
Abstract
Intraspinal microinjection of the nonspecific nitric oxide synthase (NOS) inhibitor N-nitro-L-arginine methyl ester (L-NAME) was used to determine if inhibition of NOS results in morphological changes in the rat spinal cord. Following spinal injections of 100–750 mM L-NAME (pH 7.0), 1.0–500 mM L-NAME (pH 2.5–5.4), or L-NAME + L-arginine, quantitative analysis of morphological changes revealed a positive dose-response relationship between L-NAME and neuronal loss. This effect was blocked by L-arginine and was inversely related to spinal levels of NOS enzyme activity. Results of this study have shown the importance of basal NOS activity in maintaining the structural integrity of spinal neurons. It is proposed that the effects of L-NAME on nitric oxide (NO) production leads to decreased blood flow, secondary to vasoconstriction, and a hypoxic–ischemic reaction in spinal tissue. The results suggest that a potential contributing factor to neuronal damage in pathological conditions such as spinal cord injury may be the decreased production of nitric oxide.
Recent studies have shown nitric oxide (NO) to be involved in the excitotoxic effects of glutamate and/or glutamate agonists (Dawson et al., 1991; Kollegger et al., 1993; Reif, 1993). These results are consistent with the increased production of NO following activation of the N-methyl-D-aspartate (NMDA) receptor (Garthwaite, 1991). It follows from these reports that inhibition of NO synthesis should provide neuroprotection following injury-induced release of glutamate in the central nervous system (CNS). Support for a glutamate-NO cascade in neurotoxicity comes from studies in which inhibition of nitric oxide synthase (NOS) reduced tissue damage in models of cerebral ischemia (Moncada et al., 1992; Kuluz et al., 1993; Pelligrino, 1993; Dawson, 1994; Nishikawa et al., 1994) and provided neuroprotection in models of excitotoxic neuronal injury (Nowicki et al., 1991; Dawson et al., 1991; Reif, 1993; Monada et al., 1993). Accumulating evidence, however, has raised considerable controversy over the role of NOS inhibition in neuronal protection following spinal cord injury (SCI), cerebral ischemia, and glutamate induced excitotoxicity (Pauwles and Leysen, 1992; Greenberg et al., 1993; Dawson et al., 1992; Garthwaite and Garthwaite, 1994; Buchan et al., 1994; Cohen et al., 1994).
While the role of NO as a neuroprotectant and/or neurotoxic agent remains unresolved with regard to brain and spinal injury, there is increasing support for the involvement of NO in the regulation of cerebral blood flow (CBF) (Prado et al., 1993a, b ; Macrae et al., 1993; Dirnagl et al., 1993; Iadecola 1993; Iadecola et al., 1994; Irikura et al., 1994). While progress has been made in understanding the role of NO in CBF, little is known about the relationship between NO and the control of blood flow in the spinal cord. In a study involving long-term inhibition of NOS, Blot et al. (1994) described evidence for the importance of NO in vasomotion, structure, and permeability of spinal arteries. It is suggested by these findings, it has been suggested that altered blood flow and other vascular related events in pathological conditions of the spinal cord may be related, in part, to a decrease in basal NO production. Considering the role of diminished blood flow in the disruption of spinal cord function (Kobrine et al., 1980; Young, 1985; Fehlings et al., 1989; Tator and Fehlings, 1991), and the importance of altered blood flow in producing “secondary tissue damage” following spinal injury (Tator and Fehlings, 1991; Young, 1985), additional studies are needed to help clarify the functional relationship between NO and the regulation of spinal cord blood flow (SCBF).
The above-mentioned studies underscore the evolving significance of NO as a multifunctional messenger in the brain and spinal cord as well as a need for studies examining its role in spinal cord function. Studying the function of NO in biological systems, however, is a difficult challenge since addition of this reactive free radical to in vivo systems may cause nonspecific and/or physiologically unimportant effects. To combat this problem studies often include drugs that inhibit NO synthesis. In recent years a growing number of laboratories have used drugs that inhibit NO production to study the role of NO in SCI and in the processing of sensory information (Meiler and Gebhart, 1993; Wilcox, 1993; Wu and Li, 1993). The design of most studies, however, seldom consider the effects of decreased NO production on the structural integrity of spinal tissue. The present study was therefore carried out using the technique of intraspinal microinjection of the non-specific NOS inhibitor L-NAME to determine if acute inhibition of NOS results in morphological changes in the rat spinal cord. A preliminary description of these data have been presented (Yezierski et al., 1994).
MATERIALS AND METHODS
Male Long Evans rats (250–300 g) were anesthetized with a mixture of ketamine, acepromazine, and xylazine (0.65 ml/kg, s.c). Intraspinal injections were made in each animal at spinal levels T12–L3. After a small portion of the vertebral lamina and spinous process were removed, the dura was incised longitudinally and reflected bilaterally. At injection sites, the pia matter was carefully elevated using no. 5 Dumont forceps, and small holes were made to allow penetration of the injection pipette. Following injections, the dura was reconstructed with a piece of durafilm (Codmen and Shurtleff Inc., Randolph, MA, U.S.A.). Muscles were closed in layers, and the skin closed with wound clips. Forty-eight hours postsurgery, animals were deeply anesthetized with sodium pentobarbital and perfused transcardially with 10% buffered formalin. Spinal blocks, containing segments with injection sites, were removed, and placed in fixative for 24 h, transferred to sucrose for 24 h, and cut on a freezing microtome.
Intraspinal microinjection
Stock solutions of L-NAME (RBI, Natick, MA, U.S.A.) were made using normal saline. From these solutions, dilutions of 1.0–750 mM were made for intraspinal injection. Spinal cords were injected with 100–750 mM L-NAME (pH 7.0) buffered with sodium carbonate or sodium phosphate (Dawson, 1994; Globus et al., 1995) and unbuffered 1.0–500 mM L-NAME (pH 2.5–5.4); the amount of L-NAME in these concentrations ranged from 0.03 to 20.2 g/site. These concentrations are similar to those used in studying the effects of L-NAME on spinal sensory neurons (Meiler and Gebhart, 1993; Wilcox, 1993). L-NAME was selected as the inhibitor of NOS since it has been shown to diminish brain NOS activity in a dose and time dependent manner (Traystman et al., 1995). Unbuffered solutions of L-NAME were injected in order to evaluate the often neglected acidic characteristic of L-NAME solutions.
Intraspinal injections were made using a glass micropipette (tip diameter 5–10 μm) attached to a Hamilton microliter syringe mounted on a micromanipulator (Kopf, Tujunga, CA, U.S.A.). Injections were made at two spinal levels (opposite sides of the cord and separated by three-to-four segments). Each animal, therefore, provided two injection blocks for analysis of drug effects. This design, which enabled the maximal amount of data to be obtained from each animal, has been used successfully in previous studies evaluating the effects of excitatory amino acid (EAA) agonists and antagonists on spinal neurons (Yezierski et al., 1993; Liu et al., 1994, 1995). Injection blocks that could not be analyzed because of mechanical damage, surgical complications, or damage due to histological processing were eliminated from the study. Injections were made between the dorsal vein and dorsal root entry zone at depths of 600 and 1,200 μm below the surface of the cord. These parameters positioned injection sites in the middle of the gray matter between laminae IV-VI. At each depth, 0.1 μl of L-NAME was injected (over a 60 second time interval).
Animal groups
Four injection groups were used to evaluate the effects of different drug concentrations: group 1 included L-NAME (unbuffered, pH 2.5–5.4) at concentrations of 1 (pH 5.4), 50 (pH 5.4), 100 (pH 4.5), 250 (pH 3.6), and 500 mM (pH 2.4); group 2 included L-NAME (pH 7.0) at concentrations of 100, 250, 500 and 750 mM; group 3 included animals injected with a mixture of 250 mM L-NAME (pH 7.0) + 250 or 500 mM L-arginine (pH 7.0); and group 4 included animals injected with saline vehicle (pH 2.5, 4.5 and 7.0) or L-arginine (250 and 500 mM, pH 6.4). The above concentrations of L-arginine were selected in order to reverse the effects of L-NAME by co-administering an endogenous substrate for NO synthesis. Statistical comparisons were made using analysis of variance (ANOVA) with the Student Neuman-Keuls post-hoc correction to evaluate differences between different drug concentrations in groups 1–2. Student's t-test was used to determine differences between L-NAME alone and L-NAME + L-arginine (groups 2 and 3). In the determination of statistical significance for different L-NAME concentrations and/or drug combinations, p-values of <0.01 were used.
Measurement of cNOS activity
Spinal cords used for the measurement of the constitutive form of NOS (cNOS) activity were injected with concentrations of 10–750 mM L-NAME (pH 7.0). In this analysis, the following groups were used: normal tissue (no injection), saline injected (pH 7.0), and 10, 50, 250, 500, and 750 mM L-NAME. The technique used to make these injections and the segments injected were identical to those used in the morphological evaluation of L-NAME effects (see above).
Thirty minutes following injections, spinal cords were removed and placed in liquid nitrogen. cNOS activity was determined using a modification of the method described by Bredt and Snyder (1989). Briefly, tissue blocks containing injected material were homogenized in 400 μl of cold buffer containing 20 mM Hepes (pH 7.5), 0.5 mM EGTA, 1 mM dithiothreitol, and 0.32 M sucrose at 23,000 rpms for 40 s with a Polytron (Brinkmann, Westbury, NY, U.S.A.). Samples were centrifuged at 20,000 g for 20 minutes. One-hundred microliters of supernatant were incubated at 37°C for 45 min with a reagent buffer containing 20 mMHEPES (pH 7.4), 0.5 mM EGTA, 1 mM DDT, 0.32 M sucrose, 0.5 mM Ca+2 (1 μM free Ca+2), 200 μM NaDPH, 1 μM arginine, and 0.1 μCi/ml 3H-L-arginine. The reaction was stopped with 2 ml of ice-cold 20 mM Hepes (pH 5.5) containing 2 mM EDTA. Samples were applied to a Doewx AG 50W (Na+ form) column to remove the 3H-L-arginine. Columns were washed with 2 ml of distilled water to elute the 3H-citrulline. Radioactivity was counted in a Packard counter and cpms were converted to dpms using 3H-quenched standards. Levels of 3H-citrulline were computed after subtracting the blank, which represented nonspecific radioactivity in the absence of enzyme. Protein determinations were done using a Lowry assay. Values of cNOS activity are presented as dpm/μg protein (Zhang et al., 1995).
Histological evaluation
Morphological changes following survival periods of 48 h were evaluated from serial 50 μm cross-sections. Frozen sections were cut on a freezing microtome, mounted on gelatin-dipped slides, and stained with cresyl violet. Reconstructions of tissue damage were made with the aid of an overhead projector and camera lucida. To quantify the effects of L-NAME, an index of maximal neuronal loss was determined at the epicenter of injection sites using computerized image analysis. In previous studies, this method of epicenter analysis has proven reliable and effective in evaluating statistical differences between experimental groups injected with EAA agonists (Liu et al., 1994, 1995).
Sections were initially examined at 100–250× to determine the boundary between intact and damaged tissue. Criteria for this determination were based on the absence of neurons and the presence of degenerating neuron profiles and inflammatory cells. Neurodegeneration was characterized by pyknotic nuclei and large, spherical, darkly stained cells in a zone surrounding the necrotic area. The marginal zone around the damaged area typically contained inflammatory cells and large phagocytic cells that resembled macrophages. For the determination of neuron loss, sections were displayed on a video screen. Using a drawing tablet, the area of intact gray matter was outlined. This technique of measuring intact gray/white matter is similar to that described in previous studies evaluating the neuroprotective effects of EAA antagonists, amount of neuronal loss following ischemic injury, and effects of EAA agonists on spinal neurons (Bunge et al., 1994; Liu et al., 1994, 1995; Wrathall et al., 1994).
Areas of neuronal loss in animals injected with L-NAME alone or L-NAME + L-arginine were determined from 20 serial sections taken from the injection epicenter. Areas of neuronal loss were calculated by subtracting the intact gray matter areas of injected animals from those of uninjected control animals. In this analysis, control gray matter areas were obtained from sections from five weight-matched control animals. A standard curve was constructed for the rostral, middle, and caudal portions of spinal segments L12–S1. This enabled areas from exact cord levels where injections were made to be used in the calculation. Statistical comparisons between different drug treatment groups were made using the paired and independent Student's t-test. Additionally, when appropriate, SYST AT and SAS general linear model procedures were used in the analysis of data. Two-way ANOVA was used to compare drug effects between different treatment groups. Appropriate post-hoc tests were used to correct for multiple comparisons. For different drug doses a repeated measures of ANOVA was used to test overall ‘between subject’ effects and to assess ‘within subject’ effects.
RESULTS
Effects of L-NAME on spinal gray matter
Forty-four injection blocks from 28 rats were used for analyzing the morphological effects of different drug concentrations and/or combinations. All spinal cords injected with L-NAME concentrations of 50–500 mM (unbuffered) and 100–750 mM (buffered) had neuronal loss (Figs. 1 and 2). The area of injury was characterized by the presence of macrophages and a dense-to-moderate inflammatory cell response, including the presence of extra- and intravascular white blood cells. Blood was observed around the site of injection in 25% of the animals, suggesting damage to the microvasculature. At the border and outside the region of cell loss were scattered darkly-stained neurons of various sizes. In this transition zone were also abnormally swollen, dark, neurons with swollen dark, deteriorated nuclei, and inflammatory cell infiltration.
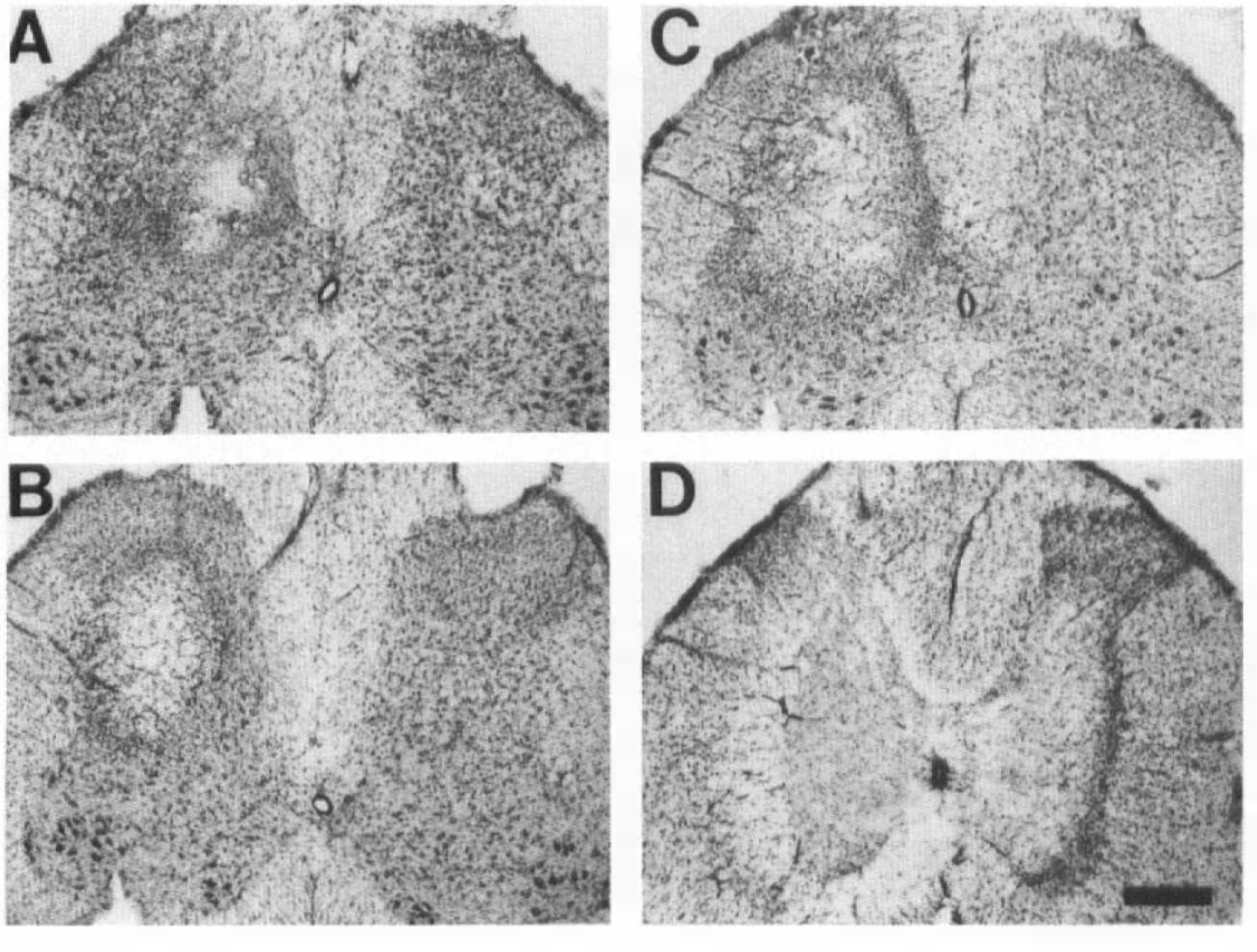
Dose-response effects of L-NAME on gray matter in the rat spinal cord. Unilateral intraspinal injections of L-NAME (unbuffered) were made with solutions of 50 (
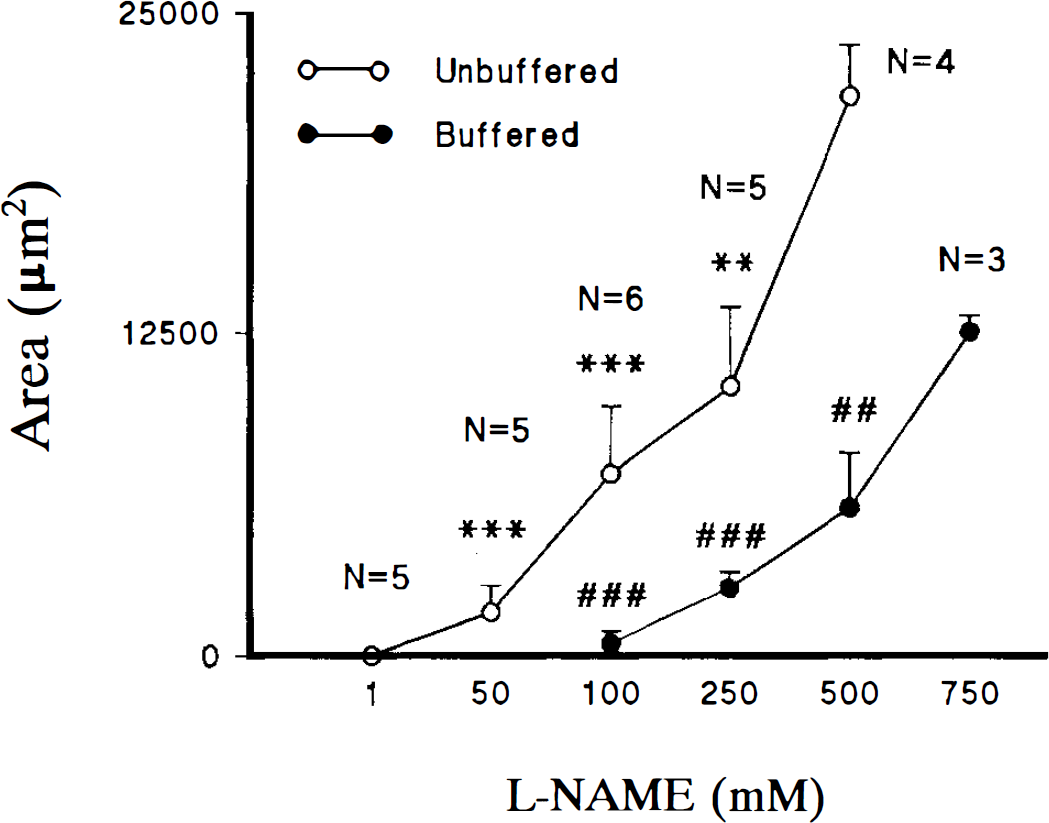
Dose-response relationship of L-NAME versus neuronal loss following intraspinal injections of buffered and unbuffered solutions. Neuronal loss (μm2) is represented on the y-axis and concentration (mM) of L-NAME on the x-axis. The number of tissue blocks (N) is indicated for each concentration of unbuffered solutions. Three tissue blocks were used for each concentration of buffered L-NAME. Five different concentrations of unbuffered (1–500 mM) and four buffered (100–750 mM) solutions of L-NAME were used in the evaluation. Error bars represent the standard deviation around the mean. Statistical comparisons for each L-NAME concentration were evaluated against 500 mM (unbuffered) and 750 mM (buffered) solutions (**, ## = p < 0.01; ***, ### = p < 0.001).
Quantitative analysis of neuronal loss provided evidence for a positive dose-response relationship between L-NAME and the area of neuronal loss. These relationships were found for both buffered and unbuffered L-NAME solutions (Fig. 2). Examples of dose-dependent neuronal loss following injections of 50–500 mM (unbuffered) L-NAME solutions are shown in Fig. 1. One-way ANOVA showed significant differences in the area of neuronal loss for the different concentrations of buffered and unbuffered L-NAME solutions (Fig. 2).
As shown in Fig. 2, the use of buffered and unbuffered solutions resulted in a shift to the right in the dose-response curve for buffered L-NAME solutions. These data suggest that pH contributed to the extent of cell loss in spinal cords injected with unbuffered L-NAME solutions. This was confirmed in animals injected with saline at pH 2.5, 4.5, and 7.0. Animals injected with saline solutions of pH 7.0 showed no signs of neuronal loss, while increased damage was found with solutions of pH 4.5 and 2.5, respectively (Fig. 3). In these latter animals, however, the extent of damage was never equivalent to that observed following injections of L-NAME (buffered or unbuffered). The main characteristic of injections with saline solutions was the presence of inflammatory cells (Fig. 3). Due to the negligible amount of neuronal loss following saline injections, quantitative evaluation of sections from these animals was not undertaken. The presence of minimal (pH 4.5) and no (pH 7.0) damage following saline injections supports the conclusion that pH contributed to the extent of cell loss in spinal cords injected with L-NAME solutions of pH < 4.5. Furthermore, the absence of neuronal loss with 1 mM L-NAME (pH 5.4) and saline (pH 7.0) indicates that the intraspinal injection technique contributed minimally, if at all, to the outcome measurement of neuronal loss.
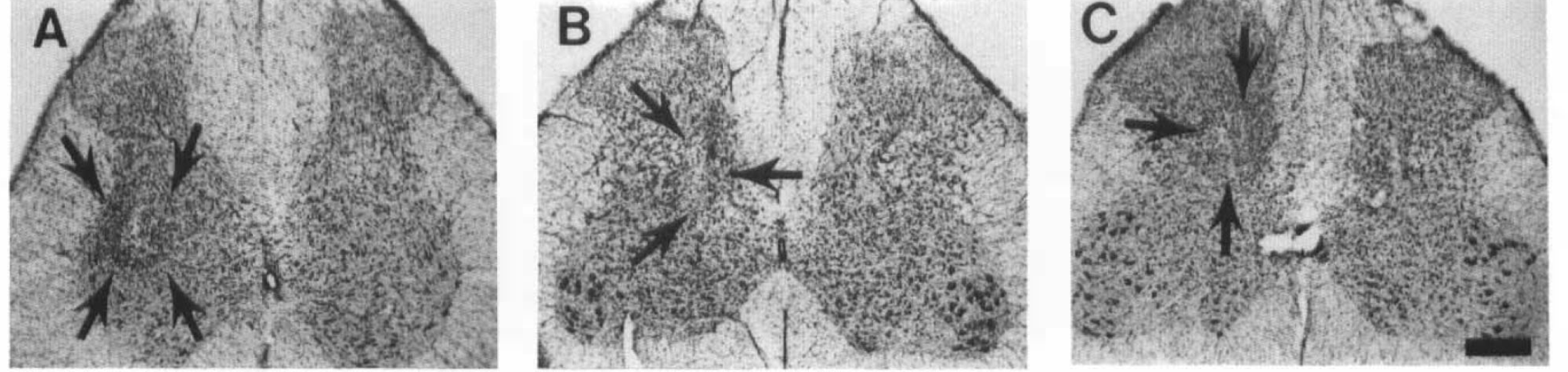
Effects of control injections of saline and L-arginine on the spinal gray matter.
Effects of L-NAME + L-arginine
When 250 mM L-NAME (pH 7.0) was combined with 250 mM L-arginine (pH 7.0), a partial block of neuronal loss was obtained (Figs. 4 and 5). Increasing the concentration of L-arginine to 500 mM resulted in an area of neuronal loss that was significantly less than that observed with 250 mM L-NAME (p < 0.01) (Figs. 4 and 5). The dose-response relationship of the L-arginine blockade of L-NAME-induced neuronal loss is summarized in Fig. 5. The effects of L-arginine at 250 mM were evaluated for eight injection blocks (four animals); 500 mM L-arginine was also evaluated for eight blocks (four animals). Examples of the effects of 250 mM L-NAME + 250 L-arginine and 250 mM L-NAME + 500 mM L-arginine are shown in Fig. 4. Finally, although there was an accumulation of inflammatory cells at injection sites, there was no evidence of neuronal loss with injections of 250 or 500 mM L-arginine (pH 7.0). These results further support the conclusion that the injection protocol itself did not contribute to the pattern of neuronal loss following intraspinal injections.
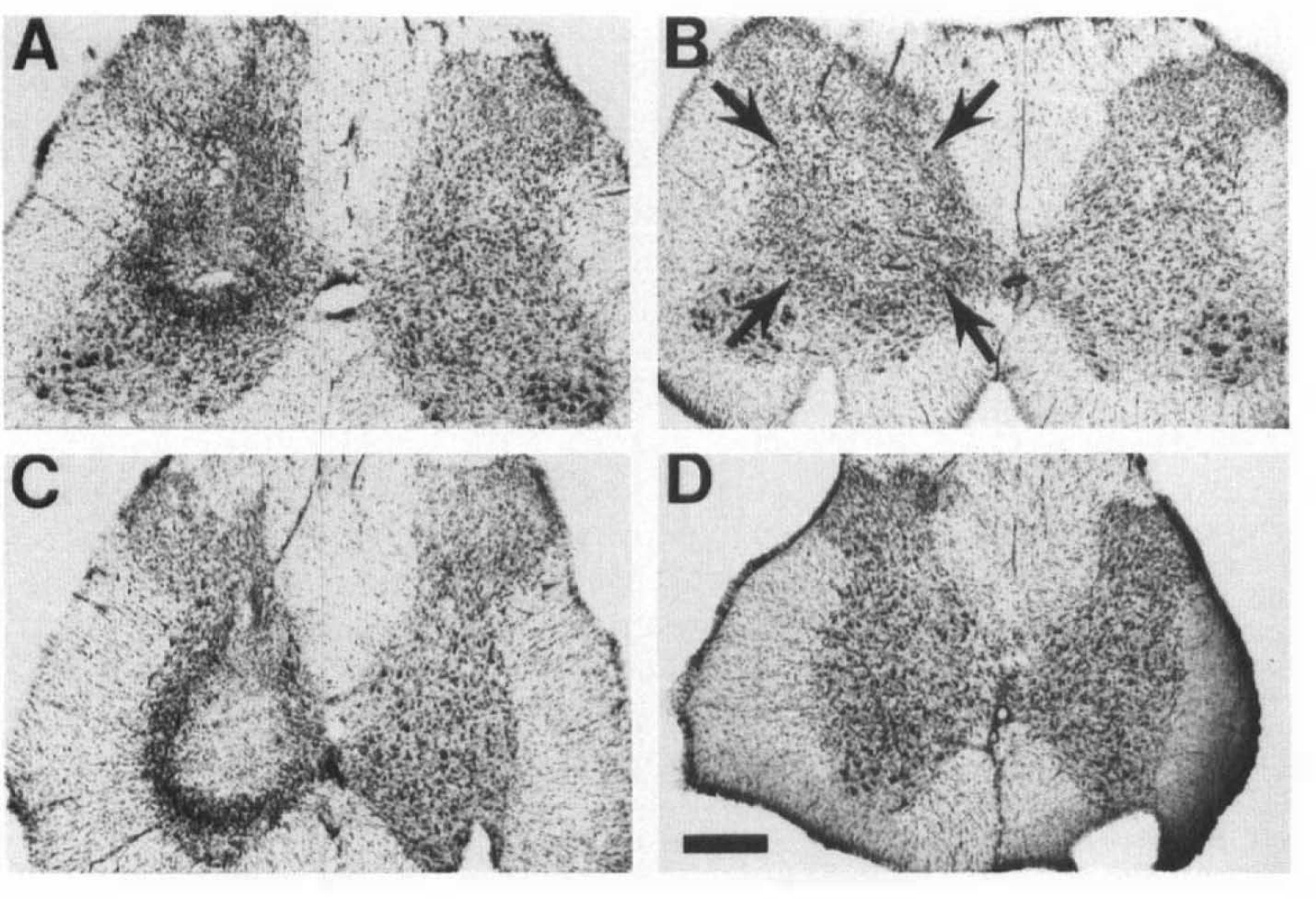
Blockade of L-NAME effects with combined injections of L-NAME + L-arginine.
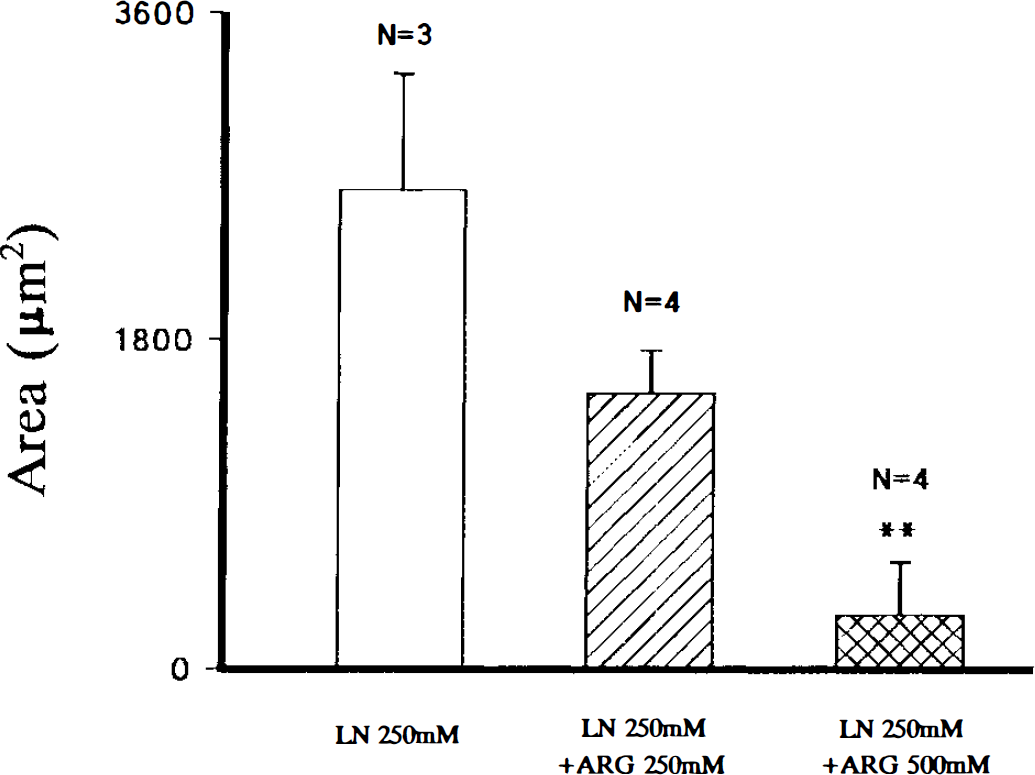
Dose-response relationship for the effects of 250 mM and 500 mM L-arginine (ARG) injected in combination with 250 mM L-NAME (LN). A partial blockade was found with 250 mM L-arginine, whereas a significant decrease (compared against 250 mM L-NAME) in the area of neuronal loss (μm2) was found with 500 mM L-arginine (** = p < 0.01).
cNOS enzyme activity
Thirty-eight spinal cord blocks (19 animals) were used in the analysis of cNOS enzyme activity. This evaluation was carried out for injected (following intraspinal injections of buffered L-NAME or saline) and uninjected spinal cord tissue. In order to ensure the consistency of measurements, special efforts were made to keep the size of tissue blocks constant. The average length of tissue blocks used in this analysis was 0.41 ± 0.06 cm. An important finding in this evaluation was the similarity of cNOS values at the two levels of the cord examined, i.e., L1–2 and L3–4. These segments were the same as those used in the evaluation of morphological damage. Additionally, no significant differences were found in cNOS levels in uninjected versus saline-injected tissue. The mean value of cNOS activity (expressed as dpm/μg protein) in spinal blocks injected with saline was 75.9 ± 4.6, while that for uninjected tissue measured 73.9 ± 15.8. Together, the mean value of cNOS activity in these animals was 74.9 ± 1.5 dpm/μg protein. Since there was no L-NAME inhibition of cNOS in these tissue blocks, these values represent baseline cNOS activity at the two levels of the cord (L1–2 and L3–4) used in the evaluation of morphological changes. The similarity of these values also supports the conclusion that intraspinal injection of fluid volumes comparable to those used with L-NAME was not responsible for changes in cNOS activity.
As shown in Fig. 6, there was a dose-dependent decrease in cNOS activity with increasing doses of L-NAME. The mean values of cNOS (expressed as dpm/μg protein) for different doses of L-NAME were 31.4 ± 7.1 (10 mM), 14.9 ± 2.6 (50 mM), 6.9 ± 2.6 (250 mM), 7.4 ± 2.2 (500 mM), and 3.4 ± 2.8 (750 mM). There were significant differences (p < 0.001) when comparing enzyme activity between baseline (uninjected and saline-injected blocks) and all L-NAME concentrations (Fig. 6).
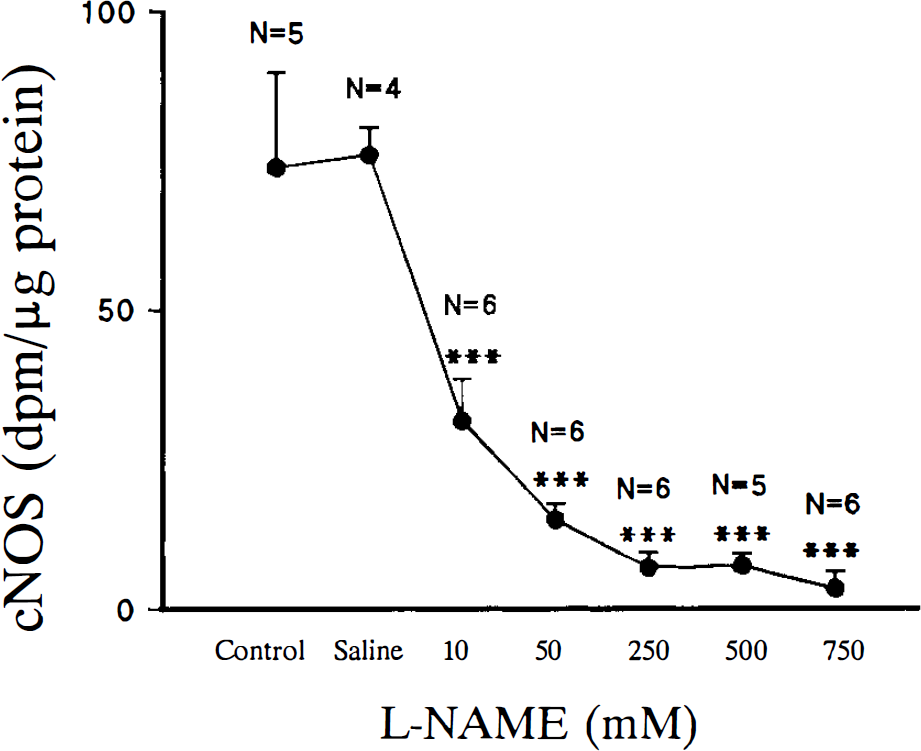
Effects of five concentrations (10–750 mM) of L-NAME on the constitutive form of the enzyme nitric oxide synthase (cNOS) measured 30 min after intraspinal injection. Enzyme activity measured in dpm/μg protein is shown on the y-axis and the concentration of L-NAME (buffered) is shown on the x-axis. The number of spinal blocks (N) used in the analysis is shown for each concentration of L-NAME. Control injections of buffered saline were made using the same injection protocol as used for L-NAME. Values from saline injected and uninjected spinal blocks were used to obtain baseline levels of cNOS activity. Significant differences (compared against saline or control blocks) were found for all concentrations of L-NAME (*** = p < .001).
DISCUSSION
Results of the present study show that intraspinal injection of the nonspecific, competitive NOS inhibitor L-NAME produces a dose-dependent neuronal loss that is blocked by L-arginine and is inversely related to spinal levels of cNOS activity. These results support the conclusion that L-NAME-induced neuronal loss is related to inhibition of NO synthesis and demonstrate the importance of basal NO production in maintaining the structural integrity of spinal neurons. Furthermore, these results are consistent with those of a recent report showing structural damage in the spinal cord following long-term ingestion of L-NAME (Blot et al., 1994).
An important question in this study was by what mechanism does L-NAME cause neuronal loss. Due to the absence of/or minimal neuronal loss following injections of control solutions, i.e., buffered saline, 250 and 500 mM L-arginine, and low concentrations of L-NAME, it is concluded that the injection technique, segmental levels injected, and the protocol used in processing spinal cords did not contribute or contributed minimally to the outcome measurement of neuronal loss. Results of the study, therefore, support the conclusion that neuronal loss is a direct result of NOS inhibition by L-NAME. Since cNOS activity was inversely related to the amount of neuronal damage, it is also likely that the dose-dependent neuronal loss is related to decreased basal levels of NO production.
Assuming the above conclusions to be correct, one explanation for the observed effects is the influence of decreased NO production on SCBF. Basal release of NO is a critical factor for vasodilation (Prado et al., 1992), blood flow (Macrae et al., 1993; Iadecola et al., 1994), and anti-aggregation of platelets (Radomski et al., 1987). Accordingly, a decrease in NO production could lead to vasoconstriction and platelet aggregation, causing a decrease in tissue blood supply, i.e. ischemia. Consistent with this hypothesis are a number of reports describing decreased CBF after systemic administration of NOS inhibitors (Pelligrino, 1993; Dawson, 1994). Alterations in CBF and morphology of arterial blood vessels leading to pathological changes consistent with focal brain ischemia have also been reported following administration of NOS inhibitors (Prado et al., 1992). Based on available evidence, it is suggested that the tissue-damaging effects of intraspinal L-NAME, a nonspecific competitive inhibitor of neuronal and endothelial NOS (Traystman et al., 1995), are likely to involve a vascular component.
As discussed above, chronic ingestion of L-NAME in the hypertensive rat was concluded to result in compromised SCBF, and NO might be a key factor in regulating vasomotor tone in the spinal cord (Blot et al., 1994). An important observation in this study was the significant difference between brain versus spinal cord damage. The incidence of brain lesions in L-NAME-treated animals was only 30% versus 100% in the spinal cord. This result suggests a differential sensitivity of spinal versus cerebral vasculature to NO regulation. Consistent with this hypothesis is the observation that acute intra-carotid infusion of L-NAME produces only subtle pathological changes in the brain (Prado et al., 1992). This is in marked contrast to the effects of L-NAME achieved with intraspinal injections (present study). An alternative explanation to this apparent dichotomy may be related to differences in the complexity of collateral circulation in the brain versus spinal cord and, thus, differences in the safety factor for compromised blood flow in these two regions of the CNS. It should be noted, however, that following cerebral ischemia, inhibition of NO synthesis increases the area of tissue damage, suggesting limitations to the safety factor provided by collateral circulation (Buchan et al., 1994; Dawson et al., 1992; Kuluz et al., 1993; Prado et al, 1993a).
In the present study, petechial bleeding was found at the epicenter of L-NAME injection sites, suggesting that decreased NO production was responsible for structural damage to the spinal vasculature. Although the presence of blood might have resulted from the intraspinal injection technique, similar findings have been rarely observed following intraspinal injections of EAA agonists (Yezierski et al., 1993; Liu et al., 1994, 1995). Consistent with the hypothesis of damage to spinal vessels, preliminary observations have shown that intraspinal injections of buffered L-NAME (250 mM) result in the breakdown of the spinal-blood brain barrier (sBBB) and evidence of neuronal damage 15 min after injection (Yezierski and Dietrich, unpublished observations). It is, therefore, possible that blood-borne agents, which get into the tissue secondary to breakdown of the sBBB, also contributed to the neuronal damage observed in the present study.
In recent years, the role of NO in neuronal injury has received support from in vitro observations that NMDA-mediated neurotoxicity is blocked by inhibition of NO synthesis (Nowicki et al., 1991; Dawson et al., 1991; Kollegger et al., 1993; Reif, 1993). In cerebral ischemia, however, systemic administration of NOS inhibitors has led to results ranging from attenuation to augmentation of brain infarction size (Prado et al., 1993a; Pelligrino, 1993; Buchan et al., 1994; Dawson, 1994). Further complicating the role of NO in brain ischemia, is the dual effect of NOS inhibitors on NMDA-mediated neurotoxicity that was described in a recent study. In that study, NOS inhibitors reduced the area of ischemia caused by high doses of NMDA, while no reversal occurred with low doses of NMDA (Globus et al., 1995). Increased tissue damage has also been reported following NOS inhibition (Kuluz et al., 1993), and L-arginine, a precursor of NO synthesis, decreases the area of ischemic brain damage providing support for NO as a neuroprotectant (Morikawa et al., 1992). Similar results were described following NMDA-induced corpus striatum damage (Prado et al., 1993a). A dual role of NO, involving hemodynamic (endothelial NO) and neurotoxic (neuronal NO) mechanisms, was recently proposed to help explain the controversy of NO in focal brain ischemia (Dalkara et al., 1994).
In many of the studies mentioned above, NOS inhibitors were administered following brain injury. In the present study however, L-NAME was micro-injected into the spinal cord in areas where only minimal tissue damage was present. The present study, therefore, examined the effects of basal NOS activity on the normal morphology of spinal neurons. The fact that inhibition of NOS activity resulted in significant damage to spinal neurons underscores a potentially important aspect of NO function, which should be considered in studies evaluating the effects of NOS inhibitors in the spinal cord.
In addition to the dose-dependant effects of unbuffered L-NAME solutions, an often neglected characteristic of L-NAME solutions, i.e. pH, was found to exert an additive effect on the morphological damage observed in the present study. Animals injected with buffered L-NAME solutions support the conclusion that decreased NOS activity, and not pH effects alone, was responsible for the neuronal loss observed in the present study. The pH-shift in the dose-response curve, however, supports the well-documented effects of low pH on brain tissue (Kraig et al., 1987; Tombaugh and Sapolsky, 1993). It can, therefore, be concluded that high concentrations of L-NAME solutions, administered without adjustments for pH, will cause tissue damage by a mechanism related to inhibition of NOS and, additionally, by the effects of low pH. These conclusions were confirmed in control experiments where saline solutions of pH 2.5–7.0 were injected into the cord parenchyma. Although tissue damage was observed, it was minimal compared to the damage following L-NAME injections. These data emphasize the importance of monitoring pH and using pH controls in studies in which L-NAME is evaluated in the spinal cord. It is also recommended that, following administration of drugs inhibiting NOS, that the morphological characteristics of tissue be examined.
In the present study, cNOS activity was measured within a 30-min window after injection, and was found to be reduced in a dose-dependent fashion with increasing doses of L-NAME. These results, using a nonspecific inhibitor of NOS, are consistent with results obtained for brain (Traystman et al., 1995). Furthermore, since significant differences in cNOS activity were not observed at the two different levels of the cord examined, support is given to the conclusion that the present results are not due to a differential segmental distribution of cNOS (at least for segmental levels L1–2 and L3–4). The distribution of neurons contributing to the spinal levels of cNOS include neurons positive for NOS-like immunoreactivity in laminae I-IV and X (Bredt et al., 1991; Dun et al., 1993; Saito et al., 1994). This distribution is consistent with NO having a role in a variety of physiological functions, including an influence on local blood flow.
In conclusion, results of the present study have shown the importance of basal NO production in the maintenance of the structural integrity of spinal neurons. Results of this initial study—examining the effects of intraspinal L-NAME injections and comparing them with available evidence from studies showing an involvement of NO in CBF—suggests that a potential contributing factor to neuronal damage in pathological conditions of the spinal cord might be the decreased synthesis of NO. Since decreased NO production has been linked to decreased levels of neuronal activity (Dirnagl et al., 1993), and decreased blood flow (Iadecola, 1993), a decrease in NO levels secondary to traumatic or ischemic spinal injury could lead to a further reduction in SCBF and exacerbation of effects from the initial injury. Preliminary results from our laboratory support this hypothesis by showing that systemic injections of L-NAME lead to dose-dependent reductions in SCBF, measured with Doppler flowmetry (Yezierski, unpublished observations). Although many factors, e.g., metabolic, biochemical, and hemodynamic, are associated with traumatic and ischemic CNS injury, the present study reveals, for the first time, that inhibition of NO synthesis can, by itself, have a significant effect on the structural integrity of spinal neurons. Future studies related to the effects of neuronal and endothelial NO on SCBF are needed to better understand the mechanism of neuronal loss following intraspinal L-NAME injection.
Footnotes
Acknowledgment:
The authors would like to thank Maurice Duharte for assistance in preparation of the manuscript, Hector Dancausse for technical assistance, Rob Camarena for photographic assistance, and Dr. Mordeci Globus for comments on the manuscript. This work was supported by funds from NS27127 (W.D.D.) and NS28059 (R.P.Y.), The Miami Project to Cure Paralysis, and by funds from the U.S. Army Research Office (DAAH04-94-0425).