
Abstract
We tested the effects of administration of a selective neuronal nitric oxide synthase (nNOS) inhibitor, ARL 17477, on ischemic cell damage and regional cerebral blood flow (rCBF), in rats subjected to transient (2 h) middle cerebral artery (MCA) occlusion and 166 h of reperfusion (n = 48) and in rats without MCA occlusion (n = 25), respectively. Animals were administered ARL 17477 (i.v.): 10 mg/kg; 3 mg/kg; 1 mg/kg; N-nitro-L-arginine (L-NA) 10 mg/kg L-NA 1 mg/kg; and Vehicle. Administration of ARL 17477 1 mg/kg, 3 mg/kg and 10 mg/kg reduced ischemic infarct volume by 53 (p < 0.05), 23, and 6.5%, respectively. L-NA 1 mg/kg and 10 mg/kg increased infarct volume by 2 and 15%, respectively (p > 0.05). Administration of ARL 17477 (10 mg/kg) significantly (p < 0.05) decreased rCBF by 27 ± 5.3 and 24 ± 14.08% and cortical NOS activity by 86 ± 14.9 and 91 ± 8.9% at 10 min or 3 h, respectively, and did not alter mean arterial blood pressure (MABP). L-NA (10 mg/kg) significantly reduced rCBF by 23 ± 9.8% and NOS activity by 81 ± 7% and significantly (p < 0.05) increased MABP. Treatment with 3 mg/kg and 1 mg/kg ARL 17477 reduced rCBF by only 2.4 ± 4.5 and 0%, respectively, even when NOS activity was reduced by 63 ± 13.4 and 45 ± 15.7% at 3 h, respectively, (p < 0.05). The data demonstrate that ARL 17477 inhibits nNOS in the rat brain and causes a dose-dependent reduction in infarct volume after transient MCA occlusion.
Nitric oxide (NO) has a multiplicity of actions and effects in physiological and pathophysiological conditions in brain and other organs (Moncada et al., 1991; Choi 1993; Bredt and Snyder, 1994; Morris and Billiar, 1994). The formation of NO is increased in brain after induction of focal and global cerebral ischemia (Kader et al., 1993; Malinski et al., 1993; Tominaga et al., 1993). Under cerebral ischemic conditions, NO may evoke contradictory effects: NO dilates blood vessels and increases blood flow and, thereby, reduces ischemic cell damage (Morikawa et al., 1994; Zhang et al., 1994); conversely NO may exacerbate ischemic cell damage by mechanisms associated with inhibition of iron-containing enzymes (Stadler et al., 1991), production of toxic peroxynitrite (Radi et al., 1991), and alteration of DNA synthesis (Wink et al., 1991). The balance between beneficial and detrimental effects of NO on cerebral ischemic cell damage may also depend upon the source of NO (Dalkara and Moskowitz, 1994). There are at least three isoforms of NO synthase (NOS): neuronal NOS (nNOS), endothelial NOS, and inducible NOS (Bredt and Snyder, 1994).
As a result of the multiple and contrasting actions of NO as well as the different sources of NOS, modulation of NO in ischemic brain has been shown to produce a diverse range of outcomes (Pelligrino 1993; Dalkara and Moskowitz, 1994; Dawson 1994). Administration of nonselective NOS inhibitors may increase or decrease cerebral ischemic cell damage. Administration of L-arginine, a precursor of NO, decreased infarction size after middle cerebral artery (MCA) occlusion by a blood flow-dependent mechanism (Morikawa et al., 1992, 1994), suggesting that endothelial NO may have a neuroprotective role. Recent studies on the transgenic nNOS knockout mouse (Huang et al., 1994) and on the rat treated with 7-nitroindazole (Yoshida et al., 1994), a nNOS inhibitor, indicated that nNOS contributes to ischemic cell damage after permanent focal cerebral ischemia. Therefore, an effective therapeutic intervention should be designed to reduce ischemic cell damage by blocking nNOS without reducing endothelial NOS activity. This model design may not be simple, in that nNOS may also regulate cerebral blood flow (CBF); suppression of nNOS to a level that reduces CBF would be detrimental.
In the present study, we tested the effect of administration of a novel selective nNOS inhibitor, ARL 17477 (Gentile et al., 1995), on ischemic cell damage and CBF in rats subjected to transient MCA occlusion. Our data indicate that this compound, when administered at a dose that does not reduce CBF, is significantly neuroprotective and reduces infarct volume by 55% after 2 h of MCA occlusion in the rat.
MATERIALS AND METHODS
All experiments were approved by the Institutional Animal Care Committee. Male Wistar rats (n = 48) weighing 270–300 g were subjected to MCA occlusion. Animals were anesthetized with 3.5% halothane for induction and 1% halothane in a 3:1 N2O/O2 (vol/vol) mixture for maintenance. The rectal temperature of the animal was maintained at 37.0 ± 0.5°C with a feedback heating pad. PE-50 catheters were placed in the right femoral artery and vein. The arterial catheter was used for continuous monitoring of mean arterial blood pressure (MABP). Arterial blood was sampled before and at 30 min after administration of pharmaceutical agents for measurements of pH, Po2, and Pco2. The venous catheter was employed for administration of pharmaceutical agents. MCA occlusion was induced for 2 h using a method of intravascular occlusion (Koizumi et al., 1986; Zea Longa et al., 1989; Chen et al., 1992). Briefly, the right common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) were isolated via a ventral midline incision. A 4–0 nylon monofilament, with its tip rounded by heating near a flame, was introduced into the ECA lumen through a puncture at the ECA and advanced into the ICA to block the origin of MCA. Restoration of MCA blood flow was accomplished by withdrawing the intraluminal suture until the tip was in the stump of the ECA. All rats exhibited left circling after MCA occlusion.
ARL 17477
ARL 17477 is one of a series of heterocyclic substituted amidines and was synthesized as described previously (Gentile et al., 1995). It possesses an IC50 equal to 35 nM versus rat neuronal NOS and is at least 100-fold selective against endothelial and inducible NOS (Gentile et al., 1995). Because of the ability of ARL 17477 to penetrate the brain and to selectively inhibit nNOS (Gentile et al., 1995), it was chosen to evaluate the contribution of nNOS to reperfusion ischemic cell damage in this model of transient focal cerebral ischemia.
Histopathology
Rats were anesthetized with ketamine (44 mg/kg body wt) and xylazine (13 mg/kg body wt) and killed 7 days after reperfusion. Animals were transcardially perfused with heparinized saline and 10% neutral buffered formalin, and brains were removed. Each brain was cut into 2 mm-thick coronal blocks in a rodent brain matrix for a total of seven blocks per animal. The brain tissue was processed and embedded, and 6 μm-thick paraffin sections were cut from each block and stained with hematoxylin and eosin (H & E) for histopathological evaluation.
Tissue volume was measured blindly using a Global Lab Image analysis system (Data Translation, Malboro, MA, U.S.A.). Each H & E section was evaluated at 2.5× magnification. Areas of infarction and both hemisphere were calculated by tracing them on the computer screen, and the volumes determined by integrating the appropriate area with the section interval thickness (Chen et al., 1992). To avoid errors associated with processing of the tissue for histological analysis, the indirect method for calculating infarct volume, in which the intact area of ipsilateral hemisphere is subtracted from the area of the contralateral hemisphere, was used (Jiang et al., 1994). The infarct volume was expressed as percentage of the infarct to the contralateral hemisphere.
Monitoring of regional CBF (rCBF) by laser Doppler flowmetry (LDF)
Male Wistar rats (n = 25) (260–300 g) were anesthetized with halothane (3.5% induction, 1.5% during surgery, and 0.8–1.0% during rCBF recordings) and allowed to breathe a 3:1 N2O/O2 (vol/vol) mixture spontaneously. These rats were not subjected to ischemia. The right femoral artery and vein were cannulated with PE-50 catheters. The arterial catheter was used for continuous monitoring of MABP and for sampling arterial blood before and after administration of N-nitro-L-arginine (L-NA) or ARL 17477. The venous catheter was employed for administration of L-NA and ARL 17477. Rectal temperature was maintained at 37.0 ± 0.5°C. Animals were mounted in a stereotaxic frame. A midline incision was performed to expose the skull. A 1.5 mm diameter burr hole was made 1.5 mm posterior and 3 mm lateral to the bregma in the right hemisphere (Paxinos and Watson, 1986). The dura was left intact. Following application of mineral oil onto the burr hole, the probe was positioned 0.5 mm above the dural surface. Regional CBF was measured using LDF (PeriFlux PF3; Perimed, Stockholm, Sweden). Regional CBF changes were recorded continuously on a multichannel chart recorder (Gould, Inc., Cleveland, OH, U.S.A.) for 10 min or 3 h after administration of pharmaceutical agents. Changes in rCBF were expressed as percentage change in flow.
Assay for NOS activity
Immediately after rCBF measurement, ∼100 mg of cortical gray matter from each hemisphere was removed and rapidly frozen. NOS activity was determined using the conversion of [3H] arginine (58.4 Ci/mmol) (NEN Dupont, Wilmington, DE, U.S.A.) to [3H] citrulline (Bredt and Snyder, 1989). Cortical samples were homogenized in 500 μl buffer containing 50 mM HEPES and 1 mM EDTA at pH 7.4. Homogenates were centrifuged at 13,000 rpm for 20 min and the supernatant used for the reaction mixture. The reaction mixture consisted of 100 μl of HEPES (50 mM), EDTA (1 mM), β-NADPH (1 mM), and Ca2+ (1.5 mM) at pH 7.4; 25 μl of 100 nM [3H] arginine (1 mCi/ml) with 18 μM unlabeled L-arginine and 25 μl of supernatant. Samples were assayed in duplicate at room temperature with a reaction time of 10 min. The reaction was stopped using 2 ml of HEPES (20 mM) and EDTA (2 mM). The mixture was then applied to 1-ml columns of Dowel AG50WX-8 (Na+ form) (Biorad, Richmond, CA, U.S.A.) and eluted with 2 ml of distilled water. [3H] Citrulline was quantified in the eluate by liquid scintillation spectroscopy at a 4 ml flow-through rate. NOS activity was expressed as percentage of control levels.
Experimental groups
To examine the effects of NOS inhibitors on infarct volume, animals (n = 48) were divided into six groups of eight rats each and administered the following (all injections were i.v.): (a) ARL 17477 (10 mg/kg body wt) was injected immediately upon reperfusion and again (5 mg/kg body wt) at 24 and 48 h of reperfusion; (b) ARL 17477 (3 mg/kg body wt) was injected immediately upon reperfusion and again (1 mg/kg body wt) at 24 and 48 h of reperfusion; (c) ARL 17477 (1 mg/kg body wt) was injected immediately upon reperfusion and again (0.3 mg/kg body wt) at 24 and 48 h of reperfusion; (d) L-NA (10 mg/kg body wt) was injected immediately upon reperfusion and again (5 mg/kg body wt) at 24 and 48 h of reperfusion; (e) L-NA (1 mg/kg body wt) was injected at 0, 24, and 48 h of reperfusion; (f) vehicle was injected at 0, 24, and 48 h of reperfusion. MABP was recorded for 30 min after administration of drugs.
To examine the effects of NOS inhibitors on rCBF and NOS activity, an additional 25 rats were subjected to rCBF, MABP, and NOS activity measurements without induction of ischemia. ARL 17477, 10 mg/kg body wt (n = 8); 3 mg/kg body wt (n = 5), 1 mg/kg body wt (n = 3); L-NA 10 mg/kg body wt (n = 4); or vehicle (n = 5) was injected (i.v.). Animals were killed at 10 min or 3 h after administration of drugs for measurement of NOS activity.
Statistical analysis
Data are presented as means ± SD and are evaluated by analysis of variance (ANOVA). Two-group comparisons of rCBF and MABP were analyzed by paired t-tests. Statistical significance was presented at the p < 0.05 level.
RESULTS
The ability of ARL 17477 to reduce infarct volume in a transient MCA occlusion rat model was investigated. As shown in Fig. 1, a low dose (1 mg/kg body wt) of ARL 17477 administered at reperfusion significantly (p < 0.05) reduced the total infarct volume by 55% measured at 7 days. Figure 2 shows area of infarction in all seven coronal sections for both the vehicle control group and the ARL 17477 (1 mg/kg body wt) group. Reduction of the infarct was evident throughout the hemisphere, and statistically significant decreases in area of infarct were detected in sections 2–6 for the ARL 17477 group. Higher doses of ARL 17477 (3 and 10 mg/kg body wt) given at reperfusion did not reduce infarct volume significantly. Treatment with the nonselective NOS inhibitor L-NA (1 and 10 mg/kg body wt) increased infarct volume by 2 and 15%, respectively (p > 0.05).
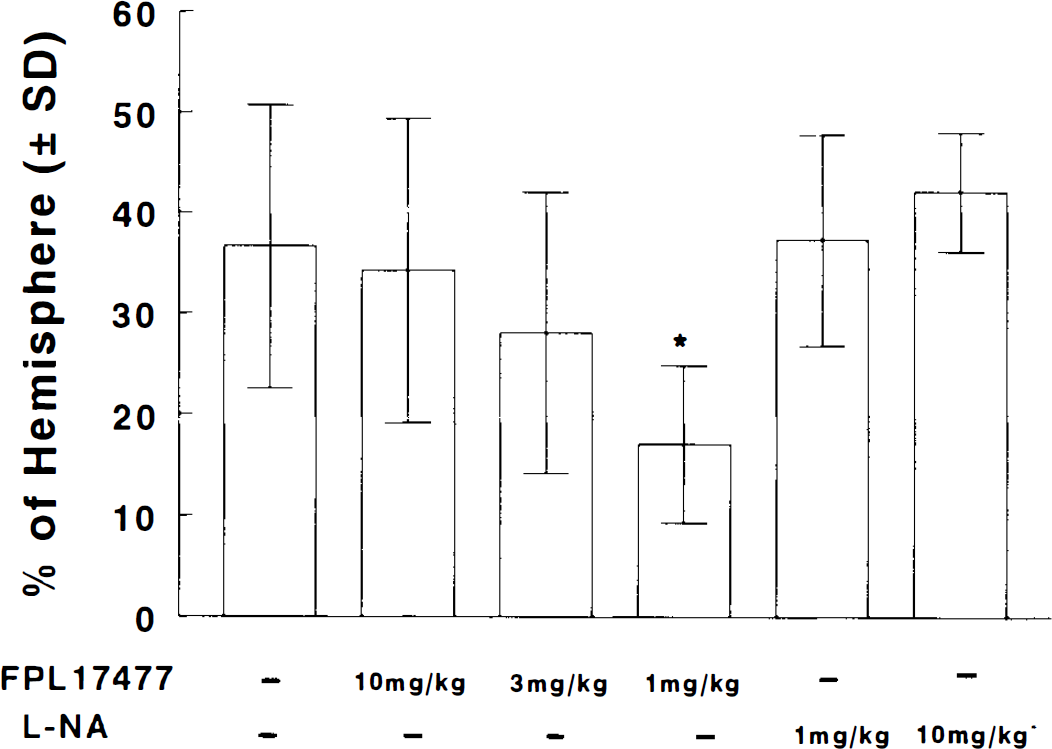
Bar graph shows the effects of ARL 17477 and L-NA on infarct volume. Values are mean ± SD. Each group includes eight rats. *p < 0.05 compared with vehicle group.
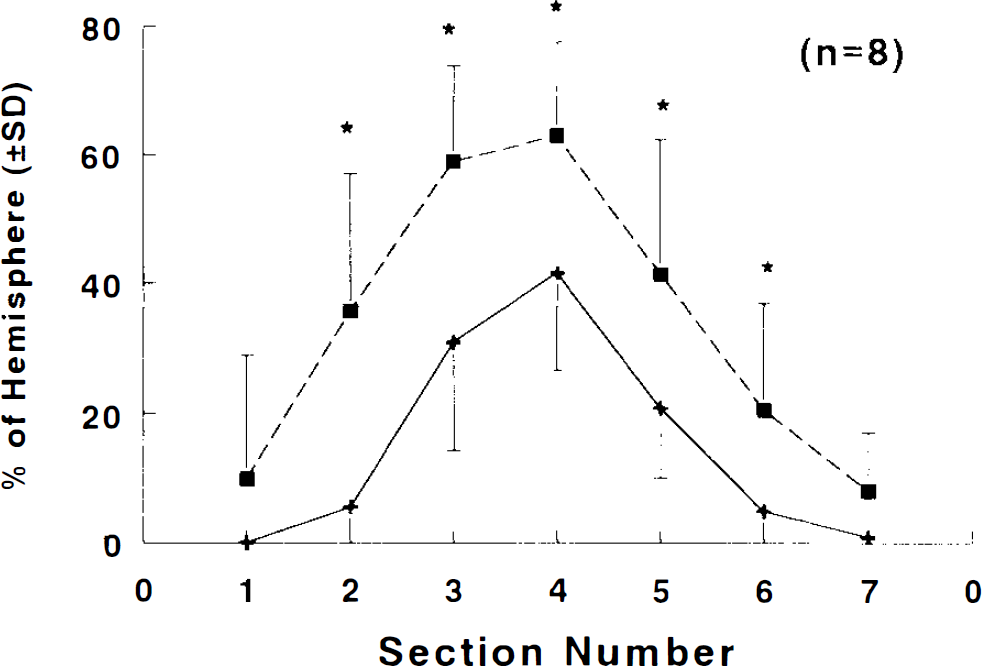
Graph shows infarct area for each coronal section (2 mm) from anterior to posterior. Values are mean ± SD. ARL 17477 (1 mg/kg body wt, +) (n = 8) significantly reduces infarct area (*p < 0.05) as compared with vehicle group (▪) (n = 8).
Blood gas parameters and MABP for all experimental groups are presented in Table 1. As shown, blood gas data were within normal physiological ranges for all groups. In addition, MABP did not change following administration of ARL 17477 (1,3, or 10 mg/kg body wt), demonstrating the absence of a detectable endothelial NOS response with this compound in vivo. Treatment with L-NA (1 mg/kg body wt) did not raise MABP. However, MABP significantly increased (98 ± 8 versus 116 ± 14 mm Hg) following administration of L-NA (10 mg/kg body wt).
Physiological variables
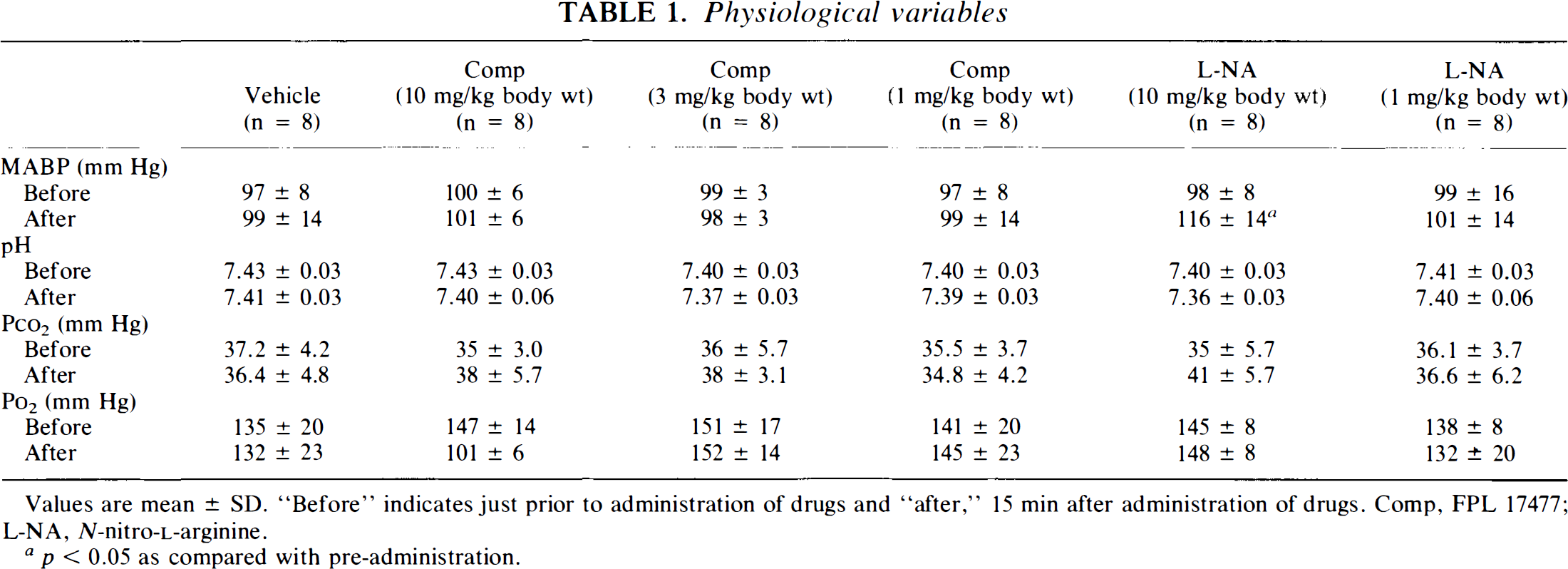
Values are mean ± SD. “Before” indicates just prior to administration of drugs and “after,” 15 min after administration of drugs. Comp, FPL 17477; L-NA, N-nitro-L-arginine.
p < 0.05 as compared with pre-administration.
Values for rCBF and cortical NOS activity at 3 h are presented in Fig. 3. Administration of ARL 17477 (10 mg/kg body wt) significantly (p < 0.05) decreased rCBF by 27 ± 5.3% (data not shown) and 24 ± 14.1% at 10 min or 3 h, respectively, and did not alter MABP in these animals (data not shown). Cortices from these rats were removed, rapidly frozen, and later assayed for remaining NOS activity. As shown, ARL 17477 significantly (p < 0.05) reduced NOS activity by 86 ± 14.9% at 10 min and 91 ± 8.9% at 3 h (n = 3) as compared to control sections. Similarly, L-NA (10 mg/kg body wt) reduced rCBF by 23 ± 9.8% and NOS activity by 81 ± 7% (data not shown). Treatment with ARL 17477 (3 and 1 mg/kg body wt) reduced rCBF by only 2.4 ± 4.5% and 0%, respectively, even when NOS activity was reduced by 63 ± 13.4% and 45 ± 15.7%, respectively, (p < 0.05).
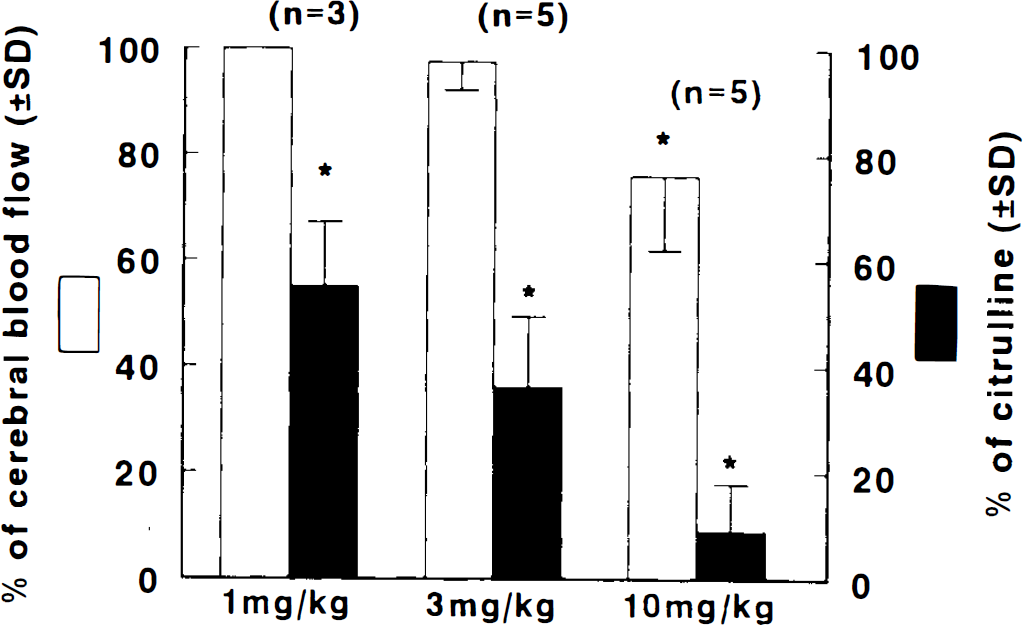
Bar graph shows the effects of ARL 17477 on rCBF (□) and NOS activity (▪). Values (mean ± SD) were obtained 3 h after administration of ARL 17477. *p < 0.05 compared with vehicle group.
DISCUSSION
Three major findings emerged from the current study. First, a low dose of ARL 17477 (1 mg/kg), with administration delayed until 2 h after onset of MCA occlusion, significantly reduces infarct volume. Second, ARL 17477 (1–10 mg/kg body wt) significantly inhibits brain NOS activity by 35–90% without raising MABP. Third, nNOS contributes to the maintenance of CBF.
The role of NO in modulating cell damage after focal cerebral ischemia is complex and controversial (Pelligrino 1993; Dalkara and Moskowitz, 1994; Dawson 1994). Some of these discrepancies may be explained by use of nonselective NOS inhibitors as well as by differences in the amount and timing (before, during, or after ischemia) of administration of NOS inhibitors or NO donors. For instance, L-NA has been shown to increase infarct volume when administered prior to MCA occlusion (Yamamoto et al., 1992). In addition, administration of L-arginine or NO donors increase basal levels of NO, increase CBF in the ischemic territory and penumbra, and can result in reduction of ischemic cell damage (Morikawa et al., 1992, 1994; Zhang et al., 1994). On the other hand, administration of a low dose of NG-nitro-L-arginine methyl ester (L-NAME) (0.1 mg/kg body wt) 30 min prior to reperfusion reduces infarct volume in transient (3 h) focal cerebral ischemia in spontaneously hypertensive rats (Ashwal et al., 1994). Similarly, a number of studies (Nowicki et al., 1991; Nafuji et al., 1992) have shown similar beneficial effects with L-NA. Unfortunately, despite all of these studies it is not possible to assign damaging or protective roles to a particular NOS isoform.
The beneficial effect of administration of L-arginine or NO donors to increase basal levels of NO after ischemia (Morikawa et al., 1992, 1994; Zhang et al., 1994) is consistent with the hypothesis that a rapid increase in NO levels at the onset of ischemia may represent an attempt by the brain to increase CBF by vasodilatation (Malinski et al., 1993). Disruption of the generation of NO at this time could markedly exacerbate the ischemic lesion. NO levels also increase above baseline during reperfusion (Malinski et al., 1993), and the increase in NO may mediate reperfusion injury. Recently, evidence has been mounting that nNOS contributes significantly to ischemic cell damage. Administration of 7-nitroindazole decreases infarct volume in permanent MCA occlusion in the rat at doses that reduce nNOS activity by 50% without affecting MABP (Yoshida et al., 1994). Also, mice deficient in nNOS activity exhibit decreased focal infarct volume 24 h after MCA occlusion (Huang et al., 1994). Results from the present study with ARL 17477 are consistent with these data and strongly suggest that nNOS contributes to reperfusion injury following transient focal ischemia in the rat.
The reason(s) for the inverse dose-response curve obtained with ARL 17477 (Fig. 1) may be related to the ability of ARL 17477 to reduce CBF at the higher doses. A reduction in CBF at the time of reperfusion will be detrimental to the outcome of the lesion (Siesjö 1992). Although we did not measure CBF changes in ischemic rats, inferences can be made from our CBF and NOS inhibition studies in nonischemic rats. We observed that doses of 10, 3, and 1 mg/kg body wt ARL 17477 reduced rCBF by 24, 2.4, and 0%, respectively, when NOS activity was reduced by 86–90, 60–70, and 35–50%, respectively (Fig. 3). A similar or even greater reduction in rCBF may occur in ischemic rats. Any beneficial effect of inhibiting nNOS would conceivably be masked by the adverse effects on rCBF. Evidenced in the present study, treatment with the nonselective NOS inhibitor L-NA (1 or 10 mg/kg body wt) did not reduce infarct volume. Reduction in CBF observed with administration of 3 or 10 mg/kg body wt ARL 17477 is likely a result of nNOS inhibition since we did not alter MABP and, thus, were not inhibiting vascular endothelial NOS. However, we cannot rule out inhibition of brain endothelial NOS at the higher doses of ARL 17477, which would be expected to have an effect on CBF.
The precise mechanisms by which NO may mediate cell death during reperfusion are unknown. One possible mechanism is that neuronally-generated NO reacts with the superoxide anion, which is also produced during reperfusion (Chan et al., 1993) to produce peroxynitrite. Peroxynitrite may then break down to a more powerful oxidant causing lipid peroxidation (Beckman 1991). Alternatively, peroxynitrite formation may lead to the irreversible nitration of critical tyrosine residues (Beckman et al., 1994), which could disrupt cellular communication and integrity. Another potential mechanism is that nNOS may form hydrogen peroxide at subphysiological levels of L-arginine or H4 biopterin (Pou et al., 1992; Cosentino and Katušić, 1995).
In summary, ARL 17477 inhibits nNOS rapidly, significantly, and selectively in the rat brain. More importantly, ARL 17477 significantly reduces ischemic cell damage after transient (2 h) MCA occlusion in the rat by selectively inhibiting nNOS when treatment is initiated at the time of reperfusion, 2 h after onset of ischemia.
Footnotes
Acknowledgment:
The authors wish to thank Denice Janus for manuscript preparation.