
Abstract
Proximate neurotoxic mechanisms during postischemic recovery may be influenced by stage of development and complicating factors such as cortical spreading depression or secondary brain insult. Using 31P nuclear magnetic resonance spectroscopy, we have monitored pH and cellular energy metabolites phosphocreatine (PCr) and ATP in the ex vivo rat cerebral cortex before, during, and after substrate and oxygen deprivation, which represents “in vitro ischemia.” There were important developmental differences in resistance and response to an ischemic insult. Twenty-one-day-old (P21) rat cortical slices had no detectable β-ATP or PCr at the end of a 20-min insult, while γ-day-old (P7) slices had 50 ± 13.7% (mean ± SD, n = 12) and 17 ± 14.8% relative to preischemia levels, respectively. Postischemic depolarization resulted in age-dependent effects on PCr (p < 0.05): In the older tissue, depolarization significantly worsened the recovery of PCr, whereas in young tissue it ameliorated recovery. This amelioration could be prevented by inhibiting nitric oxide production with methylene blue (depolarization-methylene blue interaction, p < 0.05) and enhanced by administration of the nitric oxide donor glyceryl trinitrate (GTN; p < 0.01). However, in P21 tissue, GTN further exacerbated injury (age-GTN interaction, p < 0.01). Therefore, in this vascular-independent preparation, a neuronal or glial nitric oxide-dependent mechanism appears to confer improved postischemic bioenergetic recovery in the developing brain compared with the mature brain.
In many instances of hypoxia–ischemia in infants and young children, therapeutic intervention is only practicable after the insult (Tasker et al., 1991). Of clinical concern, therefore, is not only the tolerance the young have to an insult, but also proximate postischemic neurotoxic mechanisms.
It is well known that neonatal animals display a reduced sensitivity to hypoxic–ischemic damage (Fazekas et al., 1941; Adolph, 1948, 1971) and exhibit a differential susceptibility in distribution of neuropathological sequelae when compared with adult animals (Brierley and Graham, 1984). This ontogeny-specific tolerance and difference in injury correlate well with differences in demand for oxidative metabolism (Thurston and McDougal, 1969; Duffy et al., 1975), the apparent neonatal conservation of ATP for ionic regulation during hypoxia–ischemia (Haddad and Donnelley, 1990; Xia et al., 1992; Bickler et al., 1993), and cortical synaptic maturity and sensitivity to excitatory amino acid neurotoxicity (Hamon and Heinemann, 1988; McDonald and Johnston, 1990). Importantly, in immature rats there is evidence that glutamate receptor–mediated neurotoxicity plays a major role in hypoxic–ischemic injury. The time course and histological appearance of such injury resemble those of the neurotoxic reaction to an intracerebral injection of N-methyl-D-aspartate (NMDA) (Ikonomidou et al., 1989). In addition, dizocilpine (MK-801), a noncompetitive NMDA receptor antagonist, protects against hypoxia–ischemia-induced brain damage (Hattori et al., 1989).
In some adult models of brief ischemic injury, NMDA receptor antagonists have also been effective even when administered after the end of the ischemic period (Gill et al., 1988). This postishemic protection suggests that the cellular events occurring during hypoxia–ischemia are not solely the immediate cause of death, but perhaps promote a toxic susceptibility to subsequent glutamate release, which occurs either by a spontaneous mechanism or because of neuronal activity and synaptic transmission (Szatkowski et al., 1990; Szatkowski and Attwell, 1994). Alternatively, it is possible that the postischemia protective agents are not influencing the primary ischemia-induced process, but are merely limiting secondary injury, as may occur with cortical spreading depression in penumbral neurons (Gill et al., 1992; Iijima et al., 1992), potassium release in global ischemia complicated by focal intracranial hemorrhage (Loh et al., 1979), and repeated systemic compromise (Mallard et al., 1993).
In vivo examination of cellular processes occurring during and after the ischemic period is fraught with difficulty. Consequently, much of our cellular understanding of these events comes from in vitro studies using dissociated neuronal cultures (Rothman, 1984; Choi et al., 1987; Goldberg et al., 1988; Kaku et al., 1991). To study further the developmental aspects of proximate neurotoxic mechanisms, we have used the ex vivo cortical brain slice preparation, a model that preserves intrinsic cortical anatomy and in vivo stage of development, permits controlled pharmacological interventions, and is blood flow and systemic physiology independent. In the experiments described here, we have undertaken a study using 31P nuclear magnetic resonance (NMR) spectroscopy in the superfused brain slice preparation (Bachelard et al., 1985) to look at energy metabolism following substrate deprivation, in vitro “ischemia.” We have also measured the release of taurine from the slices, using HPLC analysis of the superfusate, as an indirect indicator of regulatory cell volume changes (Pasantes-Morales and Schousdoe, 1989; Vitarella et al., 1994). By perturbing postischemic tissue with high potassium, as a model of secondary insult and sustained depolarization, we have investigated differences in postischemic neurotoxic mechanisms at two postnatal ages. Our principal observations are as follows: In cerebral cortex from 21-day-old (P21) rat pups, depolarization during recovery from ischemia worsens the recovery of high energy phosphate metabolites, but in cortex from 7-day-old (P7) rat pups, it ameliorates bioenergetic recovery via an endogenous nitric oxide (NO)–dependent mechanism, which can be reproduced with postischemic administration of an NO donor.
MATERIALS AND METHODS
Preparation of cortical brain slices and incubations
Cerebral cortical slices of 350-μm thickness from either P7 or P21 rats (three to seven animals per experiment) were prepared using a McIlwain tissue chopper. Each brain was removed from the cranial cavity within 30 s, and slices were rinsed in oxygenated Krebs–Henseleit bicarbonate (KHB) buffer before transferring them to a 20-mm NMR tube. Once in the tube, slices were perfused with KHB buffer containing 124 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 1.2 mM CaCl2, 26 mM NaHCO3, 1.2 mM KH2PO4, and 10 mM D-glucose (pH 7.4) equilibrated with O2/CO2 (19:1) for 45–60 min at 37°C and a flow rate of 20 ml/min. The perfusion apparatus (Bachelard et al., 1985) included two peristaltic pumps fitted with quick-release pump heads to make up a complete nonrecirculating perfusion circuit that would avoid the build-up of hydrostatic pressure within the NMR tube. Extracellular fluid samples of perfusion effluent could be collected after any perturbation. To monitor and utilize the intracellular phosphate (Pi) as an indicator of intracellular pH (pHi) using 31P-NMR (Taylor et al., 1983), medium phosphate was later reduced to 100 μM (substituted with equimolar amount of KCl). In vitro ischemia was induced for periods of 20, 40, or 60 min by removing 10 mM D-glucose from the superfusion medium and gassing with N2/CO2 (19:1), a procedure known to drop the oxygen tension in the superfusion chamber by >90% (Kauppinen and Williams, 1990).
In some experiments, slices were superfused with methylene blue (MB; 1 μM), glyceryl trinitrate (GTN; 100 μM), adenosine (50 μM), and theophylline (100 μM) in the recovery period after in vitro ischemia. Postischemic depolarization was induced using a high potassium perfusion medium (30 mM) by replacing an equimolar concentration of NaCl with KCl.
Reagents
Adenosine, theophylline, and MB were purchased from Sigma (Poole, Dorset, U.K.) and GTN from DuPont Pharmaceuticals Limited Multisource products (Letch-worth, Hertfordshire, U.K.).
NMR techniques
In these experiments, the metabolic status of the tissue was assessed from determinations of pHi and the relative concentrations of ATP and phosphocreatine (PCr). 31P-NMR spectroscopy was used to follow changes in pHi and metabolic recovery of high energy phosphates. Spectra were recorded using a 20-mm NMR probe in an 8.5 T vertical magnet interfaced with a SMIS console operating at 145.7 MHz. Magnetic field homogeneity was optimized by adjusting room temperature shim currents until the water proton linewidth from the preparation was <30 Hz. Throughout the experiments, 31P spectra were acquired (45° pulse, relaxation time 1.1 s, sweep width 10 kHz, 4,000 points, 280 or 500 scans) and processed as described previously (Kauppinen and Williams, 1990) with a !A sine filter over the first 2 ms of the free induction decay (to suppress broad phospholipid resonances) followed by 15-Hz line-broadening. These pulsing conditions do not allow for full relaxation of all signals (particularly the PCr) between pulses; therefore, peak area ratios do not correspond directly to concentration ratios. The data have not been corrected for this effect, but from a series of experiments comparing the acquisition described to spectra acquired with an 8-s relaxation time, the β-ATP signal is fully relaxed for all conditions. The saturation factor for the PCr signal was 64.4 ± 3.2% (mean ± SD, n = 12) and was not influenced by condition or age. Therefore, since the acquisition conditions in the experiments described were the same for all 31P spectra, and we were interested in time-dependent changes in individual metabolites during and after ischemia, the changes in peak heights of the PCr, γ-ATP, and β-ATP relative to their control values were used (Kauppinen and Williams 1990).
The pHi was calculated from the chemical shift (σ) of Pi relative to PCr at 0 ppm using the following titration curve (Taylor et al., 1983): pHi = 6.75 + log [(σ — 3.27)/(5.69 — σ)]. It is generally accepted that absolute pH determinations by NMR are accurate to within ±0.1 pH unit, but that pH changes can be measured with a precision of ±0.03 pH unit.
Biochemical analysis of taurine
HPLC was used to measure taurine in samples of effluent from the brain slice perfusion system. Without pretreatment, samples were analyzed using a Lichrospher C18 reverse phase column (Merck). Following derivatization with o-phthalaldehyde (Roth, 1971) and elution with a gradient system running from 90% NaH2PO4 (pH 6.0)/10% methanol (vol/vol) to 40% (NaH2PO4 (pH 6.0)/60% methanol over 30 min, fluorescence detection was used at excitation and emission wavelengths of 340 and 450 nm, respectively. Changes in taurine concentration during and after intervention within experiments were expressed relative to the mean of four basal levels taken at least 12 min apart during an initial control period. Comparisons between ages and conditions (in terms of percentage change) were made using statistical measures described herein.
Overall experimental design
Preliminary experiments were carried out to assess the time dependence (up to 60 min) of substrate deprivation (no oxygen, no glucose) on high energy phosphorus metabolites in P7 and P21 cortical slices, as well as metabolic stability on early recovery (up to 40 min). P21 slices were always more sensitive than P7 slices to in vitro ischemia. A 20-min insult in both P21 and P7 tissue resulted in consistent injury and stable recovery within 20 min of reperfusion with oxygen and glucose containing KHB buffer. Next, this 20-min insult was fully characterized. Subsequent studies were performed investigating the early recovery period with ionic perturbation (depolarization) and pharmacological intervention. The effects on changes in pHi and high energy phosphorus metabolites were then compared using statistical measures described.
In vitro ischemia studies
After recovery from dissection, perfusion of slices with well-oxygenated KHB buffer was continued for 45–60 min (Fig. 1). Following this period, the perfusion medium was changed to KHB buffer containing a reduced concentration of phosphate (100 μM). The washout period for medium changes in our apparatus is <5 min. Over the next 50 min, six control 31P spectra were acquired (3 × 500 scans, 3 × 280 scans), and four perfusion effluent samples were stored for analysis of basal taurine concentration. Next followed two 20-min intervals, during each of which three 31P spectra were acquired (3 × 280 scans), and at the end of each a superfusate effluent sample was collected. In the first interval, the slices were perfused with either KHB buffer lacking oxygen and glucose or normal control buffer. In the second interval (recovery after ischemia 0–20 min), slices were perfused with either control or high potassium (30 mM [K+]e) containing KHB buffer. Recovery from early postischemic perturbation in control KHB buffer was also studied, during which time three 31P spectra were acquired (3 × 280 scans) and a superfusate effluent sample was collected. Finally, the postischemic period was further studied with pharmacological interventions: During the initial recovery from ischemia (0–20 min), drugs were added to the control or high potassium (30 mM [K+]e) containing KHB buffer and then the tissue was superfused with control KHB buffer (21–40 min). 31P spectra were also acquired (3 × 280 scans), and a superfusate effluent sample was collected for each 20-min interval.
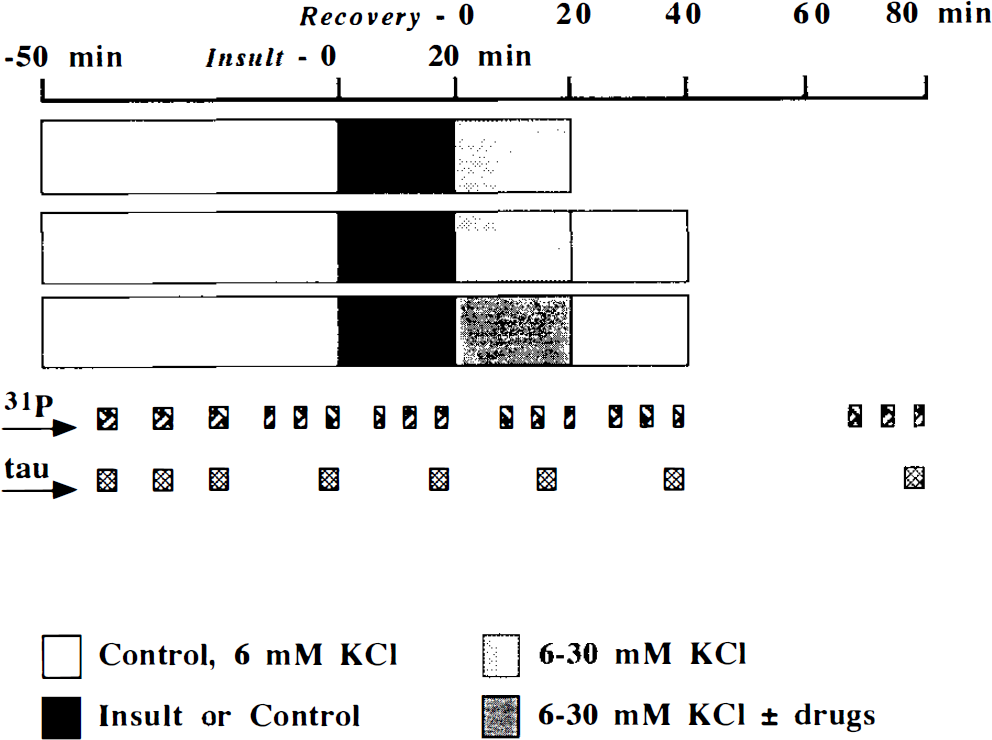
Diagram illustrating the time course of experiments after an initial 45- to 60-min equilibration period in KHB buffer. Each horizontal bar represents a series of expeiments. 31P, times when 31P-NMR spectra were acquired; tau, time when superfusate effluent samples were collected. See text for abbreviations.
Sixty separate brain slice NMR spectroscopy experiments were conducted: (a) n = 24 to evaluate the effect of cortical development on resistance to ischemia and the effect of postischemic depolarization during recovery [3 experiments × 4 conditions (control, ischemia, high potassium, ischemia followed by high potassium) for two ages (P7 and P21)]; (b) n = 18 to study the effect of a period of postischemic depolarization on recovery [(3 experiments × 4 conditions in P7 tissue) and (3 experiments × 2 conditions in P21 tissue)]; (c) n = 18 drug experiments during the postischemic period [3 experiments × 3 agents (adenosine, theophylline, and MB) during depolarization in P7 tissue], [3 experiments × 1 agent (MB) postischemia in P7 tissue], and [3 experiments × 1 agent (GTN) postischemia for two ages (P7 and P21)].
Statistical analysis
Throughout this article, quantitative results are presented as means ± SD. Statistical comparisons were made of mean relative NMR metabolite values, change in pHi, and change in effluent taurine concentrations at a priori chosen time points (Fig. 1) in the protocol (end of ischemia—20 or 40 min, recovery—20 or 40 min). We have used both nonparametric tests and an analysis of variance (ANOVA) to look for statistically significant (p < 0.05) main effects and interactions. ANOVA was performed on the logarithms of percent changes to overcome the nonnormal distribution of ratios and to comply with a linear model testing for interactions.
RESULTS
Effect of development on resistance to ischemia-induced bioenergetic failure: comparison of P7 and P21 ex vivo cortical slices
31P-NMR spectra from a P7 and P21 superfused brain slice preparation before, during, and following a 20-min in vitro ischemic insult are shown in Fig. 2. In these studies, before insult, the initial pHi was similar in P7 (7.20 ± 0.03) and P21 cortical slices (7.22 ± 0.04). At the end of a 20-min in vitro ischemic period, pHi fell significantly (n = 12, p < 0.01) in both P7 and P21 slices (–0.16 ± 0.03 and −0.11 ±0.06, respectively), but more so in the P7 slices (p < 0.05). The levels of the energy metabolite PCr were particularly sensitive to changes in the availability of oxygen and glucose. However, the effect of ischemia was different at the two postnatal ages: P21 slices retained minimal β-ATP and PCr at the end of the insult, while P7 cortical slices had 50 ± 13.7 and 17 ± 14.8% relative to preischemic levels, respectively. In fact, to induce a similar profound energy failure in P7 slices, a 40-min in vitro ischemic period was necessary.
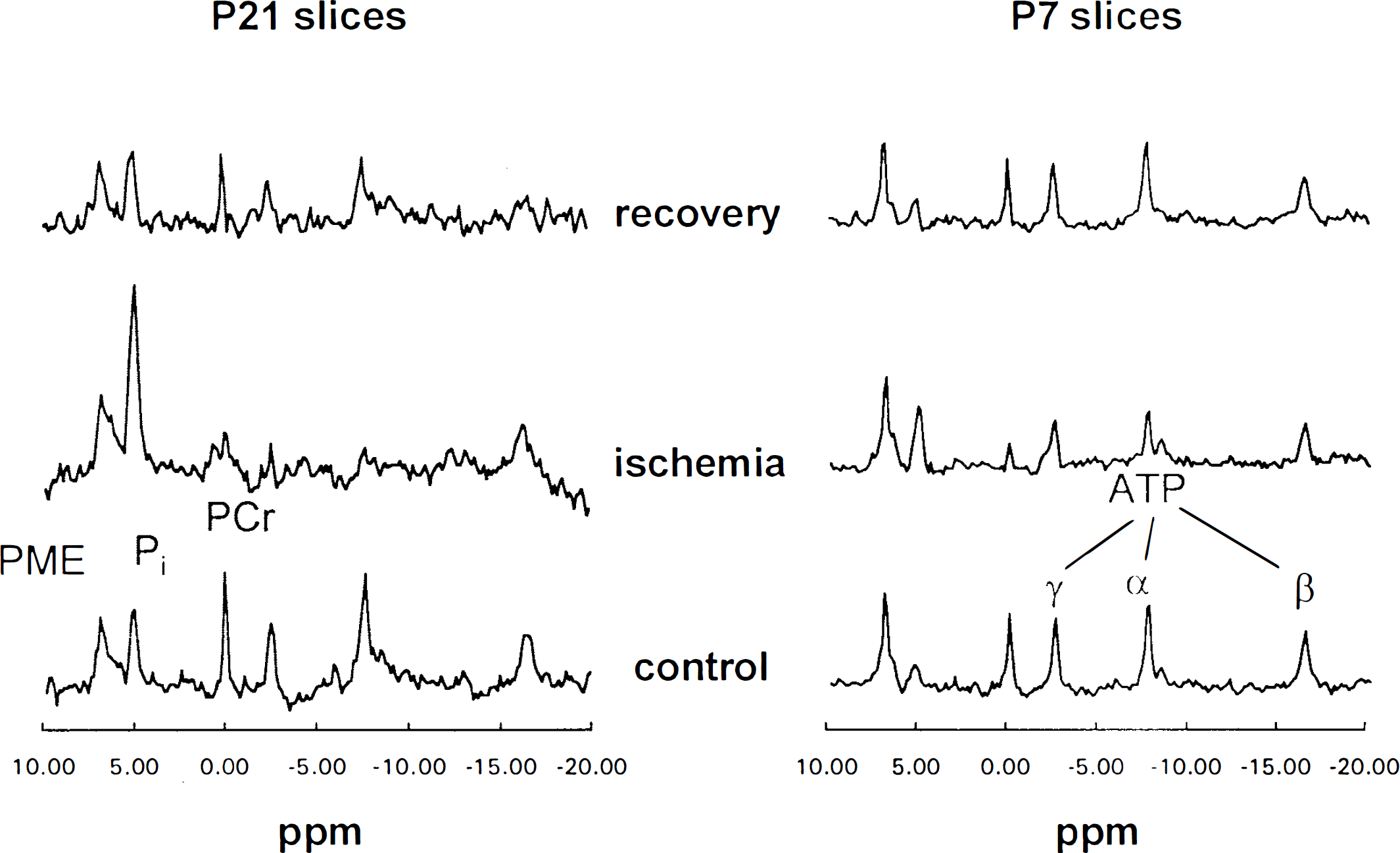
31P-NMR spectra of superfused cortical brain slices. 31P-NMR spectra from P7 and P21 rat cortical slices are shown. Each spectrum is the sum of 280 scans recorded prior to ischemia (control), at the end of the ischemic period (13–20 min), and at the end of a 20-min recovery period (13–20 min). Acquisition and processing conditions are given in Materials and Methods. PME, phosphomonoesters (mainly phosphorylethanolamine); ppm, parts per million chemical shift scale expressed relative to PCr at 0 ppm. See text for other abbreviations.
Recovery from in vitro ischemia and effect of depolarization
Following the 20-min insult period, the brain slices were allowed to recover for a further 20 min in either normal (6 mM [K+]e) or high K+ (30 mM [K+]e) buffer (Table 1). Because of the balanced design of the experiment, there is an experimental group at each age that experiences the effect of high K+ without intervening ischemia. The data in Table 1 summarize the changes in PCr and β-ATP relative to preischemic levels and the overall change in pHi (preischemia–recovery ΔpHi) at the end of the recovery period. By carrying out ANOVA on these data, we are able to evaluate independently the effects of age, ischemia, and high K+ on high energy phosphates and pHi and also determine whether factors such as high K+ and ischemia act independently or synergistically.
Summary of changes induced by 20-min in vitro ischemia observed after 20-min recovery period in presence of 6 or 30 mM KCl
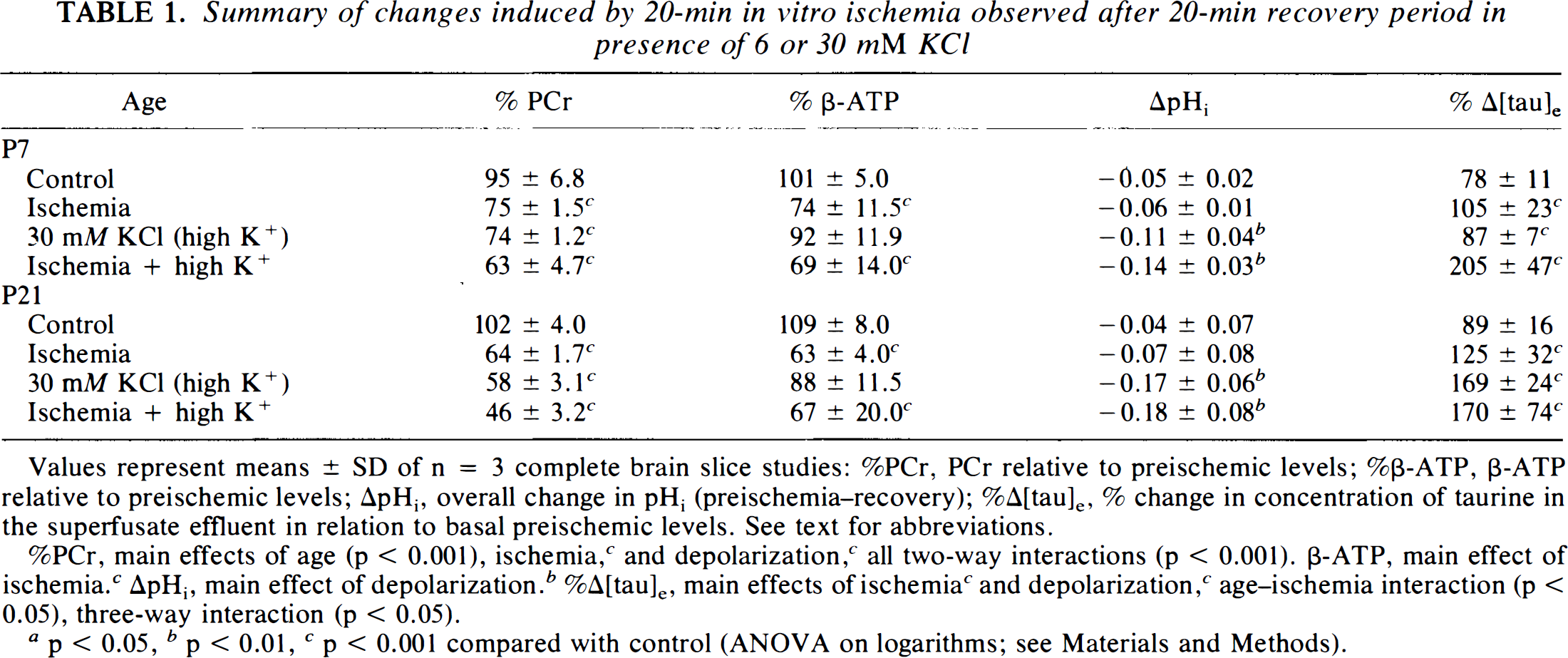
Values represent means ± SD of n = 3 complete brain slice studies: %PCr, PCr relative to preischemic levels; %β-ATP, β-ATP relative to preischemic levels; ΔpHi, overall change in pHi (preischemia–recovery); %Δ[tau]e, % change in concentration of taurine in the superfusate effluent in relation to basal preischemic levels. See text for abbreviations.
%PCr, main effects of age (p < 0.001), ischemia, c and depolarization, c all two-way interactions (p < 0.001). β-ATP, main effect of ischemia. c ΔpHi, main effect of depolarization. b %Δ[tau]e, main effects of ischemia c and depolarization, c age-ischemia interaction (p < 0.05), three-way interaction (p < 0.05).
p < 0.05, b p < 0.01, c p < 0.001 compared with control (ANOVA on logarithms; see Materials and Methods).
First, with respect to PCr, there are significant main effects for all three variables (age, ischemia, and K+) and significant interactions for each pair of variables. This indicates that PCr recovery is significantly less following either ischemia or high K+ and that the older age group recovers to a lesser extent. The interaction between ischemia and high K+ shows that these factors do not act independently. Second, it is notable that differing effects are seen on β-ATP and pHi: Only ischemia had a significant effect on β-ATP and only high K+ on pHi.
We also note by inspection of the data that the PCr/ATP ratio remains the same under conditions of ischemia only, whereas it is reduced under conditions of raised K+. The reduction in the PCr/ATP ratio by high K+ can be partly explained by induced pH changes, since via the creatine kinase equilibrium an acid shift in pH will result in a decrease in the amount of PCr. Nevertheless, it is evident that the effect of K+ on PCr is greater for the older age group, whereas there is no age dependence for the pH effect, which indicates that, at least in the older group, there is another mechanism by which high K+ influences the bioenergetic status.
The concentration of taurine in the superfusate effluent ([tau]e) increased during the period of in vitro ischemia (n = 12, %Δ[tau]e, % change in concentration of taurine in the superfusate effluent in relation to basal preischemic levels = 367 ± 150). After 20-min recovery (Table 1), the statistically significant main effects were ischemia (p < 0.001) and potassium (p < 0.001), with a three-way interaction between age, ischemia, and depolarization (p < 0.05), indicating that in postischemic P7 cortical slices, depolarization induces a greater and further rise in [tau]e.
Recovery after postischemic depolarization: differential age effects mediated by endogenous nitric oxide
It is evident that recovery of bioenergetic status and cell volume changes (as supported by changes in [tau]e) were exacerbated for both age groups if the slices were depolarized during the recovery period. To differentiate between the interactive effects of age and depolarization postischemia on energy state, we extended the recovery period by a further 20 min in normal KHB buffer. At the end of this period, pHi was not significantly different from control, and PCr had recovered somewhat toward control values (cf. Tables 2 and 3). At this time, there was an age effect (p < 0.001) and age–depolarization interaction (p < 0.05) on %PCr relative to control (i.e., 40-min recovery: 20 min after the initial 20-min postischemic recovery in either 6 or 30 mM KCl). Of note, the interaction between postischemic depolarization and age reflected an amelioration of bioenergetic recovery in P7 slices and a significant worsening of postischemic recovery in P21 cortical slices.
Summary of changes induced by in vitro ischemia observed after initial 20-min recovery period in presence of 6 or 30 mM KCl, followed by 20 min in 6 mM KCl

Values represent means ± SD of n = 3 complete brain slice studies. %PCr, PCr relative to preischemic levels; %β-ATP, β-ATP relative to preischemic levels; ΔpHi, overall change in pHi (preischemia–recovery). (t1 26 min postischemia, 6 min postdepolarization/control; t2. 33 min postischemia, 13 min postdepolarization/control; t3, 40 min postischemia, 20 min postdepolarization/control. ANOVA on logarithms (see Materials and Methods) comparing t3 %PCr ( a p < 0.01, b p < 0.001) revealed a main effect of age b and an age–depolarization interaction (p < 0.01) for the 20-min in vitro ischemic insult. On comparing %β-ATP, there was a main effect of age. a A comparison of duration of insult in P7 slices revealed no interaction between depolarization and the duration of ischemia on t3 %PCr. There were no significant changes in pHi. See text for abbreviations.
Summary of in vitro ischemia-induced changes in PCr and β-ATP modified by MB and GTN administered during initial recovery period (0–20 min) followed by further 20 min in 6 mM KCl

Values represent means ± SD of n = 3 complete brain slice studies. %PCr, PCr relative to preischemic levels; %β-ATP, β-ATP relative to preischemic levels; t3, 40 min postischemia, 20 min postdepolarization/drug administration period; MB, 1 μM; GTN, 100 μM. ANOVA (on logarithms, see Materials and Methods) comparing MB effect on t3 %PCr ( a p < 0.05, b p < 0.01) demonstrated a depolarization-MB interaction, a with an inhibition of the depolarization protective effect. The t3 % β-ATP exhibited no significant effects. An examination of GTN effects in P7 and P21 slices: t3 %PCr, age effect (p < 0.01), age–GTN interaction. b There were no significant changes in pHi. See text for abbreviations.
Experiments common to Table 2.
In relation to changes in β-ATP, there was greater recovery in P7 slices, but no interactions. As indicated in Fig. 2, the effect of 20 min with no oxygen and no glucose (in vitro ischemia) in P7 slices was different from that observed in P21 slices. The differential effect of depolarization at these two stages of cortical development may be explained by a difference in severity of underlying insult. However, when the period of ischemic insult in P7 slices was extended to 40 min, a period known to deplete all energy metabolites, depolarization did not exacerbate the expected recovery bioenergetic state, suggesting that these changes were age and not severity of insult dependent.
Since the findings in P7 slices were reminiscent of previously described ischemia- or depolarization-induced adenosine inhibitory neuromodulation (Van Wylen et al., 1986; Schubert and Kreutzberg, 1990; Dux et al., 1992), initial experiments were carried out to determine whether the seemingly early neuroprotective effect of depolarization could be mediated by adenosine release. The postischemic amelioration of energy state was neither mimicked by administered adenosine (50 μM) in the absence of depolarization nor inhibited by the nonspecific adenosine antagonist theophylline (100 μM) in the presence of depolarization (data not shown).
Next, we studied the possible mediation of this effect by the endogenous production of NO (Table 3). In P7 slices, MB (1 μM), an inhibitor of both NO synthesis and soluble guanylyl cyclase activity (Mayer et al., 1993), administered during the early postischemic recovery period (tr = 0–20 min) inhibited the ameliorating effect of depolarization on PCr recovery (t3 %PCr depolarization–MB interaction, p < 0.05). This suggested that the depolarization effect was mediated by the endogenous generation of NO. GTN (100 μM), an NO-generating compound (Ignarro et al., 1981), added to the recovery superfusate for 20 min in the absence of depolarization mimicked the effect of postischemic depolarization in both P7 and P21 slices: improving PCr recovery in the younger tissue and worsening PCr recovery in the older tissue, as indicated by the significant age–GTN interaction (Table 3).
DISCUSSION
The results of these bioenergetic studies show that the stage of postnatal cortical development has significant bearing on resistance to an hypoxic–ischemic insult, the mechanism of injury during postischemic recovery, and the postischemic response to NO.
Critique of methods
The study of energy metabolism using NMR spectroscopy in ex vivo respiring cortical brain slices has a number of methodological advantages over similar in vivo brain studies in live animals (Bachelard et al., 1985; Kauppinen and Williams, 1990; Espanol et al., 1992): NMR signals from extracerebral soft tissue and muscles can be eliminated; specific brain regions can be selected for study; spectral resolution is greater; and pharmacology is more controlled. It should be remembered, however, that the use of NMR-detected concentrations of PCr and other metabolites as a marker of cell injury is based on the premise that initial recovery of normal bioenergetic state equates with later structural and functional viability, which, although justified by other reports in ex vivo cortical slices (Kauppinen and Williams, 1990, 1991), in vivo brain (Hurn et al., 1991), and liver (Bates et al., 1988), still requires verification. Furthermore, interpretation of such changes needs to take into account the unavoidable fact that the tissue being studied has been previously compromised by the process of preparation, although after reoxygenation, absolute concentrations of adenylates and PCr in the ex vivo brain slice preparation may attain ∼80% of those found in vivo, with PCr/ATP ratios the same as in vivo (Whittingham et al., 1984).
In this context, these experiments have not looked at prolonged recovery, but rather have focused on the early postischemic recovery period (up to 40 min). Our previous work with energy failure in brain slice cultures (Tasker et al., 1992; Vornov et al., 1994) indicates that ionic and NMD A receptor–mediated events in the first 30 min of reperfusion are of major significance and thus warrant the concentration on energetics, reflecting ionic stress (Haddad and Donnelley, 1990; Xia et al., 1992; Bickler et al., 1993), during the early recovery period.
Maturational changes in response to in vitro ischemia and postischemic depolarization
The ischemic insult was the same for both age groups, yet it clearly had a more severe effect on the older tissue, in which high energy phosphate metabolites were completely depleted, thus confirming the reduced sensitivity of the postnatal brain to ischemic insult (Fazekas et al., 1941; Adolph, 1948, 1971). The P7 slices were also slightly more acidic at the end of the ischemic period. No glucose was present during the insult, and the difference in acidosis was not due to more accumulated lactate (unpublished observations), so it may represent differences in proton exchangers or intracellular buffering capacity.
Ionic perturbation of postischemic tissue with sustained depolarization induced marked effects in PCr, pHi, and taurine release. Some of the differences in PCr at this stage of recovery could be accounted for by changes in pHi: The creatine kinase equilibrium is pH-dependent, so a decrease of 0.2 pH unit will lead to a reduction in the PCr/creatine ratio of almost 40%, if there is no change in the ATP/ADP ratio or free Mg2+. The reduction in PCr following ischemia is accompanied by a parallel loss of ATP, possibly indicating a failure of some cells to synthesize normal levels of high energy phosphate. In depolarization, however, the capacity of all cells to maintain normal ATP levels is unaffected; thus, it appears that the energetic stress of depolarization is relatively mild compared to ischemia. But the absence of any interaction between ischemia, depolarization, and age on pHi, yet there being one on PCr, suggested that there was an age-dependent further bioenergetic stress induced by depolarization in postischemic cortical tissue—an interpretation confirmed by the effects and interactions we observed in changes in extracellular taurine, which correlates with regulatory cell volume changes (Pasantes-Morales and Schousdoe, 1989; Vitarella et al., 1994). These differential age responses to one level of increased [K+]e may be attributable to inherent differences in normal ambient tissue electrolyte levels (Vernadakis and Woodbury, 1962) or likely developmental changes in the Na+, K+ -ATPase system (Haglund and Schwartzkroin, 1990). We have not examined whether or not similar cellular responses occur at different ages, but with different levels of [K+]e.
Age-related altered postischemic physiology and nitric oxide
In our studies, perturbing postischemic cortex with a period of depolarization disclosed important age-related differences in proximate neurotoxic mechanisms. In P21 cortical slices, depolarization significantly worsened the recovery of PCr, whereas in P7 slices it ameliorated PCr recovery. This protective effect appeared to be particular to the young cortex, rather than to the severity of ischemia-induced energy failure, since a prolonged insult with total energy failure in P7 tissue did not make depolarization produce an exacerbation of potential energetic recovery. In view of the rapid time course of these effects, some experiments were directed toward the possibility that high potassium released an inhibitory neuromodulator (Van Wylen et al., 1986; Schubert and Kreutzberg, 1990; Dux et al., 1992). In our model, a depolarization-induced adenosine-mediated protective mechanism did not appear to be of significance.
The previously reported dual neuroprotective and neurodestructive effects of NO (Lipton et al., 1993) prompted us to investigate whether NO mediated the differential changes of depolarization on postischemic cortex. NO in different oxidation states (NO′ or in a form related to the nitrosonium ion (NO+) may have opposite effects on neurons with either the induction of toxicity in the case of NO′ (Dawson et al., 1992) or protection by NO+ reacting with free sulfhydryl groups and altering function, e.g., the redox modulatory site on the NMD A receptor (Lei et al., 1992; Lipton et al., 1993). The finding in P7 slices, that MB, an NO synthase and soluble guanylyl cyclase inhibitor, abolished the protective effect of postischemic depolarization, suggested that the depolarization protective effect was mediated by the endogenous generation of NO. Nitroglycerin, an NO-generating compound, is metabolized (in part to NO) by way of 5-nitrosothiol, an NO+ equivalent (Ignarro et al., 1981; Lei et al., 1992; Lipton et al., 1993). [At best, NO′ production is slow (<0.001 %/min), even with millimolar concentrations of GTN and available thiols.] In P7 slices, the addition of GTN postischemia resulted in an improved recovery. A detrimental response was seen in P21 cortex.
In relating these observations to in vivo models of focal cerebral ischemia: Inhibition of NO synthesis has resulted in conflicting reports concerning possible neuroprotection (Buisson et al., 1992; Yamamoto et al., 1992; Zhang and Iadecola, 1994). These apparent contradictions may be reconciled by differing effects of NO in the neuronal, glial, or endothelial compartment. Focal ischemia in mice deficient in neuronal NO synthase demonstrates that neuronal NO production appears to exacerbate acute ischemic injury, whereas vascular NO protects (Huang et al., 1994). Our findings in older tissue are entirely consistent with these observations, but they contrast with the results in postnatal tissue.
Taken together, the age-related transition between these different effects of depolarization and GTN postischemia may relate to the significance of certain inherent ontogeny-specific neuronal properties or cell functions, for instance, changes in the relative importance of NMDA or non-NMDA type of glutamate receptors (Ikonomidou et al., 1989; Constantine-Paton et al., 1990), brain redox state (Guarneri and Bonavita, 1966), intracellular calcium regulation (Bickler et al., 1993), and propensity to sustain cortical spreading depression (Bures et al., 1974).
CONCLUSIONS
In our ex vivo cortical slice model of cerebral hypoxic–ischemic insult, we have studied age-related vascular-independent proximate mechanisms of neuronal injury. Our findings indicate that postischemic depolarization (as may occur in spreading depression) and direct administration of NO generators to the developed cortex exacerbate cellular stress. However, in postnatal brain tissue, postischemic depolarization (an NO-mediated process) or administration of GTN (an NO+ donor) has a very different effect, with the amelioration of proximate bioenergetic cell stress. This mechanism of injury and potential for protection special to the developing brain suggests that the use of NO-generating agents with both beneficial vascular and neuronal or glial effects should be explored further.
Footnotes
Acknowledgment:
This work was supported by the Child Health Research Appeal Trust (R.C.T), the Wellcome Trust, and the Rank Foundation.