
Abstract
This study ascertains whether high extracellular glutamate contributes to the initiation of spreading depression (SD) by K+. Two microdialysis probes, each incorporating an electrode to record the extracellular direct current (DC) potential at the elicitation site, were implanted symmetrically in the cortex of anesthetized rats. Recurrent SD was triggered by perfusion of 130 mM K+ through the microdialysis probe for 20 min. On one side, this medium was supplemented with increasing concentrations of glutamate (0.1–1 mM) or of the selective glutamate uptake inhibitor L-trans-pyrrolidine-2,4-dicarboxylate (L-trans-PDC; 1–10 mM). The effects of L-trans-PDC on extracellular glutamate and basal DC potential were studied in separate experiments. Application of K+ for 20 min consistently elicited five to seven waves of SD. Increasing the concentration of glutamate in the perfusion medium did not alter SD elicitation. Application of L-trans-PDC concentration dependently increased the dialysate levels of glutamate (by ∼ 19-fold with 10 mM L-trans-PDC) but, unexpectedly, reduced SD elicitation. These data do not support the hypothesis that SD is elicited because high extracellular glutamate resulting from exocytosis and/or reversal of glutamate uptake depolarizes adjacent neurons. As SD elicitation requires activation of N-methyl-D-aspartate (NMDA) receptors, these results also illustrate that sensitivity of a pathological or experimental event to NMDA receptor antagonists does not necessarily imply involvement of increased extracellular glutamate. This does not rule out a selective action of glutamate, transiently released from presynaptic vesicles, on immediately juxtaposed postsynaptic receptors.
Keywords
Spreading depression (SD) is a local, transient suppression of electrical activity associated with depolarization, propagating slowly across the cerebral cortex or other gray matter regions (Leão, 1944; Burěs et al., 1974). Both experimental and clinical evidence point to cortical SD as the underlying mechanism of the migraine aura (i.e., neurological disturbances associated with the development of a migraine attack) (Lauritzen, 1994). Recurrent SD also contributes to neuronal damage, subsequent to experimental focal ischemia (Iijima et al., 1992), presumably by producing marked disruption of ionic homeostasis, acidosis, enhanced energy demand, and neurotransmitter efflux in regions where residual blood supply can only sustain basal ionic homeostasis (Obrenovitch, 1995). With regard to humans, spontaneous cortical SDs were reported in patients with severe head injury (Mayevsky et al., 1995), but SDs could not be elicited in the neocortex of patients undergoing cortical resections for intractable epilepsy, even in the absence of general anesthesia (McLachlan and Girvin, 1994).
Despite numerous studies prompted by the potential clinical relevance of SD, the mechanisms that trigger and propagate this event remain elusive. Experimental elicitation of SD appears to depend on local accumulation of extracellular K+ (Somjen, 1979; Gardner-Medwin, 1981; Reid et al., 1988), but high extracellular glutamate may be also a triggering factor (Van Harreveld and Fifková, 1970; Lauritzen and Hansen, 1992): (a) Both elicitation and propagation of SD are exclusively sensitive to competitive and noncompetitive antagonists of the N-methyl-D-aspartate (NMDA) receptor complex (Lauritzen and Hansen, 1992; Sheardown, 1993); (b) glutamate is released during SD (Van Harreveld and Fifková, 1979; Obrenovitch et al., 1995); and (c) excitatory amino acids directly depolarize glial cells (Bowman and Kimelberg, 1984) and can initiate SD in brain tissue slices (Lauritzen et al., 1988). Paradoxically, we have found that extremely high concentrations of glutamate must be applied to the rat striatum to elicit SD in vivo (Obrenovitch et al., 1994) and recently demonstrated that a high extracellular level of glutamate is not the driving force sustaining cortical SD propagation (Obrenovitch and Zilkha, 1995).
The purpose of this study was to ascertain whether high extracellular glutamate contributes to the initiation of SD by K+. Our strategy was to compare changes in the extracellular direct current (DC) potential induced by K+ alone with those produced by an identical K+ stimulus on which were superimposed controlled, gradual increases of extracellular glutamate. We demonstrate that, as for SD propagation, high extracellular glutamate is not required for SD elicitation by K+. Some of these results were published previously in abstract form (Urenjak et al., 1995).
MATERIALS AND METHODS
Animal preparation—bilateral implantation of microdialysis electrodes
Twenty-seven adult male Sprague-Dawley rats (weight 306 ± 31 g, mean ± SD; Bantin & Kingman, Grimston, Hull, U.K.) were used, with food and water available ad libitum. All animal procedures used were in strict accordance with the British Home Office Guidelines and specifically licensed under the Animals (Scientific Procedures) Act 1986. Anesthesia was induced and maintained during surgery with halothane (2.5 and 1.5–2.0%, respectively) in O2/N2O (1:1), with the animal breathing spontaneously. Concentric microdialysis probes incorporating a recording electrode in the inlet tube (Fig. 1) (microdialysis electrode ME-H2; Applied Neuroscience Ltd., London, U.K.) (Obrenovitch et al., 1993, 1994) were implanted symmetrically in the frontoparietal cortex (coordinates: 1.3 mm posterior to bregma, 2 mm lateral, and 2 mm deep from the dural surface). Unless otherwise stated, microdialysis probes were perfused with artificial CSF (ACSF) (composition in mM: NaCl 125, KCl 2.5, MgCl2 1.18, CaCl2 1.26, NaH2PO4 0.2; pH 7.3 adjusted with 1 M NaOH) at 1 μl min−1 with a syringe pump (CMA/100; CMA/Microdialysis, Stockholm, Sweden). The microdialysis probes were used to trigger repetitive SDs by switching from normal ACSF to a medium containing 130 mM K+. This perfusion medium was used by itself for SD elicitation on the control side, but was supplemented on the contralateral side with increasing concentrations of exogenous glutamate or of a selective glutamate uptake inhibitor (see Experimental Procedures). SD initiation at the elicitation sites was precisely recorded with the electrode incorporated within the microdialysis probe.
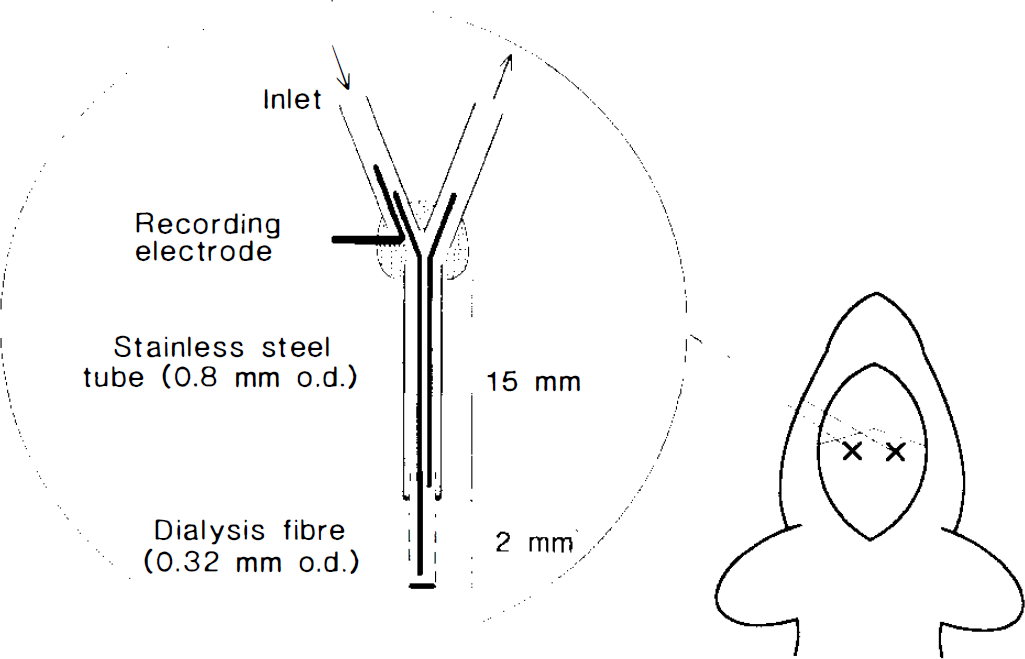
Schematic representation of the rat skull showing the bilateral location of the microdialysis electrodes used to record K+-evoked spreading depression (SD) directly from the elicitation sites. In each experiment, the 130 mM K+ medium for triggering SD was supplemented on one side with increasing concentrations of glutamate or of the selective glutamate uptake inhibitor L-frans-pyrrolidine-2,4-dicarboxylate.
A femoral artery was catheterized for continuous monitoring of arterial blood pressure and a vein for potential drug administration and induction of cardiac arrest. To minimize any possible interference of halothane anesthesia with the processes under study (Piper et al., 1991; Verhaegen et al., 1992; Saito et al., 1993), once the surgical procedure had been completed, the depth of anesthesia was carefully controlled by monitoring EEG and arterial blood pressure, and the concentration of halothane in the breathing mixture kept to a minimum (0.8–1.2%). Body temperature was maintained at 37.0°C throughout the experiment.
Recording of extracellular DC potential and EEG
The DC potential and EEG were derived from the potential between the electrode built into the probe and a chlorided silver reference electrode placed under the scalp (Obrenovitch et al., 1993). The signals were first amplified (×10) with a multichannel, high-impedance input preamplifier (NL834; Neurolog System, Digitimer Ltd., Welwyn Garden City, U.K.). With each channel, the alternating current component in the 1- to 30-Hz window, amplified 6,000–8,000 times, provided EEG and the DC component, the DC potential. A dedicated application program allowed all parameters to be continuously acquired, displayed, and stored (Obrenovitch et al., 1989). The DC potentials were converted to absolute values (millivolts) from prior calibration.
Experimental procedures
Experiments were performed at least 2 h after electrode implantation. The aim of the first series was to investigate whether perfusion of increasing concentrations of glutamate through the microdialysis probe potentiated SD elicitation evoked by K+ at the same tissue site. Four episodes of repetitive SD were produced on the control side by switching the perfusion medium from normal ACSF to a solution containing 130 mM K+ and 2.5 mM Na+ for 20 min, using a liquid switch (CMA/110). Contralaterally, the K+ medium was supplemented with increasing levels of glutamate (0, 0.1, 0.25, and 1 mM). This range of concentration was selected from previous experiments, which showed that the dialysate concentration of glutamate reached a maximum of ∼30 μM when SD was elicited by K+ (Zilkha et al., 1995) and ∼15 μM during its propagation (Obrenovitch et al., 1995). Each K+ stimulus was followed by 40 min of recovery (i.e., perfusion with normal ACSF). Preliminary experiments had shown that such a high concentration of K+ (130 mM) was required to consistently evoke periodic SD under our experimental conditions. This agrees with previous work from other laboratories (Szerb, 1991; Herreras and Somjen, 1993; Saito et al., 1993). A previous study also showed that 130 mM K+ was a submaximal stimulus for SD initiation; i.e., the frequency of SD elicitation was larger when 160 mM K+ was perfused through a microdialysis electrode (Taylor et al., 1994).
As potent uptake mechanisms may efficiently clear exogenous glutamate from the extracellular space (Erecinska et al., 1986), in the second series, endogenous glutamate was elevated at the site of SD elicitation by increasing concentrations of L-trans-pyrrolidine-2,4-dicarboxylate (L-trans-FDC; Tocris Cookson, Bristol, U.K.) as 130 mM K+ was applied. L-trans-FDC is a competitive inhibitor of the Na+-dependent high-affinity uptake of excitatory amino acids (Griffiths et al., 1994) with virtually no action on ionotropic glutamate receptors (Bridges et al., 1991). Five K+ challenges were applied in these experiments, supplemented with 0, 1, 2.5, 5, and 10 mM L-trans-FDC on one side. In both series, the control side was alternately chosen to be left or right, and all microdialysis perfusion media were isosmotic, with their pH adjusted to 7.3.
The concentration range of L-trans-FDC used in the second series had been determined from separate experiments where only a single microdialysis probe was implanted and extracellular glutamate changes continuously monitored. After a few pilot experiments, the same concentrations of L-trans-FDC as those used in the second series were perfused for 20 min. The recovery period was shortened from 40 to 15 min in these experiments because this delay was sufficient for dialysate glutamate levels to normalize. Glutamate concentration in the dialysate was determined by on-line fluorometric detection of NADH resulting from the reaction of glutamate and NAD+ catalyzed by glutamate dehydrogenase (Obrenovitch et al., 1990). In brief, a peristaltic pump (Minipulse 3, 10 μl min−1 flow rate; Gilson France, Villiers Le Bel, France) mixed the enzymatic reagent with either a standard solution of L-glutamate (20 μM) or the brain dialysate as it emerged from the implanted microdialysis probe. The enzymatic reaction developed in a polyethylene tubing (0.4-mm i.d.; Portex Ltd., Hythe, U.K.) through which the reagent/dialysate solution flowed to the fluorometer. The dead volume of this tubing permitted a reaction time of ∼10 min. Changes in NADH were detected using a fluorescence spectrophotometer with a 4-μl flow cell (F 1050, excitation 345 nm, emission 455 nm; Merck Hitachi, Darmstadt, Germany).
Data presentation and analysis
To conform to previous reports, the representative recordings of SD initiation in Figs. 2 and 4 are such that the polarity of the DC potential was reversed, with depolarization producing an upward deflection. To facilitate comparison of the data, the DC potential sequences presented in these figures were aligned by setting the 2-min period preceding changes in perfusion medium to 0 mV. As changes in the magnitude of the elicitation of recurrent K+-induced SD may be characterized by an alteration in the number and/or magnitude of SD, for each K+ stimulus, SD elicitation was quantified by calculating the cumulative area (mV min) of the corresponding peaks. All values in Results are means ± SD. Statistical analysis was by Student's t test (paired or unpaired). The log concentration-response relationships in Fig. 3 were fitted by the Gauss-Newton method (ASYST; Keithley,
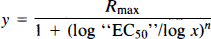
where x is dialysate concentration of drug producing responses of amplitude y, Rmax is maximal responses, “EC50” is estimated EC50 (i.e., drug concentration in the ACSF producing half-maximal responses), and n is slope parameter.
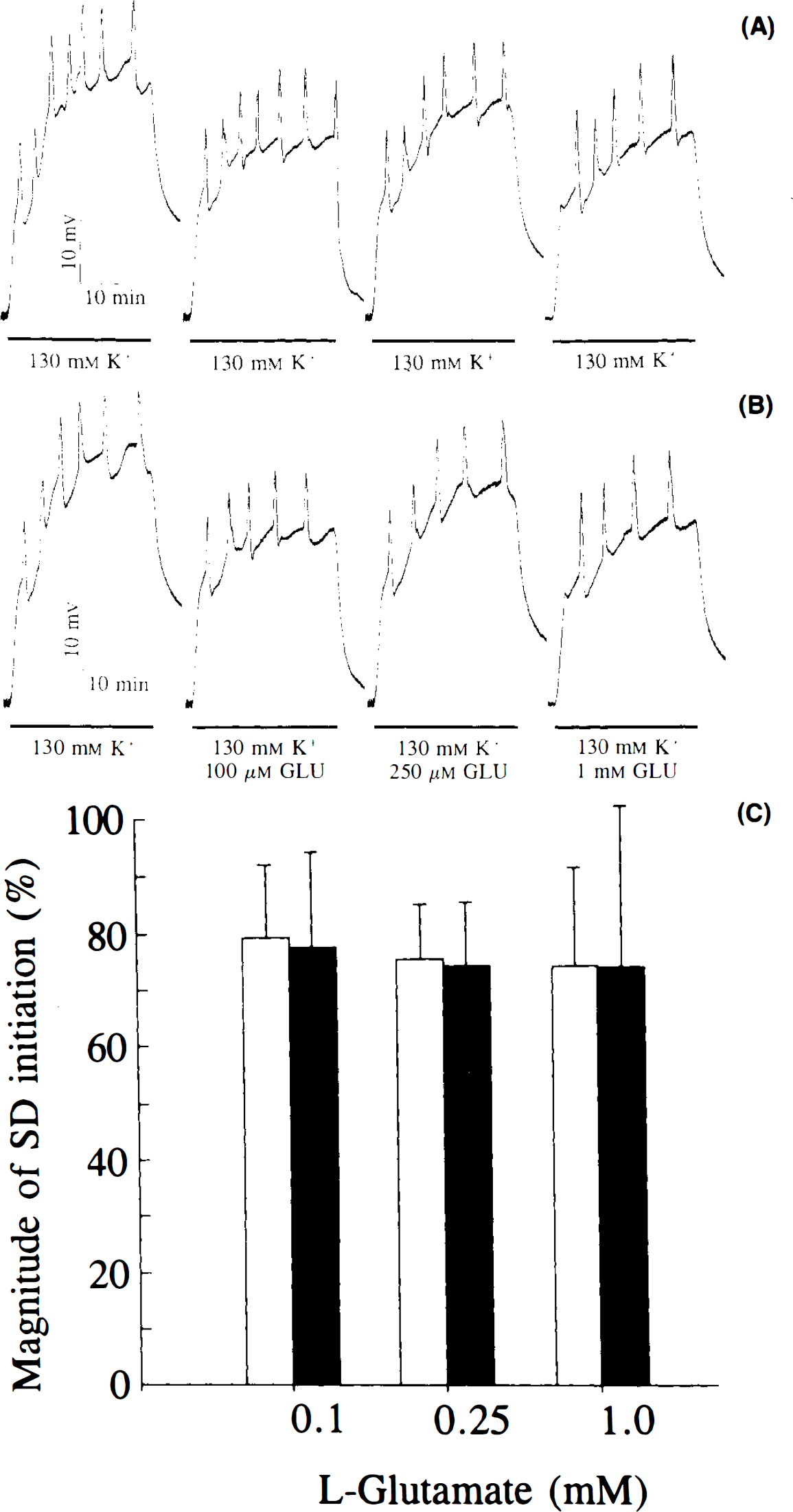
Effect of high extracellular glutamate on K+-evoked spreading depression (SD).
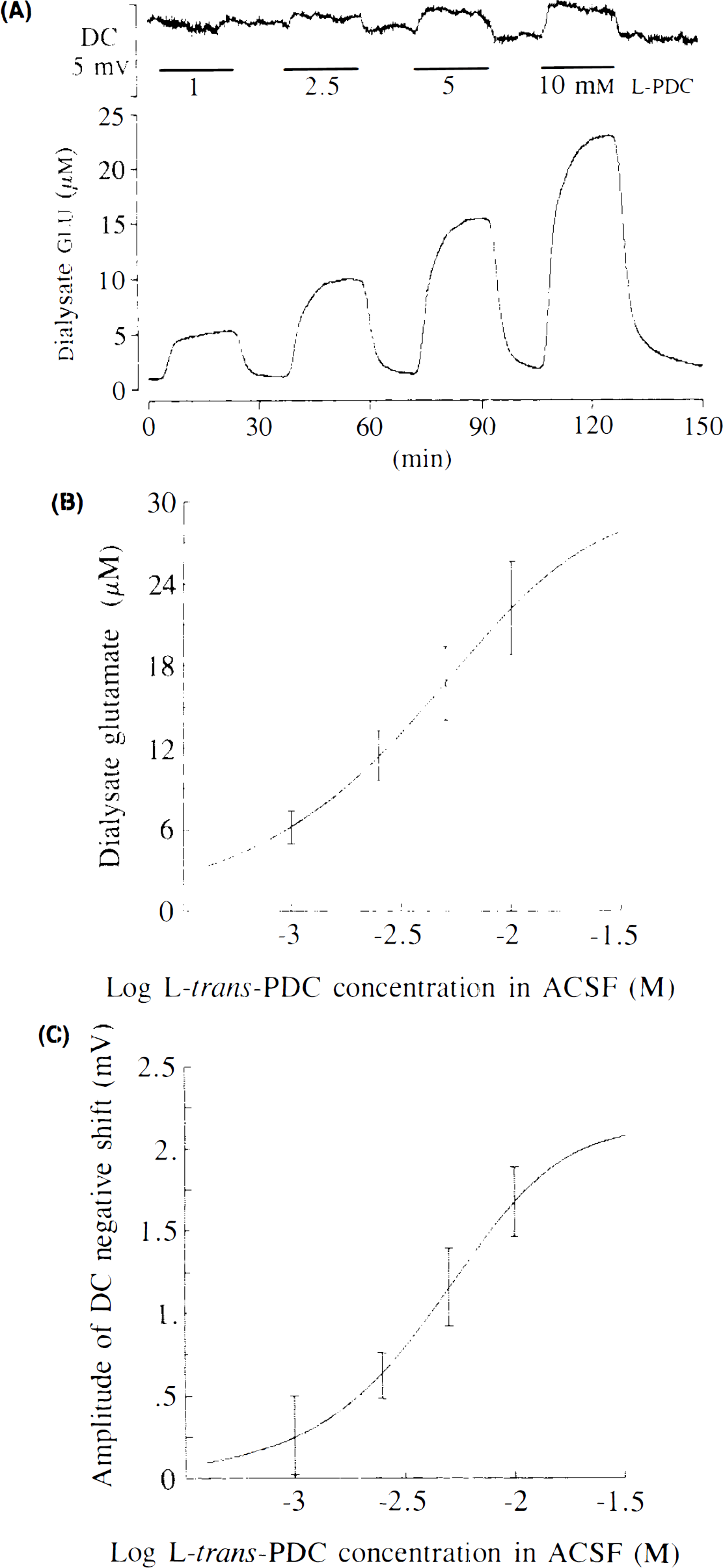
Effects of glutamate uptake inhibition produced by perfusion of increasing concentrations of L-trans-pyrrolidine-2,4-dicarboxylate (L-trans-PDC) through a microdialysis electrode implanted into the rat cortex.
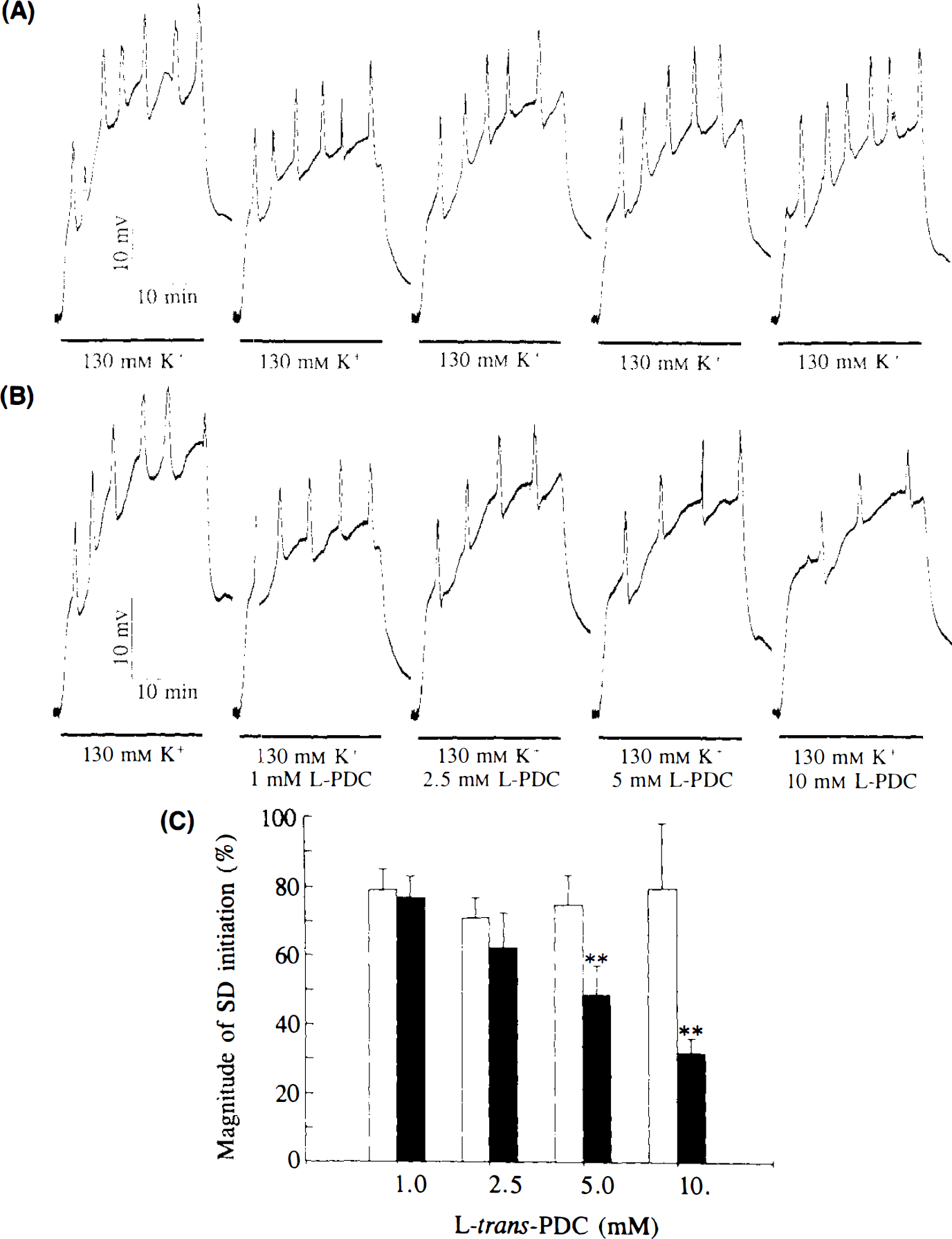
Effect of high extracellular glutamate progressively increased by inhibition of glutamate uptake with L-trans-pyrrolidine-2,4-dicarboxylate (L-trans-PDC) (see Fig. 3) on K+-induced spreading depression (SD). A: Representative changes in the direct current (DC) potential produced on the control side by five repeated perfusions of 130 mM K+ through the microdialysis probe for 20 min (horizontal bars). B: Representative changes in the DC potential, produced contralateral, by 130 mM high K+ onto which were superimposed increasing concentrations of the selective glutamate uptake inhibitor L-trans-PDC. For more details on (a) and (b), see legend to Fig. 2. C: Average changes in the magnitude of SD initiation expressed as percentage of the first challenge where no drug was applied on either side; SD elicitation was produced by 130 mM K+ alone (open bars) or together with increasing concentration of L-trans-PDC in the perfusion medium (filled bars). Cumulative areas of SD peaks for the first challenge were 21.7 ± 5.1 and 22.1 ± 4.4 mV min, respectively. Bars represent means ± SD (n = 6). **p < 0.01, compared with the control side by Student's t test. Note the unexpected inhibition of SD elicitation by L-trans-PDC.
RESULTS
Recurrent initiation of cortical SD by microdialysis application of K+
Application of 130 mM K+ for 20 min through the cortical microdialysis electrode produced a sustained negative shift of the DC potential, onto which were superimposed between five and seven peaks of further depolarization (Figs. 2A and 4A). Each of these peaks corresponded to the initiation of a wave of SD (Obrenovitch et al., 1993). On the control side (i.e., where 130 mM K+ was applied alone), the magnitude of SD elicitation was consistent throughout the experiments, except for the first K+ challenge, which triggered SD more efficiently than subsequent ones (Figs. 2A,C and 4A,C) (cumulative peak areas: 21.2 ± 4.5 and 16.8 ± 4.1 mV min for the first and second challenge, respectively; n = 12, p < 0.001, Student's paired t test). The first application of K+ also produced a larger sustained negative shift of the DC potential (Figs. 2A and 4A) (31.0 ± 2.7 and 22.8 ± 3.2 mV for the first and second challenge, respectively; n = 12, p < 0.001, Student's paired t test).
Effect of high extracellular glutamate on SD elicitation by K+
Increasing the concentration of glutamate in the perfusion medium as 130 mM K+ was applied did not alter the magnitude of SD elicitation (Fig. 2). There was also no significant difference in the amplitude of the K+-induced sustained negativation of the DC potential, even when 1 mM glutamate was added to the SD elicitation medium (25.8 ± 1.7 vs. 25.7 ± 2.2 mV; n = 6).
Effects of inhibition of glutamate uptake on extracellular glutamate levels and basal DC potential
The basal concentration of glutamate in the cortical dialysate was 1.19 ± 0.45 μM (n = 12). Under otherwise normal conditions, addition of L-trans-PDC to the perfusion medium concentration dependently increased the dialysate concentration of glutamate (Fig. 3A and B), with 10 mM L-trans-PDC increasing this variable ∼19-fold. Dialysate levels of glutamate rose rapidly from the beginning of perfusion with L-trans-PDC medium and reached a plateau within ∼15 min regardless of the concentration of L-trans-PDC tested (Fig. 3A). Normalization of dialysate glutamate after switching back to normal ACSF was even more rapid; most of the glutamate accumulated in the extracellular space was cleared from the recording site within ∼10 min (Fig. 3A).
Perfusion of L-trans-PDC through the microdialysis probe was associated with a negative shift of the DC potential (Fig. 3A), and the magnitude of this effect also increased with the concentration of L-trans-PDC applied through the microdialysis probe (Fig. 3C). However, in contrast to the marked changes in dialysate glutamate levels, the DC potential alteration was minor (1.68 ± 0.21 mV with 10 mM L-trans-PDC, n = 12; i.e., 18-fold less than the sustained depolarization produced by 130 mM K+). It is also noteworthy that the time course of the DC potential changes with L-trans-PDC did not match those of glutamate. In contrast to the changes in dialysate glutamate, the maximal effect of L-trans-PDC on the DC potential was observed early during drug application (Fig. 3A).
Effects of inhibition of glutamate uptake on SD elicitation by K+
Unexpectedly, L-trans-PDC reduced concentration dependently the elicitation of SD by K+ (Fig. 4). With 10 mM L-trans-PDC, the cumulative area of the peak was 6.9 ± 1.7 mV min in comparison with 18.9 ± 6.8 mV min on the control side (n = 6, p < 0.01, Student's t test). Inhibition of SD initiation by L-trans-PDC occurred rapidly and apparently persisted throughout the perfusion of L-trans-PDC/K+ medium (Fig. 4B). There was no evidence of a more pronounced effect toward the end of the K+ challenge, i.e., when extracellular glutamate was maximal (Fig. 3A).
DISCUSSION
Recurrent initiation of cortical SD by microdialysis application of K+
Repeated perfusion of 130 mM K+ through the microdialysis probe for 20 min consistently triggered SD, except that SD elicitation was more pronounced with the first challenge (Figs. 2 and 4). A similar phenomenon was observed with electrically evoked SD; i.e., the triggering threshold increased with the second wave of SD as compared with the first (Verhaegen et al., 1992). We concur with these authors in proposing that the reduced sensitivity to SD elicitation with subsequent challenges may be due to persistent effects of the first SD waves, possibly tissue acidosis (Mutch and Hansen, 1984; Taylor et al., 1994), which inhibits SD in vivo (Gardner-Medwin, 1981; Marrannes et al., 1985) and to which long-lasting reduction of cerebral blood flow after SD may contribute (Lauritzen, 1984; Duckrow, 1991).
Failure of high extracellular glutamate to potentiate SD elicitation by K+
One plausible explanation is that glutamate had no effect on SD elicitation because it was actively taken up by the surrounding cells as it diffused from the microdialysis fiber. Indeed, it is generally considered that glutamate applied exogenously to the intact brain may not significantly raise interstitial concentrations because of efficient uptake systems (Garthwaite, 1985; Erecinska et al., 1986; Nicholls and Attwell, 1990). However, the efficacy of Na+-dependent high-affinity glutamate uptake is dependent on the Na+/K+ gradient across the plasma membrane (Kanner and Bendahan, 1982), and the transport of glutamate in glial cells from the salamander retina was reversed by high extracellular K+ (Szatkowski et al., 1990). Furthermore, we had previously observed that perfusion of 100 mM K+ in the rat striatum markedly increased extracellular glutamate (Obrenovitch et al., 1993) and showed that the dialysate concentration of glutamate reached a maximum of ∼30 μM when SD was elicited by K+ (Zilkha et al., 1995) and ∼15 μM during its propagation (Obrenovitch et al., 1995). It is very likely, therefore, that extracellular glutamate levels were markedly increased at the site of SD elicitation when glutamate concentrations 100–1,000 times higher than normal basal levels were perfused through the microdialysis probe.
The finding that up to 1 mM glutamate did not enhance the sustained depolarization produced by 130 mM K+ is in line with our previous study, which showed that, in otherwise normal conditions, >10, mM glutamate had to be perfused through the microdialysis probe to evoke a clear depolarization (Obrenovitch et al., 1994).
Effects of glutamate uptake inhibition by L-trans-PDC
Before a discussion of the effects of glutamate uptake inhibition on SD, emphasis must be placed on two important features of L-trans-PDC action: (a) L-trans-PDC inhibits synaptosomal glutamate two- to fourfold more strongly than astroglial uptake (Rauen et al., 1992); and (b) L-trans-PDC inhibition of glutamate uptake is competitive (i.e., when applied to the extracellular space by microdialysis, L-trans-PDC is transported into intracellular compartments, competing with glutamate at the carrier recognition site) (Waldmeier et al., 1993; Griffiths et al., 1994). The latter implies that L-trans-PDC does not abolish the electrogenic effect associated with glutamate carrier function, but actually induces its own as it is transported (Urenjak et al., 1996).
Perfusion of L-trans-PDC through the microdialysis probe concentration dependently increased extracellular glutamate levels, because of inhibition of high-affinity glutamate uptake, onto which was presumably superimposed glutamate release by heteroexchange as L-trans-PDC is a transportable inhibitor of the glutamate transporter (Waldmeier et al., 1993; Griffiths et al., 1994).
Despite markedly increasing the concentration of glutamate in the interstitial space, L-trans-PDC evoked only minor depolarizations (Fig. 3C) and the time course of these changes did not match that of extracellular glutamate (Fig. 3A). This latter observation suggests that L-trans-PDC-induced depolarizations do not result from an action of increased extracellular glutamate on ionotropic glutamate receptors. If this were the case, one would expect L-trans-PDC-induced depolarizations to progressively increase with extracellular glutamate levels. Therefore, changes in the DC potential produced by L-trans-PDC probably reflect the influx of Na+ and efflux of K+ associated with the transport of this drug by the glutamate carrier. Several elements support this hypothesis: (a) The amplitude of L-trans-PDC-evoked depolarization was maximal early after drug application, i.e., when the transmembrane gradient of the drug was maximal; (b) L-trans-PDC depolarizations increased concentration dependently; and (c) their amplitude was not modified by blockade of NMDA and/or α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors (Urenjak et al., 1996).
The concentration-dependent reduction of SD elicitation by L-trans-PDC was not expected. It is certainly not linked to the associated increase in extracellular glutamate since application of exogenous glutamate had no effect on SD. A direct inhibitory effect of L-trans-PDC on NMDA receptor activation, which is essential for SD elicitation (Lauritzen and Hansen, 1992; Sheardown, 1993), can also be ruled out because this drug has virtually no affinity for these receptors (Bridges et al., 1991). Rather, inhibition of SD elicitation may be due to the fact that L-trans-PDC, by being transported through the high-affinity glutamate carrier, produces marked alterations of the gradients of major cations across the cellular membrane, ultimately leading to activation of Na+/K +-ATPase and acidosis. Prolonged electrical stimulation or seizures, which stimulate Na+/K+ pumping and anaerobic metabolism, also raise the threshold for SD and may block propagation (Burěs et al., 1975; Koroleva et al., 1985). It is interesting to note that probenecid, a competitive inhibitor of organic anion transport, inhibited the elicitation of SD by K+ in the rat striatum (Taylor et al., 1994).
Implications of these findings
These findings do not support the hypothesis that high extracellular K+ concentration triggers SD because high extracellular glutamate, resulting from exocytosis and/or reversal of glutamate uptake (Nicholls, 1989; Paulsen and Fonnum, 1989; Szatkowski et al., 1990), depolarizes adjacent neurons (Van Harreveld and Fifková, 1970; Sheardown, 1993). However, two elements suggest strongly that the initiating mechanism of SD includes exocytotic release of glutamate with activation of NMDA receptors: (a) Antagonists of these receptors completely inhibit SD propagation and markedly reduce the sensitivity to the triggering stimulus (Hernándéz-Cáceres et al., 1987; Marrannes et al., 1988; Lauritzen and Hansen, 1992; Sheardown, 1993); and (b) both K +-induced release of glutamate and SD initiation require the presence of extracellular Ca2+ (Burěs et al., 1974; Obrenovitch et al., 1993). This apparent contradiction suggests that glutamate, required for SD elicitation through NMDA receptor activation, is transiently released from presynaptic vesicles and acts exclusively on juxtaposed postsynaptic receptors. The resulting increase in extracellular glutamate in itself is not causative.
Local, high extracellular concentration of K+ remains the plausible trigger of SD. This event presumably develops whenever K+, whether applied or released by cellular injury or depolarization, can no longer be contained by reuptake, diffusion, or transport away from the active site by the glial buffering system (Reid et al., 1988). Astrocytes may be the primary instigators of SD as their depolarization coincides closely with the onset of SD, in contrast to that of neurons (Sugaya et al., 1975), and the initiating event may involve intercellular coupling through gap junction (Nedergaard and Goldman, 1995). Neuronal depolarization may be secondary and dependent on exocytotic glutamate release. Further studies with blockade of exocytosis by tetanus toxin should allow testing of the latter hypothesis.
These results illustrate that sensitivity of a pathological or experimental event to NMDA receptor antagonists does not necessarily imply the involvement of increased extracellular glutamate. Accumulating evidence from in vivo studies rather suggests that high extracellular glutamate is not a prerequisite of excessive, neurotoxic ionic fluxes through glutamate-operated channels (Massieu et al., 1995; Obrenovitch and Richards, 1995; Obrenovitch et al., 1996).
Footnotes
Acknowledgment:
This work was supported by the Thompson Fund and the Joint Research Academic Committee (National Hospitals and Institute of Neurology, London). We thank Dr. Douglas A. Richards (Department of Pharmacology, School of Pharmacy, London) for his critical comments.