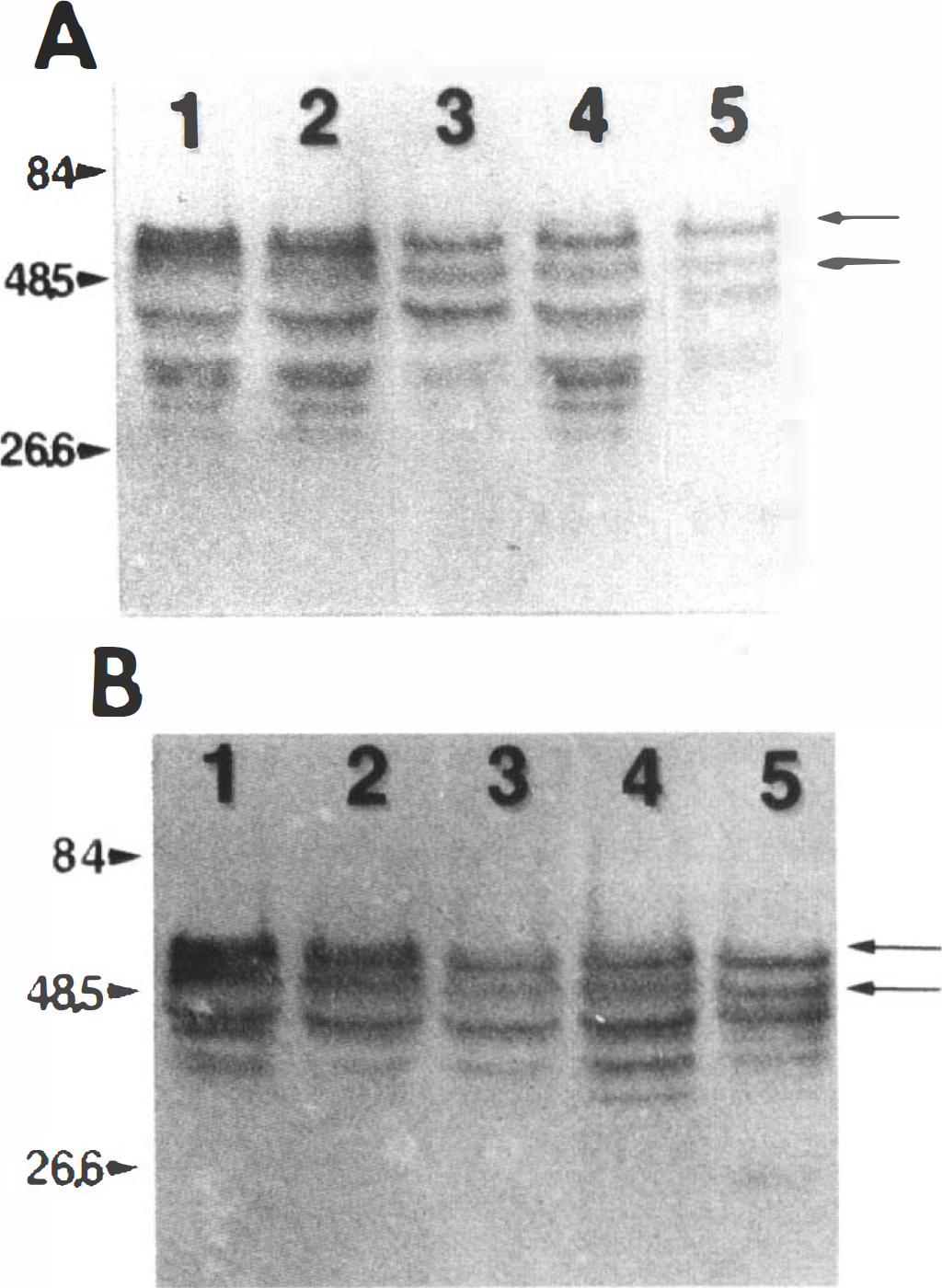
Representative Western analysis of τ protein in the same set of samples displayed in Fig. 6. The τ protein shown in
To better characterize the alterations in τ and MAP2 proteins, laser densitometry was performed on Western blot of unaffected and ischemic cortex sampled between 15 min and 24 h of MCA-CCA occlusion (Fig. 8). For this quantitative analysis, τ protein was identified only by the τ-1 antibody. Comparison of optical density values associated with MAP2 quantified in ischemic cortex showed that there was heterogeneity of group means detected by Kruskal-Wallis ANOVA (p ≤ 0.04). Post hoc testing demonstrated that the mean optical density representing MAP2 antigen in ischemic cortex sampled at 24 h was significantly lower than those obtained at earlier time points (p ≤ 0.05). The optical densities associated with MAP2 antigen in unaffected cortex remained uniform across all sampling times. There were no significant differences in optical densities associated with τ protein in unaffected or ischemic tissue.
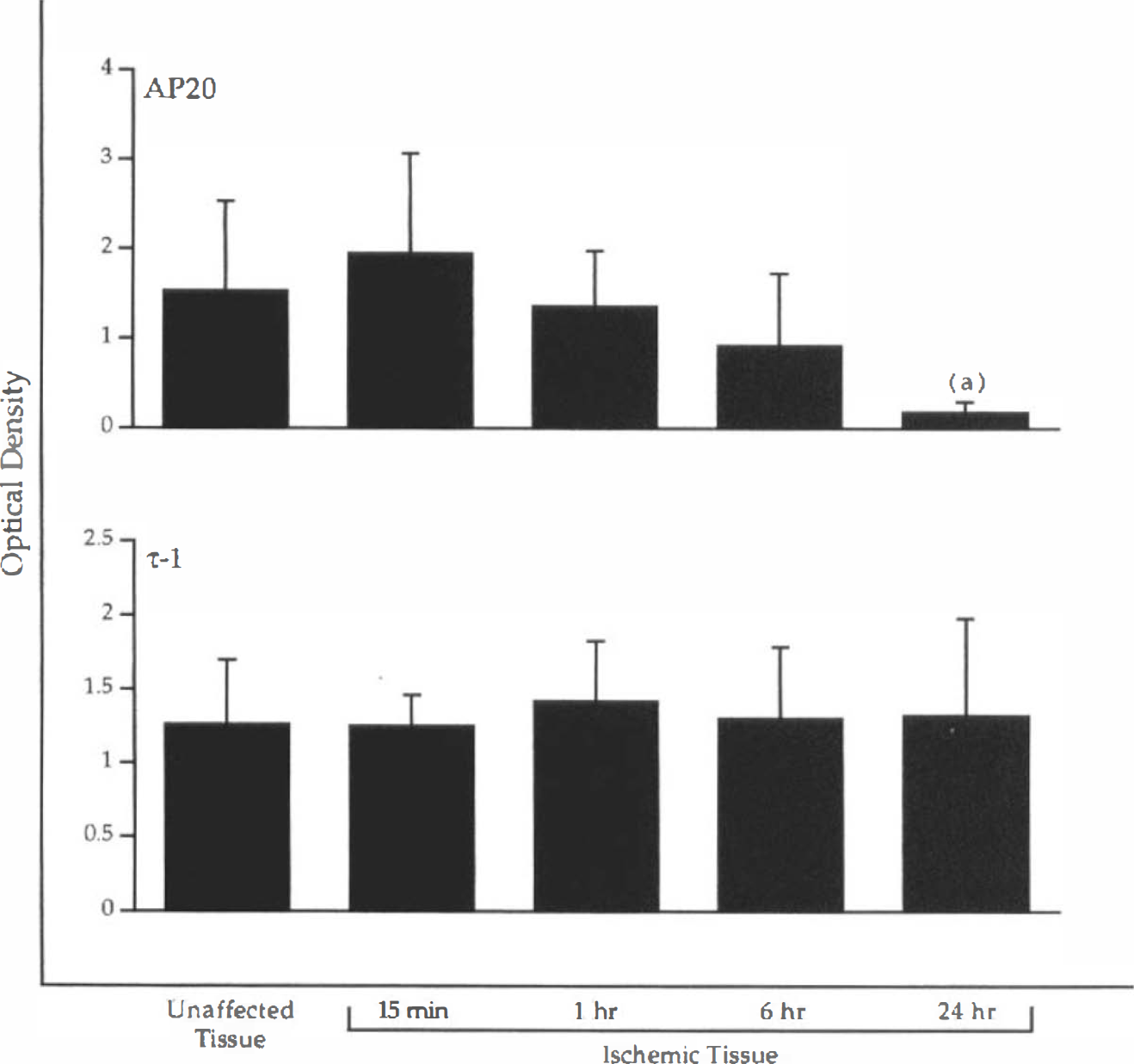
Histograms of mean optical densities (ODs) ± SD obtained from quantitative laser densitometry of Western blots similar to those shown in Figs. 6 and 7. Four animals were prepared for each duration of CCA–MCA occlusion. Note the progressive reduction of OD associated with AP20 immunostaining of MAP2 in ischemic tissue subjected to increasing duration of hypoperfusion, (a) ≤ 0.05 between mean OD at 24 h and all preceding time points.
Double-labeled immunofluorescence
To determine the temporal and spatial relationship between cytoskeletal alterations and the activation of calpain in individual neurons affected by ischemia, tissue sections taken from group A animals were incubated simultaneously with AP20 and Ab-37 complexed with fluorescently labeled secondary antibodies. All images used for this double-labeled immunofluorescence technique were taken from within the infarct, avoiding the transition zone between ischemic and unaffected tissue. As is shown in Fig. 9B, perikaryal Ab-37 immunoreactivity was already apparent after 15 min of MCA-CCA occlusion. The same region immunolabeled with AP20 (Fig. 9A) showed no somal accumulation of MAP2. To better delineate when somal accumulation of MAP2 would be seen after calpain activation occurred at 15 min, three rats were subjected to 30 min of arterial occlusion and added to those already prepared for later time points in group A. Representative photomicrographs taken of the ischemic tissue in one of these animals are shown in Fig. 9 (C and D) and demonstrate that somal accumulation of MAP2 and calpain-activated spectrin proteolysis colocalized in the same ischemic neurons. Identical findings were noted in the other two rats in the 30-min group. This colocalization of AP20 and Ab-37 immunolabeling persisted in animals subjected to 60 min of CCA–MCA occlusion (data not shown). However, prolonging cortical ischemia to 6 h produced little demonstrable perikaryal MAP2 immunoreactivity and caused fragmentation of Ab-37 immunofluorescent labeling (data not shown). Surprisingly, somatic MAP2 immunoreactivity reappeared in neurons located within the infarct after 24 h of CCA–MCA occlusion (Fig. 9E). Although there was widespread Ab-37 immunofluorescent labeling within the same area at 24 h (Figure 9F), only rarely did cells coexpress both activities. Photomicrographs of ischemic tissue obtained after 72 h of arterial occlusion were not included in Fig. 9, because the severity of necrotic damage prevented reliable interpretation of the immunofluorescent labeling.
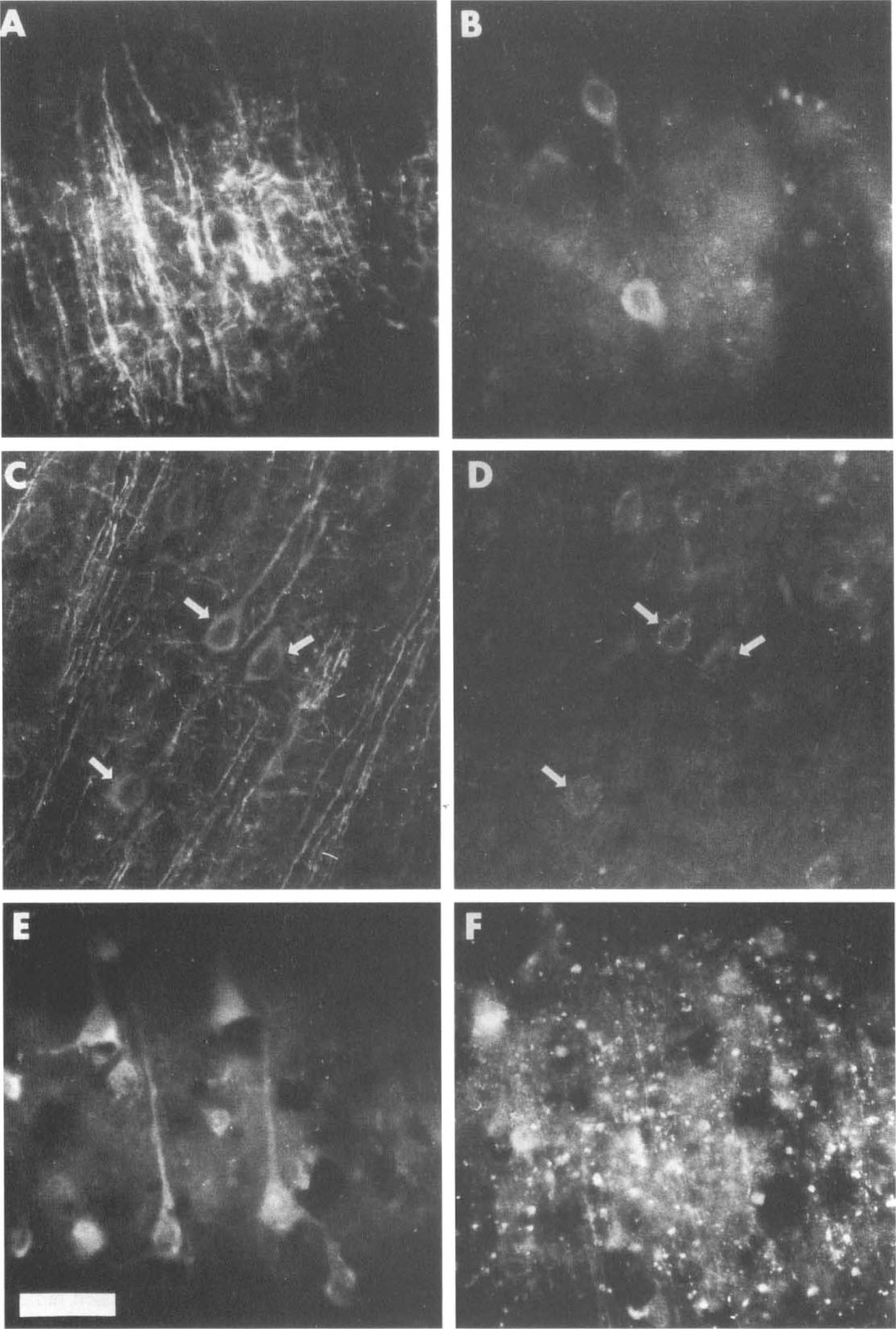
Representative photomicrographs (×40) of neurons within ischemic cortex prepared for double-labeled immunofluorescent studies of MAP2 and calpain-cleaved spectrin immunoreactivity.
DISCUSSION
The results of this study show that focal cerebral ischemia causes acute changes in cytoskeletal structure that predate histological features of neuronal death observed in a mature infarction. The alterations observed in our immunocytochemical experiments clearly preceded microscopic evidence of widespread cellular necrosis revealed by cresyl violet staining. The most sensitive indicator of ischemic neuronal change was the coiled, disfigured appearance of apical dendrites, as documented by AP20 immunostaining of MAP2 protein after 15 min of CCA–MCA occlusion. Prolongation of ischemia to 6 h caused almost complete loss of AP20 immunolabeling throughout the infarct zone, matched by the disappearance of dendritic branches that began at 1 h. The loss of AP20 immunostaining corresponded inversely to the simultaneous accumulation of calpain-cleaved spectrin breakdown products, represented by Ab-37 immunoreactivity. The immunocytochemical studies demonstrate that intraneuronal MAP2 proteolysis is a sensitive, early marker of focal ischemic injury in brain. The double-labeled immunofluorescence experiments, when combined with our immunocytochemical work, suggest that proteolytic disassembly of MAP2 coincides with calpain activation.
To our knowledge, this is the first report of temporal and spatial patterns of cytoskeletal disruption correlated with immunocytochemical evidence of calpain-mediated proteolysis in focal cerebral ischemia. The Ab-37 experiments correlate well with previous descriptions of suspected calpain activity in focal ischemia. Bartus et al (1995a) found similar evidence of calpain activation in rat cerebral tissue subjected to permanent MCA occlusion. These investigators performed Western blot studies demonstrating that spectrin breakdown products increase significantly after 1 h of ischemia, long before the development of an infarct. Their results also indicated that calpain-mediated spectrin proteolysis peaked after 12 h of permanent MCA occlusion and continued through 24 h. Although the present study did not include sampling of brain tissue at 12 h of focal ischemia, our observations of calpain activation and MAP2 proteolysis support the observations of Bartus et al. Hong et al. (1994b) reported similar findings in rats subjected to 3 h of transient focal ischemia. In this setting, Western blot analysis showed that spectrin breakdown products generated within the infarct zone reach a plateau after 3 h of reperfusion. Spectrin proteolysis in the transition zone corresponding to the ischemic penumbra followed a biphasic course, with a modest increase in breakdown products occurring after 2 h of reperfusion and a much larger elevation taking place at 24 h. The same differentiation between cytoskeletal damage within the infarct zone and the adjoining penumbral region has been shown in photo-chemically induced MCA occlusion by Yao et al. (1995), who suggested that enhanced spectrin proteolysis may result from suppression of glucose metabolic rate in ischemic tissue.
In comparison to our findings, a previous study by Inuzuka et al. (1990) failed to detect suppression of MAP2 immunoreactivity earlier than 6 h following permanent MCA occlusion. This apparent delay in reduction of MAP2 may be attributed to sampling of ischemic tissue no earlier than 4 h after occlusion of the MCA or the use of immunoblots in place of immunocytochemistry. Inuzuka et al. performed blotting assays designed to show the protein content of the entire cerebral hemisphere subjected to MCA occlusion, thereby mixing unaffected and ischemic tissue together. They were unable to observe the profound changes in MAP2 shown in our immunocytochemical experiments conducted at or before 6 h of ischemia. Our results suggest that immunocytochemical techniques are more sensitive than blot studies in detecting loss of MAP2. Both approaches were able to identify significant alterations in MAP2 immunoreactivity, but the blotting assays produced consistent results only in tissue that had been rendered ischemic for 24 h.
Our immunocytochemical experiments revealed perikaryal accumulation of MAP2 immunoreactivity in neurons within the transition zone corresponding to the ischemic penumbra. This effect was observed after no more than 1 h of ischemia. We speculate that this apparent redistribution of MAP2 antigen occurred following calpain activation and loss of dendritic branches. Our immunofluorescent studies showed robust labeling of somal MAP2 protein in isolated neurons confined to tissue that had been ischemic for 24 h. The reappearance of somal MAP2 immunoreactivity may represent the final breakdown and collapse of MAP2 fibers within the neuronal cell body, following an initial accumulation of MAP2 antigen from dendrites injured during the early stage of the ischemic injury. Another possibility is that the ischemic tissue response was heterogeneous, with small groups of neurons surviving to display perikaryal accumulation of MAP2 immunoreactivity after others had already died. Our cresyl violet studies showed a loss of 50–70% of neurons in all animals at 24 h, implying that isolated cells could have remained intact long enough to demonstrate what would otherwise be an early indicator of ischemic stress. This observation parallels the findings of Bartus et al. (1995a), who performed a time-course study of neuronal changes following permanent, focal MCA ischemia. They noted that cell loss occurs progressively in the ischemic neocortex but discovered that a limited number of small, triangulated cells remain viable even after 24 h of MCA occlusion. Lastly, it is not inconceivable that the delayed immunofluorescent labeling may represent synthesis of new MAP2 fibers to replace those lost from apical dendrites. Saito et al. (1995) showed that the mRNA transcript for MAP2c, a developmentally regulated isoform present in fetal brain, increased in the CA1 region of the rat hippocampus 1248 h after transient global ischemia produced by cardiac arrest. These results, when correlated with our double-labeled immunofluorescence work, suggest that injured neurons may reexpress a MAP2 isoform as a late postischemic response. However, our Western blot studies performed on ischemic tissue obtained at 24 h did not reveal a low-molecular-weight protein that could represent MAP2c produced under these conditions.
Tau immunostaining was also altered in response to focal ischemia, but in a manner distinct from MAP2. Tau dephosphdrylation throughout the infarct zone was evident as early as 15 min following ischemia. The perikaryal accumulation of τ preceded its disappearance, although both events were delayed as compared to MAP2. Our group has reported τ dephosphorylation and perikaryal accumulation following reversible forebrain ischemia (Geddes et al, 1994b) and in the postmortem rat brain (Schwab et al, 1994). In the present study, perikaryal τ accumulation was most evident in the ischemia penumbra, but was not observed in all neurons that were destined to become necrotic in the core of the infarction. This is consistent with our previous discovery that perikaryal τ accumulation is a sensitive marker of neuronal stress caused by reversible forebrain ischemia but is not absolutely predictive of neuronal death (Geddes et al, 1994b). Using the τ antibody, it is difficult to estimate relative τ levels as the intensity of staining is dependent upon both τ protein levels and the degree of phosphorylation at the τ-1 epitope. The phosphorylation-independent τ antibody, 8C11, confirmed that τ is resistant to proteolysis following focal ischemia, as compared to MAP2. This finding contrasts with in vitro studies, in which τ and MAP2 are both sensitive to calpain-mediated proteolysis (Johnson et al., 1989, 1991). The vulnerability of τ to calpain-mediated proteolysis is influenced by its localization, phosphorylation, and its association with microtubules. Highly phosphorylated τ protein in human paired helical filaments and bovine brain is more resistant to calpain proteolysis than normal τ (Litersky and Johnson, 1992; Yang and Ksiezak-Reding, 1995). However, enzymatic dephosphorylation of normal τ does not further enhance vulnerability to calpain proteolysis (Litersky and Johnson, 1992; Litersky et al, 1993). In situ, τ dephosphorylation may increase τ binding to microtubules (Lindwall and Cole, 1984), increasing its susceptibility to calpain-mediated proteolysis (Johnson et al., 1989). The relative resistance of τ observed following focal ischemia may reflect its predominantly axonal localization, in contrast to the somatodendritic localization of MAP2 and calpain (Binder et al, 1986; Perlmutter et al., 1988).
In summary, the results of this study support the role of calpain in microtubular proteolysis associated with focal cerebral ischemia, but demonstrate that MAP2 and τ are affected differently. Calpain-induced microtubular proteolysis is an early marker of ischemic neuronal stress, preceding cell death by several hours. Our data are consistent with previous reports suggesting that calpain inhibition is neuroprotective in cerebral ischemia. Several investigators have shown that pretreatment with calpain inhibitors will preserve selectively vulnerable neurons in rodent models of reversible forebrain ischemia (Lee et al., 1991; Rami and Krieglstein, 1993). Bartus et al. (1995b) found that postischemic, intrahippocampal injection of a calpain inhibitor protected CA1 neurons in the four-vessel occlusion model. Others have demonstrated similar effects with calpain inhibitors administered before or after the onset of focal brain ischemia (Hong et al, 1994a; Bartus et al, 1994a,b). Our findings, when correlated with these published results, suggest that calpain inhibitors hold great promise for ameliorating cerebral injury caused by stroke.
Footnotes
Abbreviations used
Acknowledgment:
A portion of this work was presented in abstract form before the XVII International Symposium on Cerebral Blood Flow and Metabolism held in Cologne, Germany on July 2–6, 1995. Financial support was provided by NIH/NINDS Clinical Investigator Development Award NS01505. (L.C.P.), R01 NS33773 (L.C.P.), ROI AG10678 (J.W.G.), Alzheimer's Disease Research Center grant P50 AG05144 (J.W.G.), the Kentucky Affiliate of the American Heart Association (L.C.P.), and the American Health Assistance Foundation (S.L.M.). We thank Michael Liebman and Sherry Williams for editorial assistance.