
Abstract
Ischemic cell death occurs when extracellular glutamate levels increase, causing tissue depolarization and an excessive rise in intracellular calcium concentrations. The relative occurrence of the depolarization events and the changes in glutamate concentration in ischemia have not been studied. In a model of focal cerebral ischemia in the rat, three measurements were made simultaneously in vivo: cerebral blood flow (CBF) by the H2-clearance method, extracellular glutamate concentration by microdialysis, and activation of the voltage-sensitive calcium channel (VSCC) by its binding to [3H]nimodipine. Effects of probe implantation on these measurements were accounted for. The CBF to control ratio obtained during the experiments spanned the range of 1.08 to 0.07. Binding to [3H]nimodipine became significantly activated when CBF fell to ∼0.49 of its control value while extracellular glutamate concentrations increased significantly only at a CBF ratio of <0.33. Activation of the VSCC at this high CBF ratio may be due to ischemic depolarization, which has been shown to activate the binding to [3H]nimodipine. It may be useful to define a CBF threshold of 50% of normal in focal ischemia for opening of the VSCC. The same threshold has been linked to an overall depression of protein synthesis and to activation of a number of molecular responses.
Falling cerebral perfusion leads to a rise in glutamate concentration in the extracellular space and activation of the voltage-sensitive calcium channel (VSCC). [3H]nimodipine, a VSCC ligand, has been used to study regional tissue vulnerability in focal ischemia, and its binding to brain was reported to be proportional to the severity of ischemia and anticipated histologic outcome (Hakim and Hogan, 1991). More recent studies have shown that binding of [3H]nimodipine is responsive to the depolarization induced by spreading depression (Osuga et al., 1993) and, in the setting of ischemia, may be an indicator of the response of the VSCC to ischemic depolarization. The relationship of depolarization to the neuronal damage resulting from ischemia is not straightforward. Kawahara et al. (1994) showed that spreading depression applied prior to the imposition of forebrain ischemia is neuroprotective, and Matsushima et al. (1995) observed a similar protective effect of spreading depression during subsequent focal ischemia. In contrast, depolarization occurring in the setting of focal ischemia may aggravate outcome. Mies et al. (1993a) reported that the number of peri-infarct depolarization events correlates with the volume of infarct, and decreasing their occurrence will reduce ischemic damage (Chen et al., 1993). Thus, the influence of depolarization on ischemic neuronal survival would seem to depend upon the temporal relationship of the depolarization to the ischemia and, probably, the energy state of the tissue. Glutamate, in addition to its determining influence on ischemic outcome, plays an important role in initiating and sustaining depolarization. Glutamate antagonists reduce or prevent the propagation of spreading depression waves (Lauritzen and Hansen, 1992; McLachlan, 1992). On the other hand, blockade of the VSCC decreases the ischemia-induced rise in glutamate (Serieller et al., 1993). Thus, glutamate and VSCC modulate each other as well as influence ischemic outcome, but the sequence of their activation and relative intensity are not known.
The purpose of this study was to define in vivo the relationships among increase in extracellular glutamate, binding of [3H]nimodipine, and cerebral blood flow (CBF), measured simultaneously in a model of focal cerebral ischemia in the rat. It was hoped that defining the relative intensity and timing of changes in extracellular glutamate concentrations as well as activation of the VSCC in the ischemic setting would lead to a more precise formulation of the pathophysiologic events that result in ischemic damage. Based upon available literature (Obrenovitch et al., 1990; Jing et al., 1993), the hypothesis was formulated that VSCC activation is consequent to the rise in extracellular glutamate concentration. Contrary to the expected outcome, results of this work suggest that VSCC activation occurs at a higher postischemic CBF level than does the rise in extracellular glutamate concentration.
MATERIALS AND METHODS
Animal preparation
Eight Sprague-Dawley rats weighing 275–300 g were anesthetized with 2% halothane and a 30% oxygen/70% nitrous oxide (vol/vol) gas mixture. Two femoral arteries were cannulated with polyethylene catheters for blood pressure monitoring and arterial blood sampling. One femoral vein was also cannulated for later [3H]nimodipine injection. Body temperature was controlled at 37°C by a thermal blanket with feedback control (Ealing Scientific, Quebec, Canada). Endotracheal intubation was performed with a 14 gauge intravenous catheter, and ventilation was controlled artificially at a respiratory rate of 35–40 breaths/min (tidal volume 2.5–3.5 ml) by using a Harvard Small Animal Ventilator (Ealing). Paco2 and Pao2 were adjusted in each animal within the physiologic range. Animals were placed on a stereotaxic instrument. A sagittal skin incision was followed by drilling 2.0 mm (diameter) burr-holes at six different sites (Fig. 1): (a) right parietal [Anteroposterior (A-P), −0.3, lateral, 6.0], (b) left cingulate (A-P, −0.3; lateral, 1.2), (c) left frontal (A-P, + 1.2; lateral, 3.5), (d) left parietal (A-P, −0.3, lateral, 6.0), (e) left parietooccipital (A-P, −4.5, lateral, 3.8), and (f) left occipital (A-P, −8.3, lateral, 5.5), after which the dura was carefully removed. A thermocouple probe was placed on the left skull surface adjacent to the left parietal burr-hole, and brain temperature was maintained as close to 37°C as possible by using an infrared lamp. Anesthesia was maintained with 1–1.5% halothane during the entire experiment.
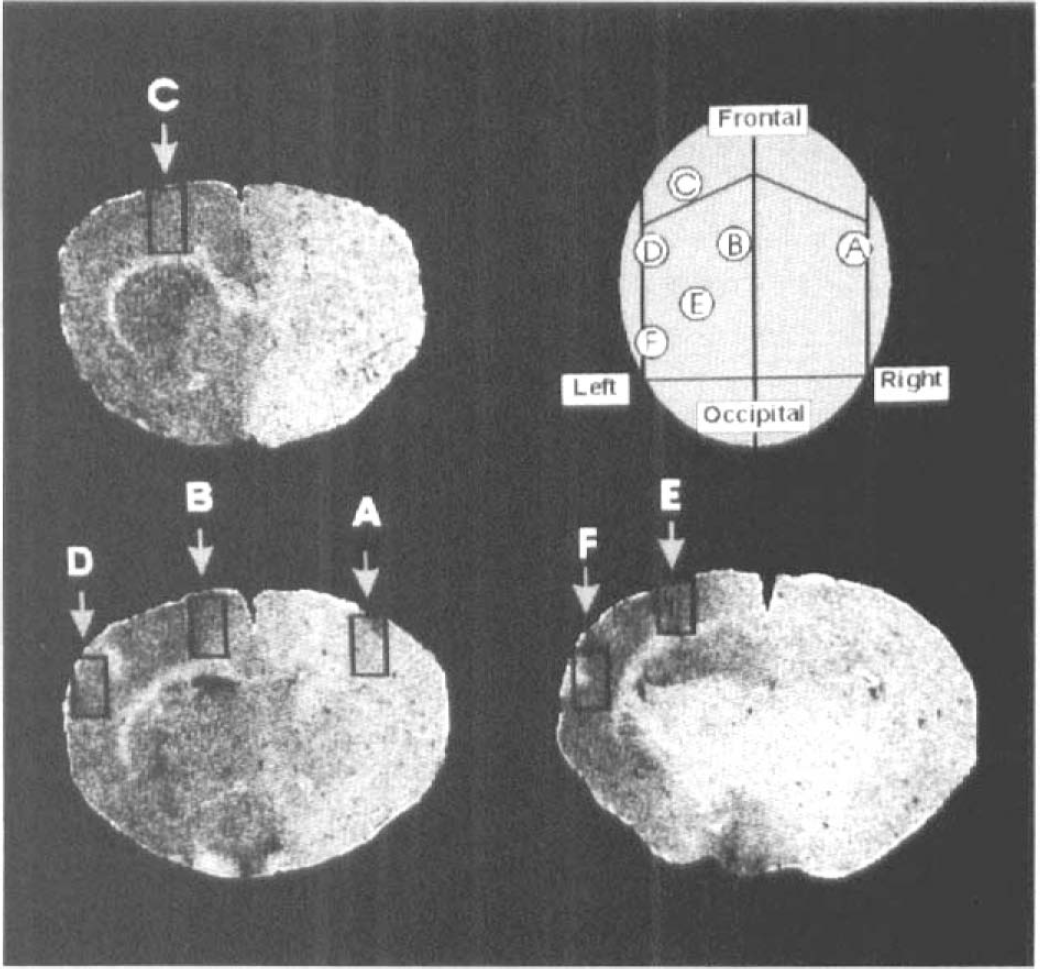
Positions of implantation of microdialysis probes and platinum wire for CBF measurements, shown as a superior view of the skull and in coronal sections.
Microdialysis
Microdialysis probes were made by a similar method to that of Van Wylen et al. (1986) (0.3 mm diameter, service membrane length 2 mm, molecular weight cut-off 5,000 Da). Probes were stereotaxically implanted so that the tips were 3 mm below the brain surface. Platinum wires (Teflon-coated, 0.13 mm diameter, 2-mm bare portion) (A-M Systems, Everett, WA, U.S.A.) were positioned 1 mm from the microdialysis probe and used for CBF measurements by means of H2 clearance. They were placed in all six cortical regions. A reference electrode attached to a stainless steel screw was placed on the right frontal skull. Microdialysis probe/platinum wire assemblies were fixed to the skull using dental cement. Microdialysis probes were continuously perfused, with Krebs-Ringer-bicarbonate solution (pH 7.4) at a rate 2 μl/min, starting at 30 min before implantation to the end of the experiment, by using a 10-channel infusion microprocessor-controlled syringe pump (Stoelting, model number 53220, Chicago, IL, U.S.A.). Dialysates were collected in 20 μl fractions at 10-min intervals. Samples were stored at −80°C for high-performance liquid chromatography (HPLC) analysis.
The ischemia model
The “thread model” of Nagasawa and Kogure (1989) was used to produce ischemia in the territory of the middle cerebral artery (MCA). A left postauricular axial skin incision was made after implantation of the microdialysis probes. The left common, external, and internal carotid arteries were dissected free. Common and external carotid arteries were ligated with 4–0 silk sutures as were the left occipital and pterygopalatine arteries. A 4–0 nylon thread coated at the tip with enamel paint (diameter 0.3–0.5 mm) was prepared. Occlusion of the internal carotid was performed 180 min after microdialysis probe implantation. A small incision was made with a 28 gauge needle 1.5 mm proximal to the bifurcation of the external and internal carotid arteries and the thread introduced through the incision, advanced 1.2–1.5 cm until it occluded the MCA, and maintained at the same position throughout the experiments. Occlusion was confirmed by blood flow measurement using H2 clearance (see below). Animals were killed by decapitation 30 min after occlusion. The brain was immediately excised, frozen in isopentane, and stored at −80°C for later sectioning.
HPLC measurement
Glutamate concentration in the dialysate was measured by HPLC, utilizing baseline software (Waters Chromatography Division, Millipore Corporation, Milford, MA, U.S.A.). Fifteen μl of dialysate was mixed with 20 μl of 50 μM norvaline internal standard solution. Fifteen μl of this solution was automatically reacted with 20 μl of O-phthaldialdehyde for 60 s just prior to injection. A glutamate calibration line based on the ratio of glutamate/norvaline peak areas was obtained by measurement of five amino-acid standard solutions (Sigma Chemical Co., St. Louis, MO, U.S.A.). Glutamate concentration in the dialysate was calculated by reference to the standard dose-response line.
Local CBF measurement
CBF was measured by the H2 clearance technique. The platinum wire was polarized to 500 mV (DC) against the reference electrode and 5–15% hydrogen in a 30% oxygen/70% nitrous oxide (vol/vol) mixture was combined with the anesthetic gas mixture for 1–2 min. The current between the two electrodes was monitored by an electric circuit similar to that described by Pasztor et al. (1973). The hydrogen curve was recorded on a chart recorder (Windograph, Gould, OH, U.S.A.). Hydrogen clearance was calculated from the time, t, required for hydrogen levels to fall from H1 to Hi + t according to the formula
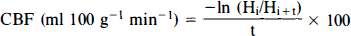
Hi and Hi + t were usually 80% and 40% of the maximum hydrogen level except where CBF was extremely low (<5 ml 100 g−1 min−1).
[3H]Nimodipine autoradiography
We used a previously published method to determine the activity of the VSCC in vivo using [3H]nimodipine (Hogan et al., 1991). For this determination, 200 μCi of [3H]nimodipine (1 mCi/ml ethanol, specific activity 130 Ci/mmol) (NEN/Dupont, Mississauga, Ontario, Canada) was diluted in 400 μl of carrier (Bay e9736 placebo, Miles Pharmaceuticals, Etobicoke, Ontario, Canada). Immediately after MCA occlusion, this solution was injected intravenously over a 1-min interval using a constant flow infusion regimen. A plasma sample was obtained 1 min prior to death, and tritium concentration was determined by using single-labeled liquid scintillation counting with appropriate quench correction.
Frozen brain was sliced into 20 μm sections in a cryostat; ∼100 slices were produced from each brain. Brain sections, along with 10 standards made of brain homogenates containing known concentrations of tritium, were exposed to Hyperfilm (Amersham, Oakville, Ontario, Canada) for 60 days. Exposed films were digitized by using a microcomputer-based image analyzer (Imaging Research Inc., St. Catharines, Ontario, Canada) to obtain actual activities of tritium. The region of interest was a rectangle (2 × 1 mm) placed on the site of microdialysis probe implantation. All tritium measurements were corrected for metabolism of nimodipine using previously determined metabolite correction factors (Hogan et al., 1991).
Effect of microdialysis probe implantation on glutamate concentration and [3H]nimodipine binding
Before combining microdialysis, CBF measurement by the H2 clearance method and [3H]nimodipine autoradiography, effect of probe implantation on [3H]nimodipine binding, and extracellular glutamate concentration were investigated. Six normal rats were treated identically to experimental animals but without vascular dissection or occlusion. Six microdialysis probes were implanted in different sites in each animal 20, 30, 60, 120, 180, and 240 min before death. [3H]nimodipine (same dose as above) was injected intravenously 30 min before death, and twenty μl of dialysate was collected at 10-min intervals from each microdialysis probe starting with [3H]nimodipine administration. Animals were killed by decapitation and the brains stored for autoradiography. Glutamate concentration in the dialysate was measured by HPLC as above.
Statistical analysis
Two complementary statistical approaches were adapted for analysis of the relationship between extracellular glutamate concentration and [3H]nimodipine binding as a function of CBF. The first approach involved identifying the CBF intercept at which these two functions rise significantly using simple bivariate linear regression. A logarithmic transformation was performed on all three measurements and an independent t-test comparison of the slopes of the fitted regressions was performed. The second approach involved a modification of the moving average procedure. It was reasoned that if the two processes tracked similarly, then testing a sliding window of datum points (n = 7) against a mutually exclusive reference set might reveal a significant differential sequence of outcomes. The seven data points of each process at the highest CBF ratio, at which both extracellular glutamate levels and VSCC function were expected to be at baseline, were chosen as the reference set. If one process were to increase significantly above its specific reference when set at a higher CBF value than the other process, then it could be argued that the processes were fundamentally different. The sliding window was advanced (away from the reference set) sequentially over three datum points. Thus, three pairs of independent one-tailed t-tests were performed.
RESULTS
Physiologic measurements
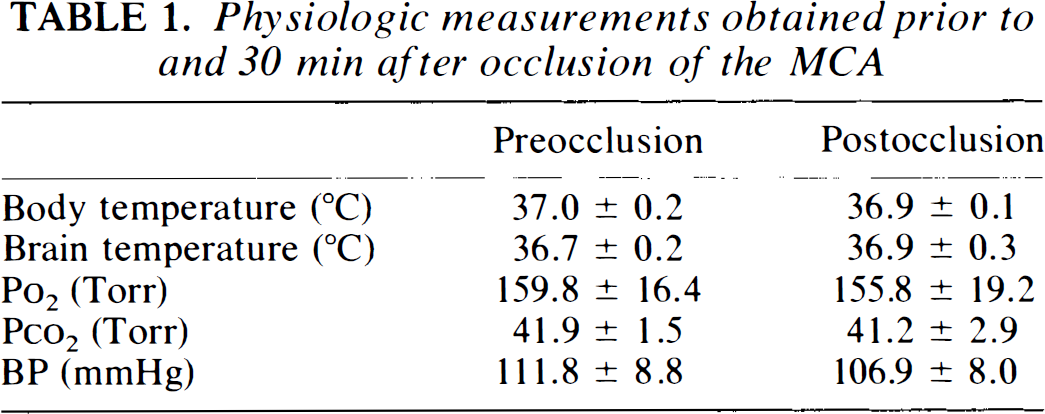
Table 1 shows mean body and brain temperatures and arterial blood gases just prior to MCA occlusion and at 30 min postischemia (mean ± SD). There were no significant differences in these values.
Physiologic measurements obtained prior to and 30 min after occlusion of the MCA
Effect of probe implantation on VSCC activity and glutamate concentration
The data obtained during this experiment were normalized by dividing the VSCC activity and the glutamate concentration values by their respective levels at 240 min and compared by using the analysis of variance (ANOVA) post hoc test (Fig. 2). Glutamate concentrations in the dialysate and VSCC activity at 20 and 30 min after probe implantation were significantly higher than the levels at 240 min, but, by 60 min and all subsequent times, both measurements became indistinguishable from control values. Since occlusion of the MCA was performed 180 min after probe implantation, both VSCC activity and glutamate concentration are presumed to have been at baseline levels at the onset of ischemia.
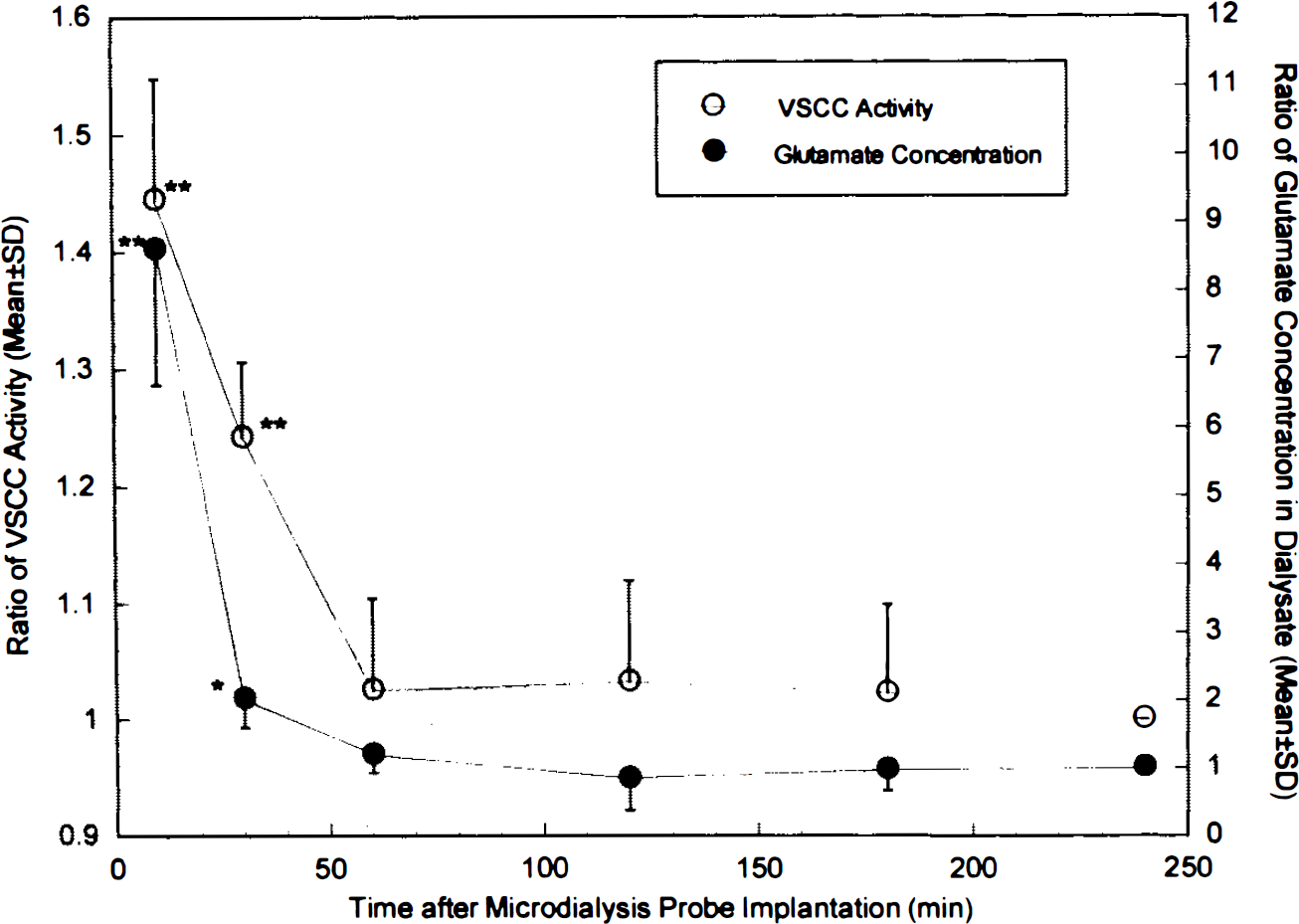
The effect of microdialysis probe implantation on glutamate concentration in the dialysate (•) and on VSCC activity measured by [3H]nimodipine binding (○). All values are ratios to the measurements at 240 min after implantation and are compared to them. *p < 0.05, **p < 0.0001.
Dependence of glutamate concentration and VSCC activity on CBF by probe location
Table 2 shows the mean preocclusional CBF values as well as those at 30 min of occlusion identified by the probe locations indicated in Fig. 1. The ratio of CBF in the ischemic regions to preocclusion control values obtained during these experiments spanned the range of 0.07 to 1.08. Preocclusional glutamate concentration in the dialysate varied between 1.27 ± 1.37 and 2.02 ± 0.76 μM. The average percent recovery of glutamate was measured at 7.89%. Table 2 also shows, by probe location, glutamate concentration between 20–30 min of occlusion as well as the mean ratio of relative VSCC activity at 30 min of occlusion compared to preocclusion values. The rise in both values in the ischemic regions is evident.
Preocclusion CBF values and CBF, glutamate concentrations and VSCC activity measured 30 min after occlusion of the MCA
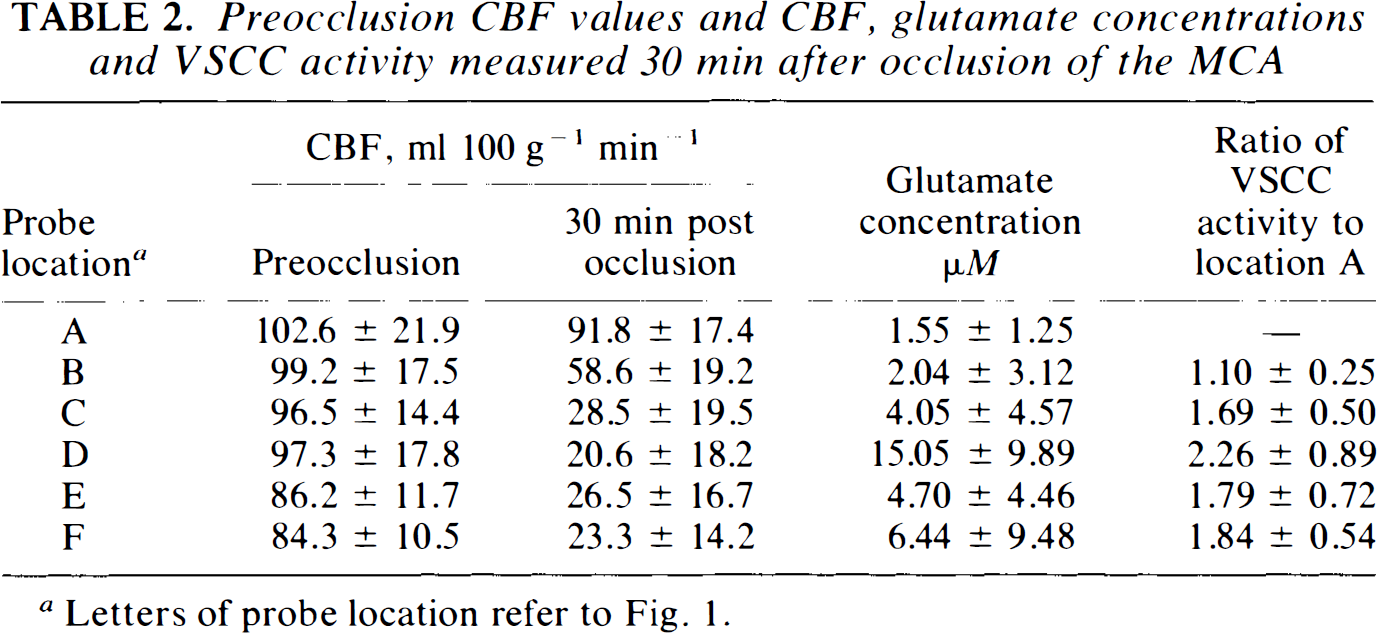
Letters of probe location refer to Fig. 1.
Dependence of glutamate concentration on CBF
All glutamate concentrations were normalized by dividing individual values by mean preocclusional measurements at all six probe sites. Measured CBF values were also normalized with respect to preocclusional values at each site in each animal. Figure 3 shows that the concentration of glutamate in the dialysate between 20 and 30 min after occlusion rose abruptly when the ratio of CBF fell to <0.3 of its preocclusional value. Below this CBF ratio, the best fit line for glutamate concentration was y = 13.04 − 39.44 × (r = −0.58, p < 0.01), which suggests an intercept with the CBF axis at 0.33. Above this CBF ratio, there was no significant correlation between glutamate concentration ratios and CBF.
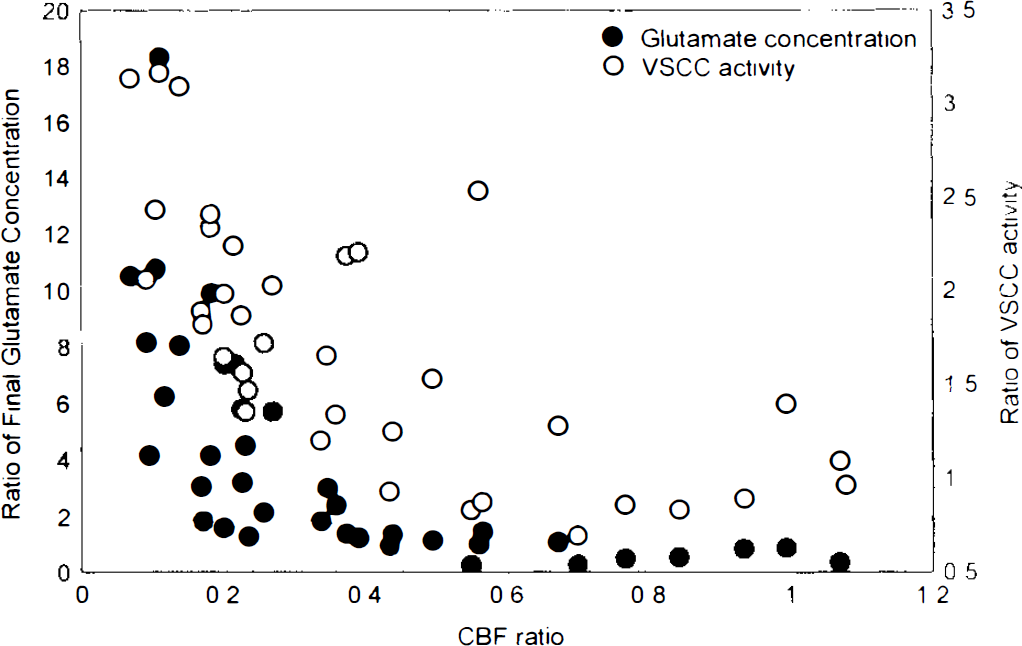
Extracellular glutamate concentration and VSCC activity measured 30 min after MCA occlusion and their relationship to CBF. All values are ratios to their preocclusion values.
Dependence of VSCC activity on CBF
Figure 3 also shows the variation in normalized VSCC activity with the CBF ratio. The best fit line for VSCC activity at a CBF ratio of <0.3 was y = 3.36 − 6.86 × (r = −0.76, p < 0.005), implying an x-axis intercept at a CBF ratio of 0.49. At a CBF ratio of >0.3, VSCC activity could be fit to the straight line y = 1.83 — 0.85 × (r = 0.51, p < 0.01).
Comparison of CBF dependence of VSCC activity and extracellular glutamate concentration
The slopes of logarithmically-transformed glutamate and VSCC activity versus CBF were significantly different. This indicated that the two data sets behaved differently with respect to CBF, but also prevented a test of the significance in CBF intercepts of the two functions by this method (Pedhazur, 1982). Thus, differences in the dependence of the two measurements on CBF could be tested only by the method of moving averages. Table 3 shows the results of this “sliding t”-test, whereby the ratios of glutamate and VSCC determinations at the highest CBF values were used as a baseline against which groups (n = 7) of determinations obtained at lower CBF values were tested. It is evident that VSCC activity begins to rise significantly above baseline at a CBF ratio of 0.464, whereas glutamate begins to rise significantly only at a CBF ratio of 0.384. These results were not affected by a change in the size of the groupings. Figure 4 shows the relationship between VSCC activity and increasing glutamate concentration. After an initial slow rise in glutamate concentration with activation of VSCC, there is a suggestion that glutamate concentration rises sharply when the ratio of VSCC activation to control reaches 2.
Comparison of glutamate and VSCC determinations at progressively lower CBF values to the seven measurements at the highest CBF values a
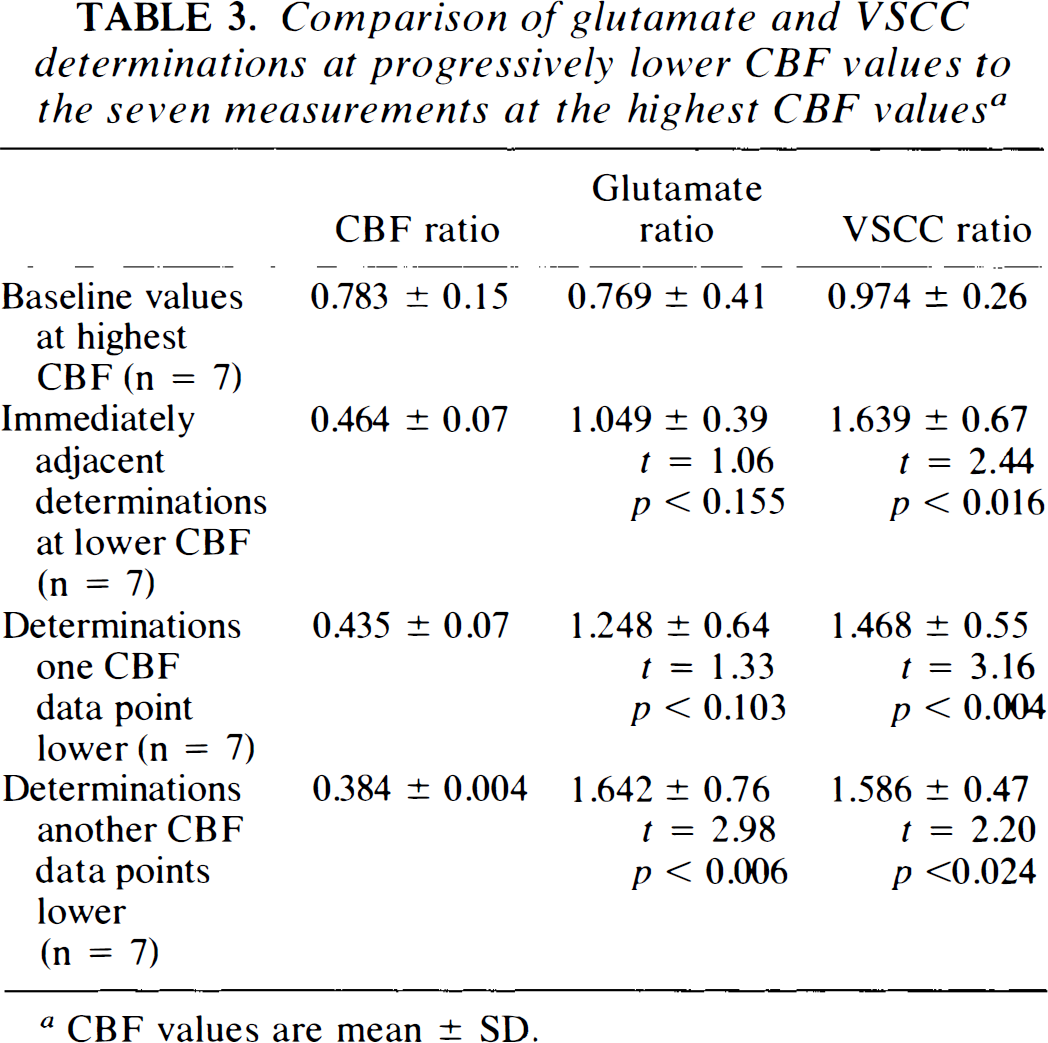
CBF values are mean ± SD.
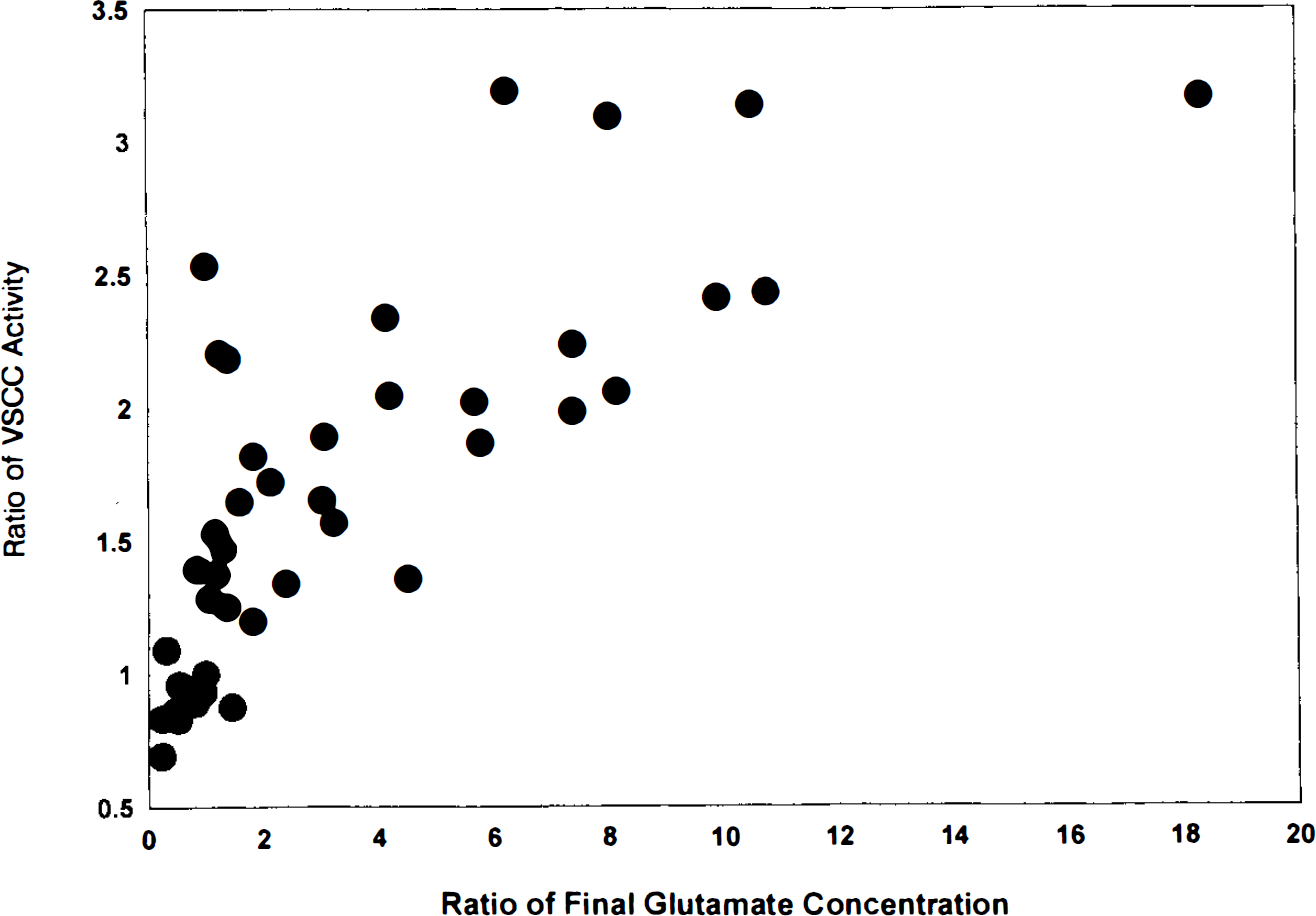
Variation in relative VSCC activity with increasing glutamate concentration.
Thus, two analysis procedures are concordant in suggesting that VSCC activation occurs at higher CBF values than those coinciding with a rise in extracellular concentrations of glutamate.
DISCUSSION
Many neurochemical changes have been shown to accompany the imposition of anoxia and ischemia (Haddad and Jiang, 1993). Two of these—excitotoxicity due to excessive glutamate in the extracellular space and activation of the channels that regulate the intracellular movement of calcium—have been shown to play important roles in determining ischemic outcome. In this and in previous studies, measurement of in vivo binding of [3H]nimodipine—aligand of the VSCC—has been used as an index of depolarization. This channel makes up only 20–25% of the overall calcium current elicited by depolarization (Bean et al., 1993), but the importance of the VSCC may derive not so much from its role in allowing calcium transients but rather from its location at the proximal end of dendrites and next to the cell body, where channels are in close proximity to other receptors (Westenbrock et al., 1990). Hogan et al. (1991) have shown that [3H]nimodipine binding to brain is saturable and specific. In the nonischemic setting, binding to this ligand has been used as an index of tissue depolarization (Osuga et al., 1993), and, when used as a drug, nimodipine reduced the rise in intracellular calcium during depolarization (Chisholm et al., 1993). In focal cerebral ischemia, [3H]nimodipine binding increased earlier in more severely ischemic structures than in those suffering more moderate reductions in perfusion (Hakim and Hogan, 1991) and increased [3H]nimodipine binding declined significantly with reperfusion only in those regions spared infarction (Hogan and Hakim, 1992). Thus, in addition to being an index of depolarization, the VSCC ligand, in vivo in ischemia, has allowed identification of vulnerable brain regions.
A review of the literature suggests that an interplay exists between VSCC function and rise in glutamate concentration in the ischemic setting. Excessive extracellular glutamate is neurotoxic (Choi, 1987) and regional vulnerability in focal ischemia is related to total exposure to glutamate (Osuga and Hakim, 1994). Shimada et al. (1989) showed, in a cat model of focal cerebral ischemia, that glutamate rises when CBF falls <20 ml 100 g−1min−1. Neurons exposed to glutamate succumb if calcium influx is not prevented by substitution in the milieu (Choi, 1987), and activation of the glutamate-driven N-methyl-D-aspartate (NMDA) receptor leads to a rise in intracellular calcium, which may occur at a remote time point from exposure to glutamate (Randall and Thayer, 1992). Choi (1988) and others (Graham et al., 1993) have shown that blockade of glutamate release or its receptors can be neuroprotective. Conversely, depolarization is essential both for glutamate release and for activation of the NMDA receptor by glutamate. Extracellular glutamate concentrations in global ischemia are reduced by pre-ischemic treatment with a VSCC blocker (Serieller et al., 1993), and activation of the NMDA receptor requires both the presence of glutamate and depolarization to remove the magnesium block of the receptor (Mayer et al., 1984). Thus, while both glutamate concentrations and VSCC activation of the tissue affect the outcome from ischemia, to our knowledge, they have never been studied simultaneously—in vivo and related to CBF.
The most important observation in this study is that binding to [3H]nimodipine becomes significantly elevated at a higher CBF threshold than that coinciding with a significant rise in extracellular glutamate. Although we ascertained (Fig. 2) that VSCC activity and glutamate levels had returned to baseline values prior to the onset of ischemia, it remains possible that the probes needed for this study, either directly or through the tissue injury they induced, incited ischemic depolarization after ischemia was imposed. Nonetheless, the ratio of VSCC activation to its pre-ischemic value rises to 2 before glutamate concentrations begin to rise significantly (Fig. 4). The reasons for this can only be speculated upon. Glutamate is the most important excitatory neurotransmitter in the brain, and the “steady state” glutamate values reported here, obtained between 20 and 30 min after occlusion, do not preclude earlier surges in glutamate that may have activated the VSCCs. Such transients, however, have not been noted in other studies in which glutamate levels were closely monitored after focal ischemia (Wahl et al., 1994). It is also possible that in our model, VSCCs may be differentially activated by factors that do not substantially increase glutamate release. The application of K+ to brain increases glutamate release, but the efflux of this excitotoxin decreases rapidly despite persistence of the ionic depolarization (Wahl et al., 1994). Thus, moderately ischemic brain regions may be depolarized without significant rises in glutamate concentrations. It is also possible that the binding affinity of VSCC to [3H]nimodipine may increase in the ischemic setting (Kokubun et al., 1986) before a significant rise in glutamate occurs. Thus, in the ischemic setting, a number of factors may differentially stimulate binding to VSCC and extracellular concentration of glutamate.
The present study shows that [3H]nimodipine binding increases when CBF falls to ∼50% of its control value. This is a surprisingly high CBF threshold for depolarization, but is close to the CBF threshold that defines flow to the penumbra zone (Hakim, 1987). This threshold is also very close to the one reported by Mies et al. (1991) as coinciding with suppression of cerebral protein synthesis. The same group reported a direct correlation between the onset of ischemic depolarization and decreased protein synthesis (Mies, 1993) and showed that suppression in protein synthesis is reversed by the NMDA receptor antagonist MK-801 in parallel with the suppression of peri-infarct depolarization (Mies et al., 1993b). Thus, a new CBF threshold may be defined at which extracellular glutamate concentrations have not risen measurably, but at which synthetic processes are halted in the setting of depolarization.
With the onset of ischemic depolarization at a CBF ratio to control of 0.5, neurons may activate potentially protective responses such as induction of immediate early genes (Herrera and Robertson, 1990) and expression of growth factors (Bonthius and Steward, 1993). Synthesis of these factors must proceed despite the overall depression in protein synthesis, and survival of the cell may then depend upon its ability to use these factors rapidly.
Despite differences in species and models of ischemia, it should now be permissible to refer to three defining ischemic thresholds: one at a CBF of ∼50% of control at which ischemic depolarization becomes evident, synthetic processes stop, and protective mechanisms may be activated; another at a CBF of 20–18 ml 100 g−1min−1, when rises in extracellular glutamate concentrations become significant; and, finally, a low CBF threshold of 10–12 ml 100 g−1min−1, when [Ca2+]i rises and tissue is damaged irreversibly (Uematsu et al., 1989; Harris et al., 1981). Further exploration of these thresholds and their associated events may allow us to exploit them therapeutically.
Footnotes
Acknowledgment:
The technical assistance of Georgette Roy and the clerical help of Robin Millbank and Rose Moore are gratefully acknowledged. This work was supported by a grant from the Heart and Stroke Foundation of Ontario and London Life.