
Abstract
Unlike adults, hyperglycemia with circulating glucose concentrations of 25–35 mM/L protects the immature brain from hypoxic–ischemic damage. To ascertain the effect of hyperglycemia on cerebral oxidative metabolism during the course of hypoxia–ischemia, 7-day postnatal rats underwent unilateral common carotid artery ligation followed by exposure to 8% O2 for 2 h at 37°C. Experimental animals received 0.2 cc s.c. 50% glucose at the onset of hypoxia–ischemia, and 0.15 cc 25% glucose 1 h later to maintain blood glucose concentrations at 20–25 mML for 2 h. Control rat pups received equivalent concentrations or volumes of either mannitol or 1 N saline at the same intervals. The cerebral metabolic rate for glucose (CMRglc.) increased from 7.1 (control) to 20.2 μmol 100 g−1 min−1 in hyperglycemic rats during the first hour of hypoxia–ischemia, 79 and 35% greater than the rates for saline- and mannitol-injected animals at the same interval, respectively (p < 0.01). Brain intracellular glucose concentrations were 5.2 and 3.0 mM/kg in the hyperglycemic rat pups at 1 and 2 h of hypoxia–ischemia, respectively; glucose levels were near negligible in mannitol- and saline-treated animals at the same intervals. Brain intracellular lactate concentrations averaged 13.4 and 23.3 mM/kg in hyperglycemic animals at 1 and 2 h of hypoxia–ischemia, respectively, more than twice the concentrations estimated for the saline- and mannitol-treated littermates. Phosphocreatine (PCr) and ATP decreased in all three experimental groups, but were preserved to the greatest extent in hyperglycemic animals. Results indicate that anaerobic glycolytic flux is increased to a greater extent in hyperglycemic immature rats than in normoglycemic littermates subjected to cerebral hypoxia–ischemia, and that the enhanced glycolysis leads to greater intracellular lactate accumulation. Despite cerebral lactosis, energy reserves were better preserved in hyperglycemic animals than in saline-treated controls, thus accounting for the greater resistance of hyperglycemic animals to hypoxic–ischemic brain damage.
An age-specific paradox exists pertaining to the effect of glucose on hypoxic–ischemic or ischemic brain damage. Research conducted many years ago demonstrated that pretreatment of perinatal animals with glucose prolongs their survival when they are subjected to systemic hypoxia, asphyxia, or cerebral ischemia (Himwich et al., 1942; Britton and Kline, 1945, 46; Stanfford and Weatherall, 1960) and might reduce the severity of permanent brain damage as well (Adamsons et al., 1964; Dawes et al., 1964). However, despite the increased hypoxic–ischemic resistance of glucose treated-immature animals, experiments in adult animals, including humans, have demonstrated that glucose supplementation actually accentuates ischemic brain damage (Myers and Yamaguchi, 1977; Kalimo et al., 1981; Pulsinelli et al., 1982; 1983; Berger and Hakim, 1986). More recently, we have shown, in an immature rat model of perinatal hypoxic–ischemic brain damage, that hyperglycemia to blood glucose concentrations of 35–40 mM/L entirely prevents the occurrence of brain damage (Vannucci and Mujsce, 1992). Accordingly, glucose has a paradoxical role in hypoxia–ischemia, prolonging survival and reducing brain damage during hypoxia–ischemia in immature animals while increasing brain damage in adults.
To ascertain the underlying mechanisms that account for the protective role of glucose in perinatal hypoxic–ischemic brain damage, experiments were devised to determine those alterations in cerebral glucose and energy metabolism that occur during the course of hypoxia–ischemia in our immature rat model (Rice et al., 1981). At the onset of hypoxia–ischemia, immature rats received either glucose, saline, or mannitol, following which their brains were prepared for analysis of labile metabolites as well as measurement of cerebral metabolic rate for glucose (CMRglc). The findings are discussed in relation to known metabolic alterations that occur in adult animals subjected to hyperglycemic cerebral hypoxia–ischemia.
MATERIALS AND METHODS
Pregnant, dated Wistar rats were purchased from a commercial breeder (Charles River, Wilmington, MA, U.S.A.), housed individual cages, and fed standard laboratory chow ad libidum. Offspring, delivered vaginally, were maintained with their dams until the day of experimentation. Immediately following birth, litter size was reduced to 10 animals to ensure adequate and comparable nutrition and weight gain.
Induction of hypoxia–ischemia
Seven-day postnatal rats were subjected to unilateral cerebral hypoxia–ischemia via a modification of a previously described technique (Rice et al., 1981). Specifically, individual rat pups underwent permanent ligation of the right common carotid artery under light halothane anesthesia, following which they recovered with their dams for 3 h. To induce systemic hypoxia, rats were not placed in jars as previously described (Rice et al., 1981), but, rather, were individually positioned head first within the barrel of a 20 cc syringe. The barrels themselves were aligned in parallel and adjacent to each other and attached to a plexiglass platform (Fig. 1). The smaller aperture of each barrel was attached to polyethylene tubing that, in turn, was attached to a gas tank containing either room air or 8% oxygen-92% nitrogen. Each rat pup was snugly positioned within the barrel via the larger aperture for 15 min, following which they were exposed to either room air (controls) or the hypoxic gas mixture. The larger aperture of the barrel also served as the outlet for the gas. Immature rats, when exposed to a gentle stream of gas, will orient their heads toward the source of gas flow, thereby maintaining themselves within the confines of the barrels. The internal temperature of each barrel was intermittently monitored with a microthermistor probe connected to a servo-controlled heating lamp positioned 2 ft directly above the barrel apparatus to maintain an environmental temperature of 37°C. Rat pups were exposed to the gas mixture for varying durations up to 2 h, following which they were removed from the barrels for further experimentation.
Syringe barrel apparatus for the production of cerebral hypoxia–ischemia in immature rats.
To produce hyperglycemia, immature rats were injected subcutaneously with 0.2 cc 50% glucose at the onset of systemic hypoxia followed by 0.15 cc 25% glucose 1 h later; these injections were designed to maintain blood glucose concentrations at 20–25 mM/h (360–450 mg/dl) for 2 h. Other rat pups received equivalent concentrations and volumes of mannitol at the same intervals. Control rat pups received equivalent volumes of 1 N saline at the same intervals. All injections were administered while the animals were maintained within the barrels.
We have previously demonstrated that unilateral common carotid artery ligation combined with exposure to 8% oxygen produces a spectrum of brain damage predominantly in the territory of the middle cerebral artery of the cerebral hemisphere ipsilateral to the carotid artery occlusion (Rice et al., 1981; Towfighi et al., 1991). Damage does not occur in the cerebral hemisphere contralateral to the carotid artery ligation or in either hemisphere when the animal is exposed to hypoxia or ligation alone.
Measurement of glycolytic intermediates and high-energy phosphate compounds
Immature rats from each experimental group (glucose, mannitol, saline) were quick frozen in liquid nitrogen after either 1 or 2 h of exposure to hypoxia–ischemia. Littermate controls underwent neither carotid artery ligation nor hypoxia. Each frozen brain was dissected from its skull in a cold box maintained at −20°C and the cerebral hemispheres separated from each other. A tissue specimen (60–100 mg) taken from the distribution of the middle cerebral artery of the cerebral hemisphere ipsilateral to the carotid artery occlusion was powdered under liquid nitrogen and weighed on a microanalytical balance. The contralateral cerebral hemisphere was not sampled, since it is not damaged by the hypoxic exposure (Rice et al., 1981). Perchloric acid (PCA) extracts were then prepared from each powder, as previously described (Vannucci and Duffy, 1974). Concentrations of glucose, lactate, phosphocreatine (PCr), creatine, ATP, ADP, and AMP were analyzed by specific enzymatic, fluorometric techniques (Lowry and Passonneau, 1972; Vannucci and Duffy, 1974).
To estimate intracellular glucose and lactate concentrations in brain, carotid artery-ligated rats from each experimental group were subjected to hypoxia–ischemia for either 1 or 2 h. Immediately thereafter, an aliquot (∼20 μl) of cerebrospinal fluid (CSF) was obtained from the cisterna magna of each rat pup using a previously described technique (Vannucci and Duffy, 1974), following which each animal was decapitated and blood obtained from the severed neck vessels. A precise aliquot (20 μl) of CSF or blood was diluted 1:10 (vol/vol) in 0.5 M PCA and the specimen frozen until time of analysis for glucose and lactate concentrations, as described above.
Intracellular glucose and lactate concentrations in brain were estimated from the levels of the metabolites in brain, blood, and CSF assuming (a) a brain blood volume of 3% (Buschiazzo et al., 1970); (b) an extracellular fluid (ECF) compartment of 20% (Ferguson and Woodbury, 1969); and (c) a brain water content of 87% of whole brain weight (Rice et al., 1981). Cisternal CSF glucose or lactate was considered equivalent to the concentration in ECF (Bito and Davson, 1966).
Accordingly

Since tissue concentrations of both glucose and lactate prior to and during hypoxia–ischemia are comparable in the two cerebral hemispheres, despite ligation of one carotid artery (Welsh et al., 1982; Yager et al., 1991), any potential unequal contribution to CSF glucose or lactate from the contralateral hemisphere was ignored.
Measurement of CMRglc
In additional experiments, we determined CMRglc in control 7-day postnatal rats and in glucose, mannitol, or saline injected littermates exposed to hypoxia–ischemia. A modification of the original Sokoloff et al., 1977 technique [14C]2-DG was used to measure CMRglc, using 2-deoxy-[14C]-glucose ([14C]2-DG) as the radioisotope (Vannucci et al., 1989,1994; Palmer et al., 1990). Specific details regarding the cerebral hemispheric measurement of CMRglc have been previously published in the journal (Palmer et al., 1990; Vannucci et al., 1994). Rat pups received [14C]2-DG at either 15 or 75 min of hypoxia–ischemia, and the isotope was allowed to circulate for up to 45 min. Blood was collected from separate animals following decapitation at either 2, 5, 10, 20, 30, or 45 min for isotopic counting and glucose determination. The heads of the rat pups killed at 45 min postinjection of the isotope were immediately frozen in liquid nitrogen. PCA extracts were prepared from tissue samples of the ipsilateral cerebral hemisphere, as described above, portions of which were used for analysis of glucose content, isotopic counting, or passage over an ion-exchange column to ascertain the amount of free (non-phosphorylated) 2-DG. From the data, the percentage of total 2-DG in brain that was metabolized to 2-DG-6-phosphate was calculated. The lumped constant was calculated using a nomogram for adult rat brain (Pardridge et al., 1982) that allows for calculation of individual lumped constant values determined by the concentration of glucose in brain relative to that of plasma. CMRglc was then calculated using a formula that incorporates the percentage of 2-DG metabolized rather than the rate constants K1*, K2*, and K3* (Palmer et al., 1990; Vannucci et al., 1994).
Measurement of brain water content
Another group of glucose-, mannitol-, or saline-injected rat pups were exposed to hypoxia–ischemia for either 1 or 2 h, following which animals were decapitated and their brains immediately removed from their skulls. A tissue specimen (75–100 mg) was dissected from the ipsilateral cerebral hemisphere in the distribution of the middle cerebral artery and weighed in a tared 5-ml glass vial. The tissue specimen then was desiccated at 70°C for 72 h. Reweighing of the vial ascertained the hemispheric dry weight and, by subtraction from the total weight, the water content of the tissue sample was obtained. Water content, as a reflection of brain edema, was determined as a percentage of the total hemispheric weight according to the formula.
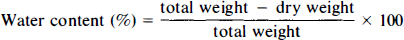
Statistical analyses
Statistical analyses of data included the unpaired Student's t-test, analysis of variance (ANOVA), and linear regression analysis.
Institutional approval
The experiments described herein were reviewed by the Animal Care and Use Committee of The Pennsylvania State University College of Medicine and approved on October 22, 1993.
RESULTS
Glucose and lactate concentrations
The double injection of glucose into immature rats produced hyperglycemia with mean blood glucose concentrations of 26 and 20 mM/L at 1 and 2 h of hypoxia–ischemia, respectively (Table 1). In contrast, a mild hypoglycemia occurred in those rat pups injected with either saline or mannitol. Hyperglycemia was associated with increased CSF and brain tissue glucose concentrations despite hypoxia–ischemia, whereas tissue glucose decreased to very low levels in saline- and mannitol-treated animals. Estimated brain intracellular glucose concentrations were increased 100–140% above the control value during the course of hypoxia–ischemia in hyperglycemic rat pups (Fig. 2).
Blood, SF and brain glucose and lactate concentrations during hypoxia–ischemia in the immature rat
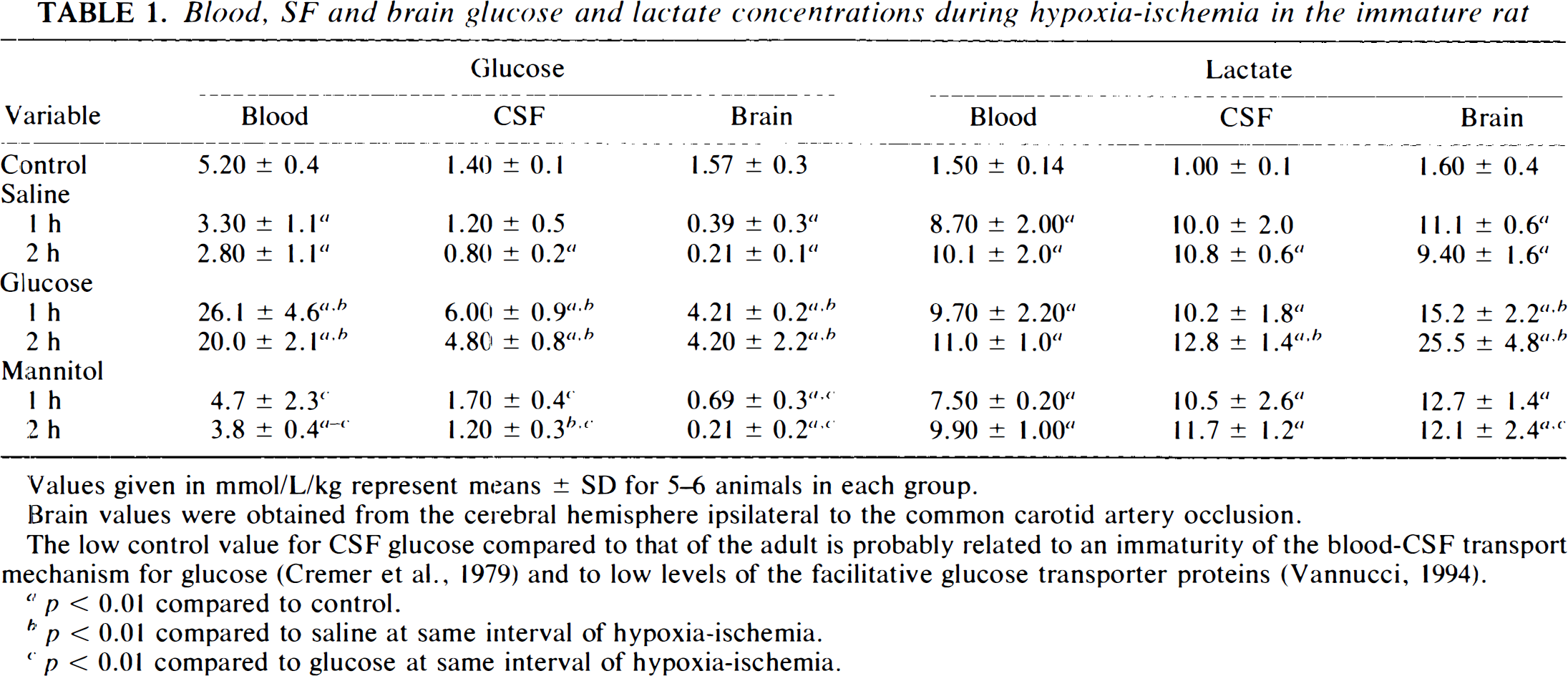
Values given in mmol/L/kg represent means ± SD for 5–6 animals in each group.
Brain values were obtained from the cerebral hemisphere ipsilateral to the common carotid artery occlusion.
The low control value for CSF glucose compared to that of the adult is probably related to an immaturity of the blood-CSF transport mechanism for glucose (Cremer et al., 1979) and to low levels of the facultative glucose transporter proteins (Vannucci, 1994).
p < 0.01 compared to control.
p < 0.01 compared to saline at same interval of hypoxia-ischemia.
p < 0.01 compared to glucose at same interval of hypoxia-ischemia.
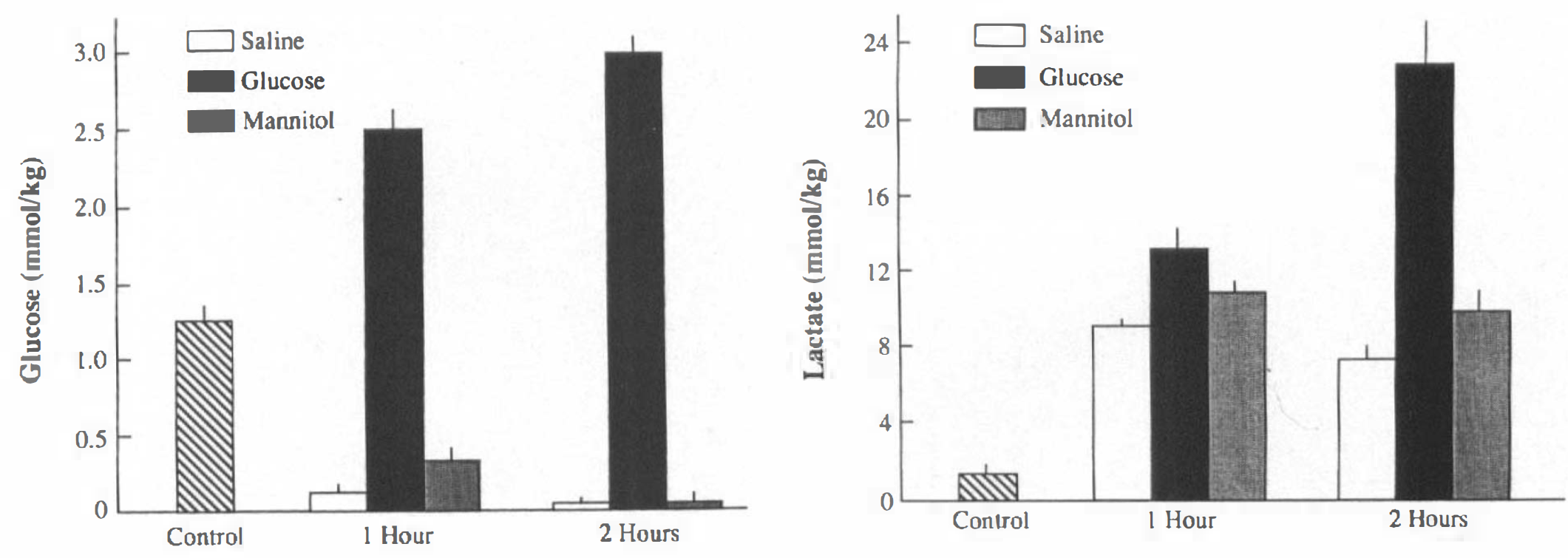
Estimated brain intracellular concentrations of glucose and lactate during hypoxia–ischemia in the immature rat. Brain intracellular concentrations were estimated from values in blood, CSF, and brain tissue, as described in Materials and Methods section and shown in Table 1. Bars represent means; vertical lines denote 1 SD.
Hypoxia-ischemia was associated with increased blood lactate concentrations to comparable values in the three experimental groups (Table 1). Mean CSF lactate levels were similar among the three groups, and averaged 1.5–3.0 mM/L higher than respective blood levels, despite different brain tissue concentrations, among the three groups. As a result, intracellular lactate levels of the hyperglycemic rat pups at 2 h of hypoxia–ischemia were more than twice the concentrations estimated in the saline- and mannitol-treated littermates (Fig. 2).
High-energy phosphate compounds
During the course of hypoxia–ischemia, PCr decreased in all three experimental groups, the lowest level occurring in saline-treated animals (Table 2). Associated increases in creatine occurred to the least extent in saline-treated animals, such that at both 1 and 2 h of hypoxia–ischemia, the sum of PCr and creatine was significantly lower than that of control animals. ATP also decreased during hypoxia–ischemia, the greatest decrease occurring in saline-treated animals. ATP decreased to comparable values in glucose- and mannitol-injected animals. Proportionate increases in ADP and AMP occurred in glucose- and mannitol-treated rat pups, such that the total adenylate nucleotide pool (ATP + ADP + AMP) in these groups was not different from that of the control group. In saline-treated animals, total adenine nucleotides were decreased by 52% at 2 h of hypoxia–ischemia.
High energy phosphate compounds during hypoxia–ischemia in the immature rat
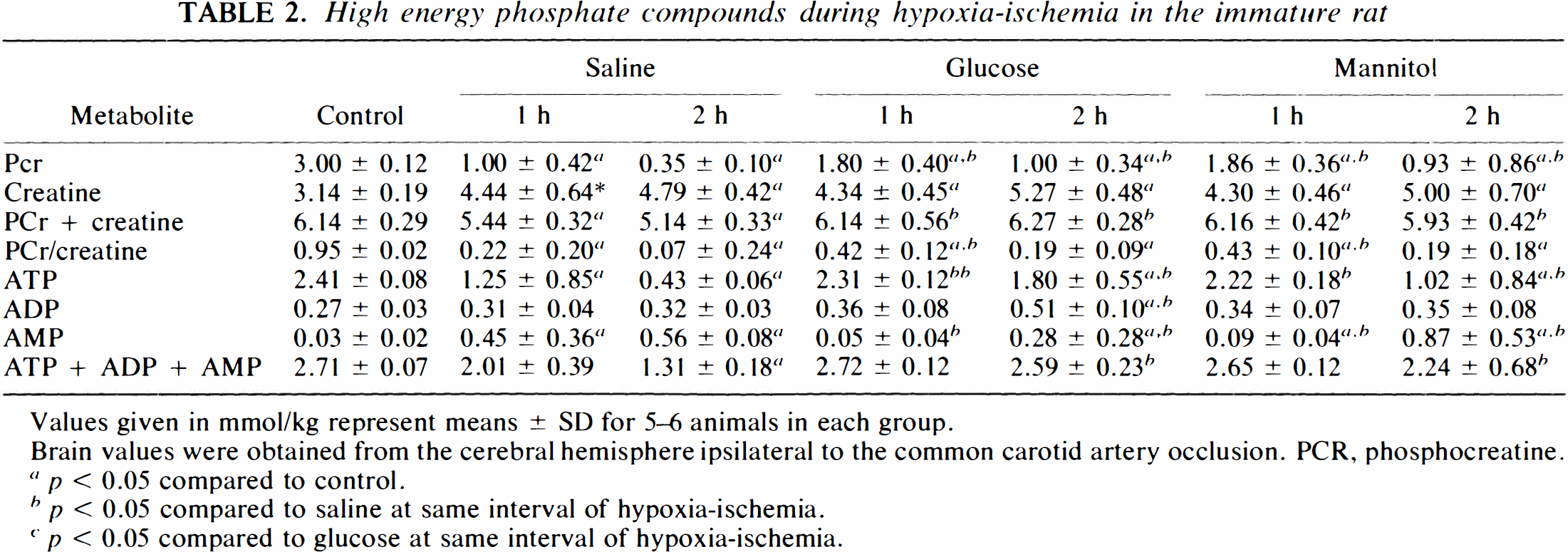
Values given in mmol/kg represent means ± SD for 5–6 animals in each group.
Brain values were obtained from the cerebral hemisphere ipsilateral to the common carotid artery occlusion. PCR, phosphoereatine.
p < 0.05 compared to control.
p < 0.05 compared to saline at same interval of hypoxia-ischemia.
p < 0.05 compared to glucose at same interval of hypoxia-ischemia.
To ascertain the association between brain tissue glucose and ATP concentrations, regression analyses were performed for each of the three experimental groups, combining the data from 1 and 2 h. In all three experimental groups, a positive direct correlation was obtained (p < 0.05); specifically, the higher the tissue glucose concentration, the higher the ATP level.
CMRglc
CMRglc increased from 7.1 (control) to 20.2 μM/100 g/min in hyperglycemic animals during the first hour of hypoxia–ischemia, 79 and 35% greater than the rates for saline- and mannitol-injected animals, respectively, for the same interval (Fig. 3). CMRglc was essentially unchanged in glucose- and mannitol-treated animals between 1 and 2 h, but increased significantly (+ 57%) in saline-treated rat pups (p < 0.05 compared to 1 h). At both measured intervals of hypoxia–ischemia, CMRglc of hyperglycemic rat pups was significantly greater than were rates of saline- and mannitol-injected animals.
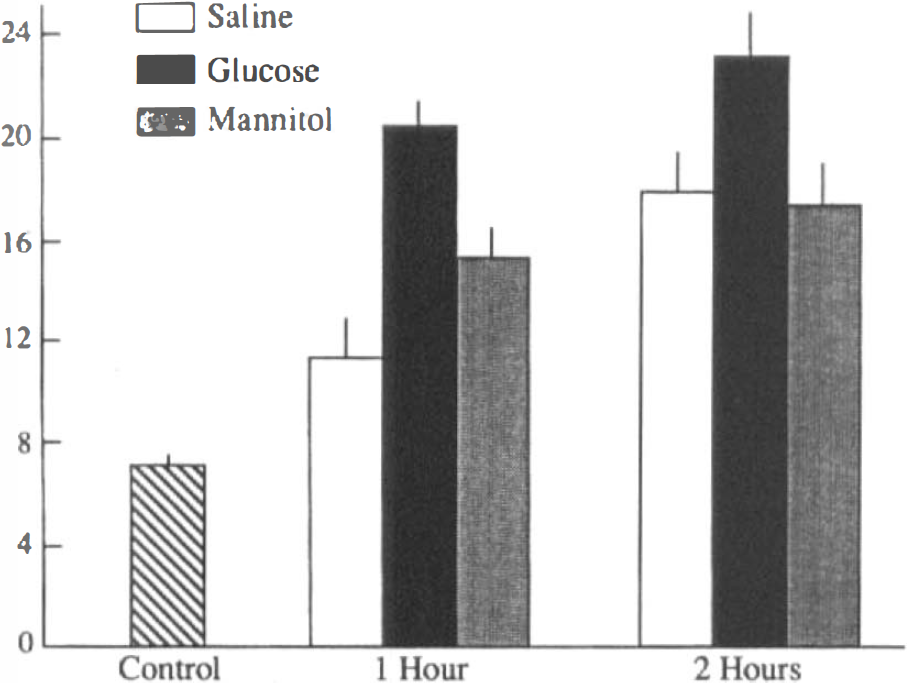
Cerebral metabolic rate for glucose (CMRglc during hypoxia–ischemia in the immature rat. Bars represent means of 6–8 animals in each group; vertical lines denote 1 SD.
Brain water content
Figure 4 depicts brain water contents of saline-, glucose-, and mannitol-treated immature rats during hypoxia–ischemia, expressed as a percentage of the control value. As previously demonstrated (Vannucci et al., 1993), brain edema, reflected in an increased water content, actually occurred during the course of hypoxia–ischemia in those immature rats injected with saline. In contrast, the hyperosmolar effect of both glucose and mannitol was associated with comparable decreases in tissue water content at both 1 and 2 h of hypoxia–ischemia. This finding indicates that the anticipated cerebral edema, which occurs during the course of hypoxia–ischemia, was entirely prevented by both glucose and mannitol. The blunting of cytotoxic edema formation during the course of hypoxia–ischemia might have contributed to the beneficial effect of both sugars on the severity of the ultimate brain damage (Vannucci and Mujsce, unpublished data).
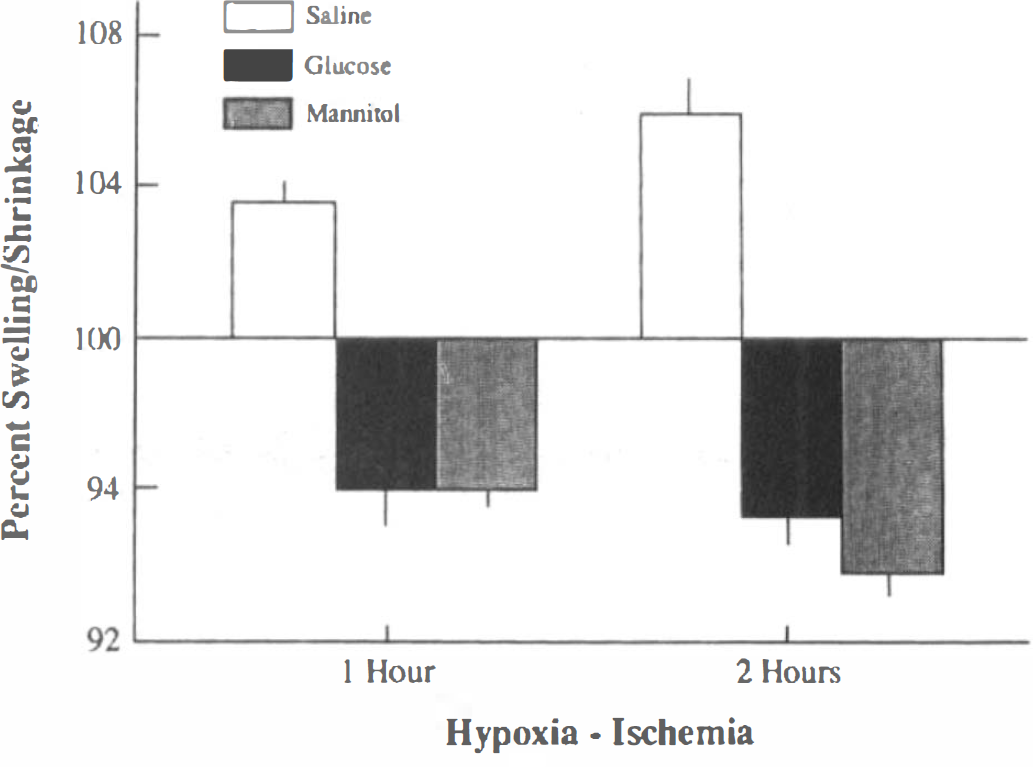
Percent change in brain tissue volume during hypoxia–ischemia in the immature rat. Bars represent mean changes in tissue volume (swelling or shrinkage) calculated from brain water contents of control and experimental animals. Vertical lines denote 1 SD. Brain water content of control animals averaged 87.7 ± 0.1% of total brain weight.
DISCUSSION
As mentioned, it is now firmly established that hyperglycemia accentuates the brain damage that occurs as a consequence of hypoxia–ischemia in adult animals. The deleterious effect of glucose has been demonstrated in adult animal models of incomplete global cerebral ischemia (carotid artery ligation + hypotension; four-vessel occlusion), complete global cerebral ischemia (cardiac arrest), and incomplete focal cerebral ischemia (common carotid, internal carotid, or middle cerebral artery occlusion ± hypotension) (Myers and Yamaguchi, 1977; Kalimo et al., 1981; Pulsinelli et al., 1982; Prado et al., 1988; Hoffman et al., 1990; Nakakimura et al., 1990; Väzquez-Cruz et al., 1990; Dietrich et al., 1993). In contrast, mild hyperglycemia (blood glucose = 15–20 mM/L) is not deleterious to immature rat brain undergoing hypoxia–ischemia, and moderate hyperglycemia (350 mM/L) is highly protective (Voorhies et al., 1986; Vannucci and Mujsce, 1992). In our immature rat model of perinatal hypoxic–ischemic brain damage, the focal cerebral ischemia is incomplete, with cerebral blood flow (CBF) to the vulnerable structures ranging from 15 to 35% of control values (Vannucci et al., 1988; Ringel et al., 1991). Therefore, hyperglycemia would preserve and probably even enhance glucose delivery (CBF × blood glucose) to the ischemic cerebral hemisphere despite moderate-to-severe tissue hypoperfusion.
Despite the age-related paradox of the effect of glucose on hypoxic–ischemic brain damage, the metabolic perturbations that occur during the course of the insult are, to at least some extent, comparable in immature and adult animals. Brain tissue lactate concentrations in hyperglycemic animals are invariably increased over levels in normoglycemic animals (Rehncroná et al., 1980; 1981; Welsh et al., 1980; Gardiner et al., 1982; Hoffman et al., 1990, 1994), and it was originally presumed that the enhanced tissue lactacidosis or associated derangement in pH homeostasis contributed to the increased brain injury (see also Myers, 1979; Siesjö, 1981). Furthermore, injection of lactic acid into the cerebral cortex of adult rats leads to histologic alterations resembling ischemic infarction, an injury that does not occur following injection of other organic acids of comparable pH (Kraig et al., 1987; Petito et al., 1987). In the present investigation in immature rats, hyperglycemia during 2 h of hypoxia–ischemia was associated with a tissue lactate level greater than those found in normoglycemic animals, although not to the extent typically seen in adult animals rendered similarly hyperglycemic.
In the present investigation, high-energy phosphate compounds—PCr, ATP, and ADP—were better preserved during hypoxia–ischemia in hyperglycemic immature rats than in saline-injected controls. The less severe depletion in cerebral energy reserves in hyperglycemic animals presumably served to reduce the severity of the ultimate brain damage (Vannucci and Mujsce, 1992). Studies in adult animals indicate that hyperglycemia typically is associated either with no deleterious or with an even beneficial effect on cerebral energy stores during ischemia (Rehncroná et al., 1981; Hoffman et al., 1990; Ekholm et al., 1993; Tyson et al., 1993; Wagner and Lanier, 1994), although they are not as well preserved as those in immature rats (see Table 2). That cerebral energy reserves are equally or better maintained during ischemia in hyperglycemic compared to normoglycemic adult animals, despite greater brain damage in the former group, suggests that mechanisms other than or in addition to cerebral lactacidosis or energy failure contribute to the ultimate tissue injury.
If the metabolic perturbations that occur during cerebral hypoxia–ischemia are at least somewhat comparable in hyperglycemic immature and adult rats, then the mechanisms underlying the age-related paradox between hyperglycemia and tissue injury require further explanation. Postischemic recovery (reperfusion) events combined with age-specific differences in blood-brain barrier (BBB) function might explain the phenomenon. In adult animals, hyperglycemia delays or entirely prevents restoration of previously depleted energy stores (Rehncroná et al., 1980, 1981; Welsh et al., 1980; Gardiner et al., 1982), owing probably to a lingering tissue lactacidosis (Welsh et al., 1980; Chopp et al., 1987; Haraldseth et al., 1992; Tyson et al., 1993). The persisting tissue acidosis adversely affects resumption of mitochondrial function, thereby preventing production of ATP or its transfer into the cytosol (Siesjö, 1992). A lingering tissue acidosis also would adversely affect restoration of ionic homeostasis, contribute to persisting intracellular calcium accumulation, and enhance edema formation (Hillered et al., 1985; Hakim and Shoubridge, 1989; Siesjö, 1992). In the immature rat recovering from hypoxia–ischemia, a lingering tissue acidosis does not occur, with normalization of pHi by 10 min of recovery and of tissue lactate concentrations by 4 h of recovery (Palmer et al., 1990; Yager et al., 1991). The relatively rapid normalization of tissue lactate presumably relates to its prompt oxidation to pyruvate, which enters the tricarboxylic acid cycle, and to the transport of lactate from brain into blood. BBB transport for lactate is especially well developed in the immature rat (Cremer et al., 1979).
An additional mechanism for the production of greater ischemic brain damage in hyperglycemic adult animals compared to normoglycemic animals might relate to the persistence of high circulating glucose concentrations during the early recovery period. In this regard, Tyson et al. (1993) showed, in adult rats, that hyperglycemia during and following, or only following, forebrain ischemia causes a delayed recovery of pHi and of any previously depleted PCr and ATP, measured by magnetic resonance (MR) spectroscopy, compared to normoglycemic and hypoglycemic animals. Postischemic administration of insulin improved the rate of pHi. PCr, and ATP recovery in previously hyperglycemic animals. The findings suggest that postischemic hyperglycemia contributes substantially to the accentuation of brain damage, probably through the mechanisms described above (mitochondrial dysfunction, ion disruption, calcium accumulation, edema formation) (see also Pulsinelli et al., 1982; Warner et al., 1987; Dietrich et al., 1993) as well as via continued anaerobic glycolysis of glucose, which is readily available to brain tissue (Tyson et al., 1993). In the immature rat recovering from hypoxia–ischemia, brain glucose concentrations are rapidly restored and actually increase slightly above control levels, owing primarily to a suppression of glucose utilization, since previously accumulated tissue lactate becomes the preferred fuel for oxidative metabolism (Palmer et al., 1990; Yager et al., 1991; Vannucci et al., 1994). However, in hyperglycemic rat pups, energy stores, especially ATP, are well preserved during the course of hypoxia–ischemia (see Table 2); accordingly, any secondary depletion during the recovery phase would not be expected to occur.
Additional biochemical support for the finding that glucose supplementation is beneficial rather than deleterious to immature rat brain exposed to hypoxia–ischemia stems from a prior investigation in our laboratory. Yager et al. (1991) subjected immature rats to cerebral hypoxia–ischemia, after 3 h of which their brains were prepared for the determination of cytosolic and mitochondrial oxidation-reduction (redox) states. As anticipated, the cytoplasmic redox state was reduced relative to controls, whereas the mitochondrial redox state was more oxidized. Concurrent measurements of brain glucose, pyruvate, and α-ketoglutarate indicated that the upward shift in mitochondrial NAD+/NADH was the result of a depletion in substrate supply to the mitochondria. An analogous situation occurs in adult brain during severe hypoglycemia, during which cellular oxidation is observed (Bryan and Jöbsis, 1986). The alterations in the mitochondrial redox state of immature rat brain during hypoxia–ischemia mimics state-II respiration, in which substrate, rather than oxygen supply, is deficient (Chance and Williams, 1955). It follows that substrate (glucose, ketone bodies) supplementation should improve the neuropathologic outcome arising from hypoxia–ischemia in the immature rat, as shown in previous investigations (Vannucci and Mujsce, 1992; Yager et al., 1992).
In summary, the age-related paradox of the beneficial effect of hyperglycemia on hypoxic–ischemic brain damage in immature rats compared to its deleterious effect in adults likely relates to better preservation of tissue energy stores and less extensive tissue lactacidosis in the immature animals. A more rapid clearance of lactic acid via metabolism and transport in immature rats is probably a contributing factor and is presently being investigated.