
Abstract
The purpose of this study was to evaluate amino acid neurotransmitter dynamics in the reperfusion phase after transient cerebral ischemia. In vivo microdialysis was used to measure extracellular amino acid levels in a rabbit model of focal ischemia. During 30 min of transient ischemia (n = 5), small but significant (p < 0.05) increases in glutamate, aspartate, γ-aminobutyric acid (GABA), and taurine were noted. These elevations rapidly returned to baseline levels upon recirculation and remained constant for up to 5.5 h of reperfusion. In rabbits subjected to 2 h of transient ischemia (n = 5), two phases of amino acid release were seen. During ischemia, large (5- to 50-fold) elevations in glutamate, aspartate, GABA, and taurine occurred, as expected. These elevations rapidly normalized upon unocclusion. However, significant (p < 0.05) secondary elevations in glutamate, aspartate, and GABA occurred after 2–4 h of reperfusion. Regression analysis demonstrated significant correlations between primary (ischemic) and secondary (reperfusion) efflux. In permanent ischemia (n = 5), amino acid levels remained elevated throughout the entire experiment. Secondary elevations in excitatory amino acids may further contribute to the excitotoxic cascade during reperfusion.
Glutamate excitotoxicity has been demonstrated to mediate neuronal death in cell cultures subjected to hypoxia or glucose deprivation (Rothman, 1984; Choi et al., 1987; Monyer and Choi, 1988). In vivo microdialysis studies in various animal models of cerebral ischemia have clearly shown that large elevations in excitatory amino acids occur (Benveniste et al., 1984; Hillered et al., 1989). The degree of ischemic damage is positively correlated with the amount of glutamate released during ischemia (Butcher et al., 1990). However, recent studies have provided pathophysiologic evidence for the continued progression of damage following transient focal cerebral ischemia (Selman et al., 1991; Yang and Betz, 1994). Several indirect lines of evidence suggest that intraischemic glutamate injury alone may not account for all the excitotoxic damage that occurs. Although current data demonstrate that glutamate levels rapidly normalize upon unocclusion (Benveniste et al., 1984; Hagberg et al., 1985; Globus et al., 1988; Andine et al., 1990; Sciotti et al., 1992; Torp et al., 1993), treatment with glutamate antagonists continues to be effective in some studies when given during the recirculation phase (Steinberg et al., 1988, 1989; Nellgard and Wieloch, 1992; Nishikawa et al., 1994). Furthermore, calculation of an “excitotoxic index” based on relative amounts of glutamate, γ-aminobutyric acid (GABA), and glycine suggested that postishemic excitotoxicity may play a critical role in the maturation of injury at 2–3 h following multiple transient forebrain ischemia (Lin et al., 1992).
Some evidence already exists to support this concept of additional excitotoxicity after reperfusion. In the retina, reperfusion induced higher (seven-fold) increases in glutamate compared with the increase during ischemia (two- to threefold) (Louzada-Junior et al., 1992). In the rabbit brain, seizure-induced glutamate and GABA levels were significantly increased when the animals were previously subjected to transient hypoxia (Young et al., 1992). Finally, a study using in vitro KCl-evoked release in excised brain tissue showed that glutamate efflux was significantly increased after 6-h recirculation in a rat global ischemia model (Simms, 1991). Augmented excitatory dynamics during times when metabolic states are already compromised may contribute to the development of damage during reperfusion injury.
In this study, we used in vivo microdialysis to map the temporal profile of amino acid neurotransmitters following different periods of ischemia–reperfusion. We chose a model where minimal damage is known to occur after 30 min of transient occlusion, whereas maximal damage is known to occur after both 2-h as well as permanent occlusions. We explored the hypothesis that secondary alterations in amino acid profiles occur during late phases of recirculation and that additional elevations in excitatory amino acids may contribute to ongoing processes of damage after transient focal ischemia.
MATERIALS AND METHODS
Animal model
All experimental protocols followed NIH Guidelines for the Care and Use of Laboratory Animals and were approved by the Massachusetts General Hospital Subcommittee on Research Animal Care. Male New Zealand rabbits, weighing 2.5–3.5 kg, were anesthetized with 3–5% halothane inhalation. Polyethylene catheters were inserted into an ear vein and a femoral artery. These procedures allowed infusion of fluid and drugs, monitoring of blood pressure, and sampling of arterial blood during the experiment. Body temperature was monitored through a rectal probe. Following tracheal cannulation after tracheostomy, pancuronium bromide (0.2 mg/kg) was administered intravenously, and the animals were ventilated mechanically with 5% oxygen/95% air with a mixture of 1–2% halothane. Supplemental doses of pancuronium were administered as required. Sodium bicarbonate was administered to correct metabolic acidosis as needed. The head was fixed with a stereotaxic frame. Focal cerebral ischemia was induced by transorbital three-vessel occlusion as previously described (Lo and Steinberg, 1992). The intracranial portion of the internal carotid artery, the proximal portion of the middle cerebral artery, and the anterior cerebral artery were occluded with mini aneurysmal clips. Recirculation was performed by releasing all clips. Restoration of blood flow was verified under the microscope in every case. At the end of all experiments, rabbits were killed with 5 ml sodium pentobarbital i.v. Brains were removed, cut into 3-mm coronal slices, and subjected to staining by immersion with 2% 2,3,5-triphenyltetrazolium chloride. Lesion area was quantified with computer-based image analysis.
Microdialysis procedure
After transorbital craniectomy for arterial occlusion, the dorsal cranium was exposed and drilled at 4 mm anterior to the bregma and 6 mm lateral (left) to the sagittal suture. Another burr hole was drilled in the contralateral side to insert a 33-gauge temperature probe for monitoring brain temperature. A microdialysis probe (CMA10, 4-mm membrane length, 0.5-mm diameter) was positioned into the left cerebral cortex at a depth of 6 mm from the brain surface. This location has been previously confirmed to represent the central region of the ischemic distribution for this model (Steinberg et al., 1988, 1989, 1991; Lo and Steinberg, 1991). The probe was perfused with artificial CSF (NaCl, 125 mM; KCl, 2.5 mM; CaCl2, 1.2 mM; NaH2PO4•H2O, 0.5 mM; Na2HPO4, 5 mM; MgCl2•6H2O, 1 mM; ascorbic acid, 0.2 mM) at a flow rate of 3.0 μl/min using a microinfusion pump (Harvard Apparatus), and samples were collected every 10 min. After an equilibrium period of 60 min, nine samples were collected before occlusion and continued for 6 h following occlusion. The following groups of rabbits were examined: Group 1 (n = 5), 30-min occlusion and 5.5-h reperfusion; Group 2 (n = 5), 2-h occlusion and 4-h reperfusion; Group 3 (n = 5), 6-h occlusion without reperfusion; Group 4 (n = 3), sham-operated controls (craniectomy and microdialysis without occlusion).
Amino acid measurement
In addition to microdialysate samples, CSF, plasma, and brain tissue homogenates were analyzed. CSF samples were collected from the subarachnoid space close to the left internal carotid artery and plasma was taken from arterial blood during ischemia–reperfusion. Brain biopsies (∼1-g samples) were obtained from contralateral hemisphere and homogenized in 0.1 N HCl. Amino acids were determined with the o-phthalaldehyde procedure (Lindorth and Mopper, 1979), but with the following modifications (Newcomb and Palma, 1994). The reagent was made by mixing ∼100 mg o-phthalaldehyde (Sigma), 500 ml ethanethiol (Fluka), and 30 ml 0.5 M sodium borate, pH 10.0–10.2. Elution used an aqueous buffer made with 2 g/L of sodium dihydrogen phosphate and 0.25 g/L disodium hydrogen phosphate (anhydrous; Sigma). The organic eluant consisted of methanol with 2–5% acetonitrile. Modifications in buffer hydrogen ion and solvent acetonitrile concentration were made to effect optimal resolution on different columns and also for compound identification. A typical gradient profile was from 40 to 50% organic solvent over 5 min, followed by a 50–60% increase over 15.5 min, started at sample injection. Fluorescent detection was performed using a Gilson 121 detector.
Statistical analysis
All data are expressed as means ± SD. For analysis of temporal changes in amino acid content or other parameters, analysis of variance with Dunnett's test or Scheffé's F test was used. Linear regressions were performed to calculate correlation coefficients between various parameters. Differences were considered to be significant at p < 0.05.
RESULTS
Physiological variables
Physiological variables were generally kept within normal limits throughout all experiments, and there were no significant differences between the various experimental subgroups and periods (Table 1).
Physiological variables
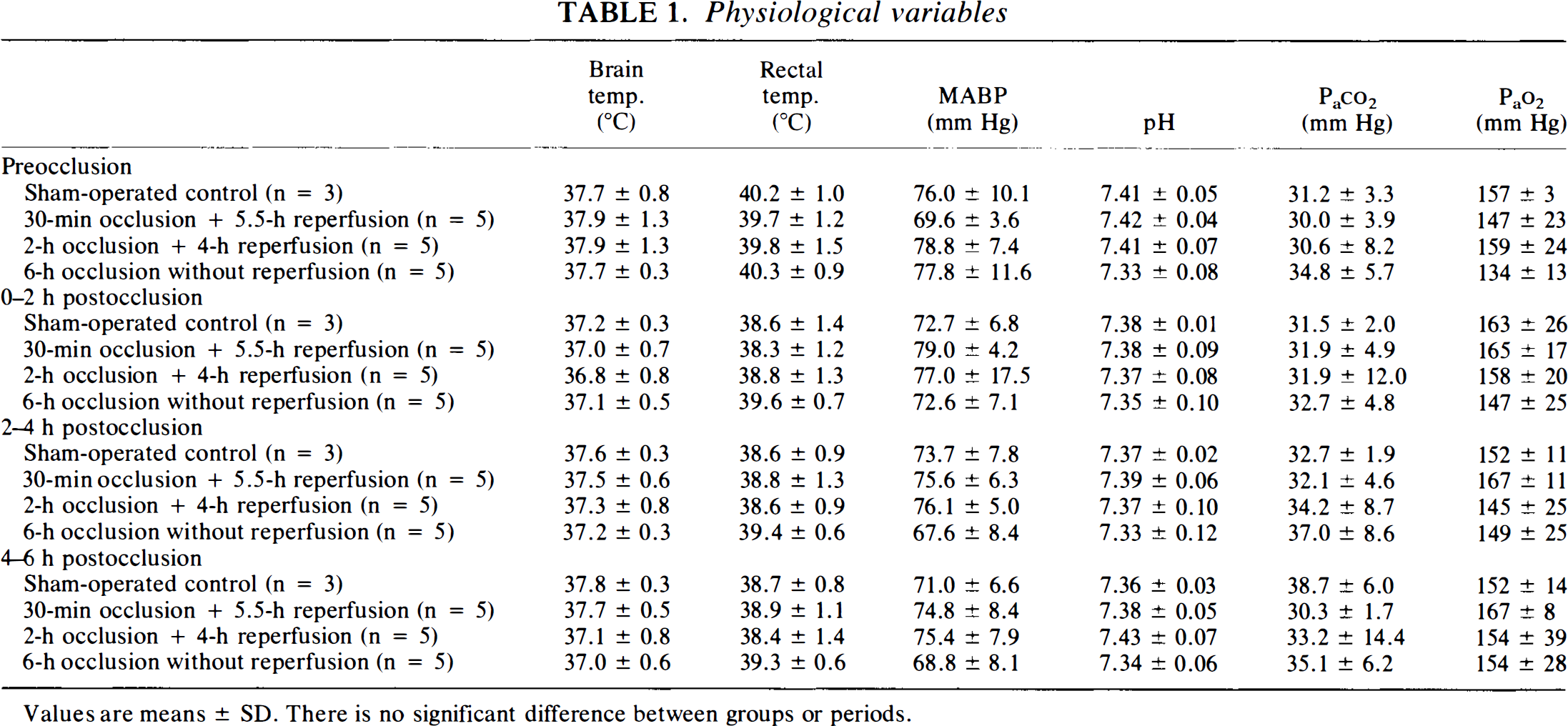
Values are means ± SD. There is no significant difference between groups or periods.
Changes in extracellular amino acids
Basal levels of amino acids in dialysate prior to ischemia remained relatively constant in all groups: glutamate, 1.19 ± 0.82 mM; aspartate, 0.06 ± 0.06 mM; GABA, 0.02 ± 0.08 mM; taurine, 1.41 ± 0.98 mM; alanine, 2.75 ± 3.07 mM; glutamine, 16.67 ± 9.67 mM. In sham-operated controls, there were no changes in dialysate concentrations during 8 h of measurement, demonstrating the stability of the model (data not shown).
In rabbits subjected to 30-min occlusion followed by 5.5-h reperfusion, transmitter amino acids (glutamate, aspartate, GABA) were increased during ischemia but rapidly returned to normal levels upon recirculation and remained constant for the duration of reperfusion (Fig. 1). Taurine levels also increased during ischemia and normalized after reperfusion (Fig. 1).
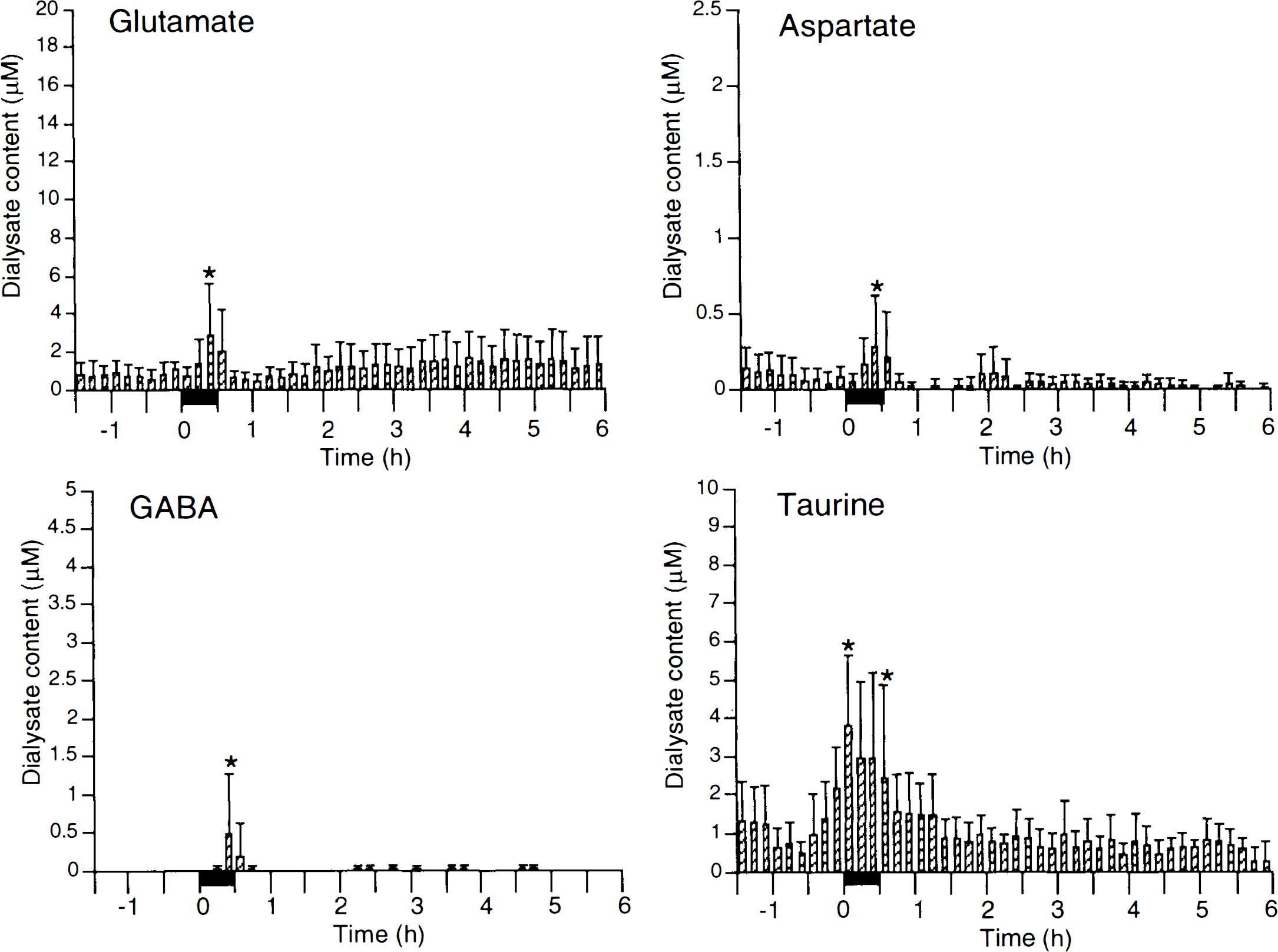
Amino acid concentration in dialysate (mean ± SD) in 30-min occlusion followed by 5.5-h recirculation. Solid bars designate occlusion periods. *p < 0.05 vs. preocclusion basal levels using multifactor analysis of variance with Dunnett's t test for multiple comparison. GABA, γ-aminobutyric acid.
In the 2-h occlusion group, concentrations of glutamate, aspartate, GABA, taurine, and alanine were significantly elevated during ischemia compared with preischemic baselines (Fig. 2). Shortly after reflow, glutamate, aspartate, and GABA concentrations briefly normalized. Thereafter, these amino acids began to increase again and were significantly higher than baseline by 2–3 h into reperfusion (Fig. 2). These secondary elevations reached levels that were comparable with or even larger than the primary (intraischemic) elevations. Taurine levels decreased slightly after recirculation but remained significantly higher than baseline for ∼3 h into reperfusion. Alanine was continuously elevated from the onset of occlusion throughout the entire measurement period. Glutamine did not show significant changes.
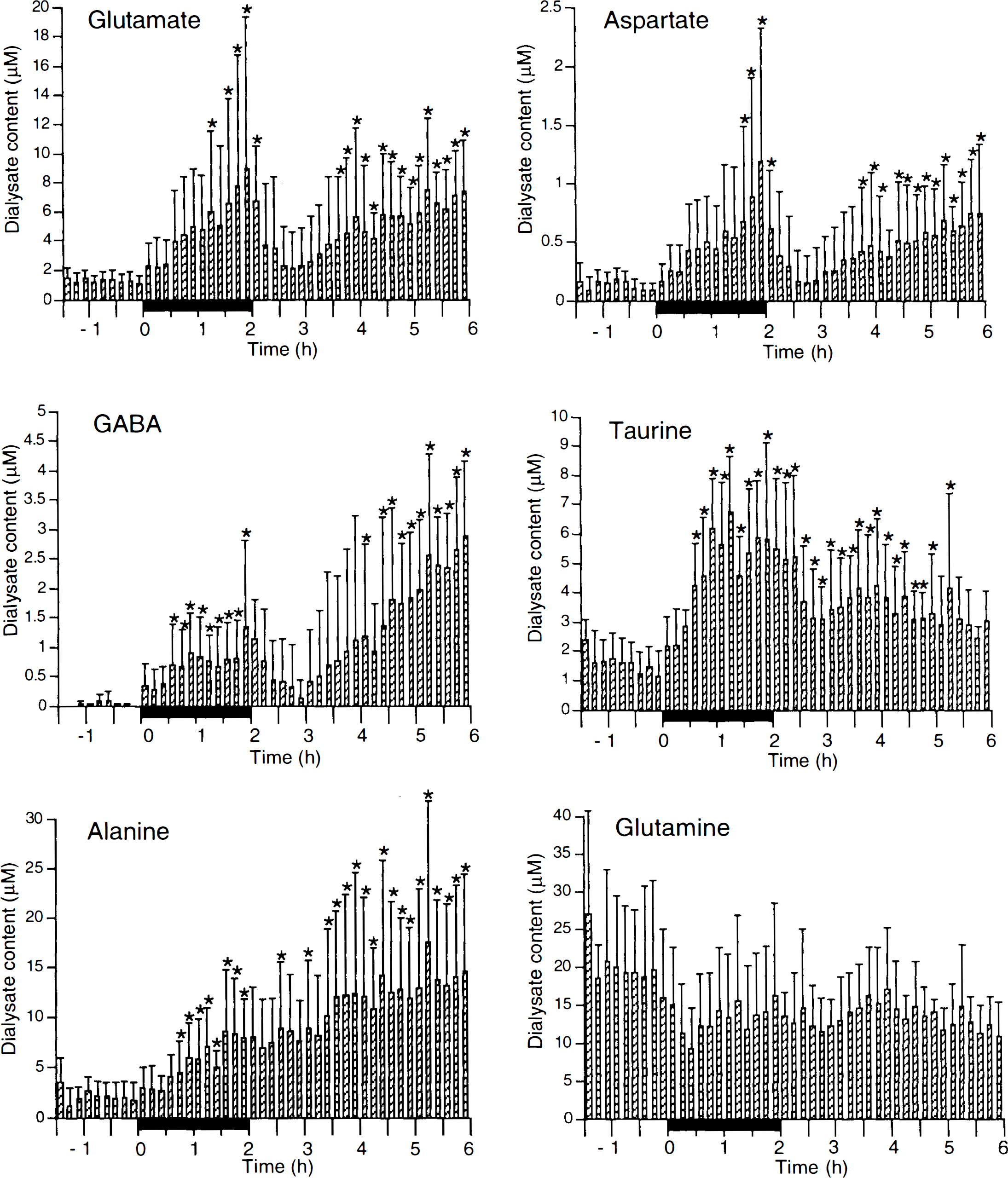
Amino acid concentration in dialysate (mean ± SD) in 2-h occlusion followed by 4-h recirculation. Solid bars designate occlusion periods. *p < 0.05 vs. preocclusion basal levels using multifactor analysis of variance with Dunnett's t test for multiple comparison. GABA, γ-aminobutyric acid.
In rabbits subjected to permanent ischemia, dialysate concentrations of all measured amino acids (except glutamine) were higher than preischemic baselines by 2–3 h after occlusion and remained elevated until the end (Fig. 3).
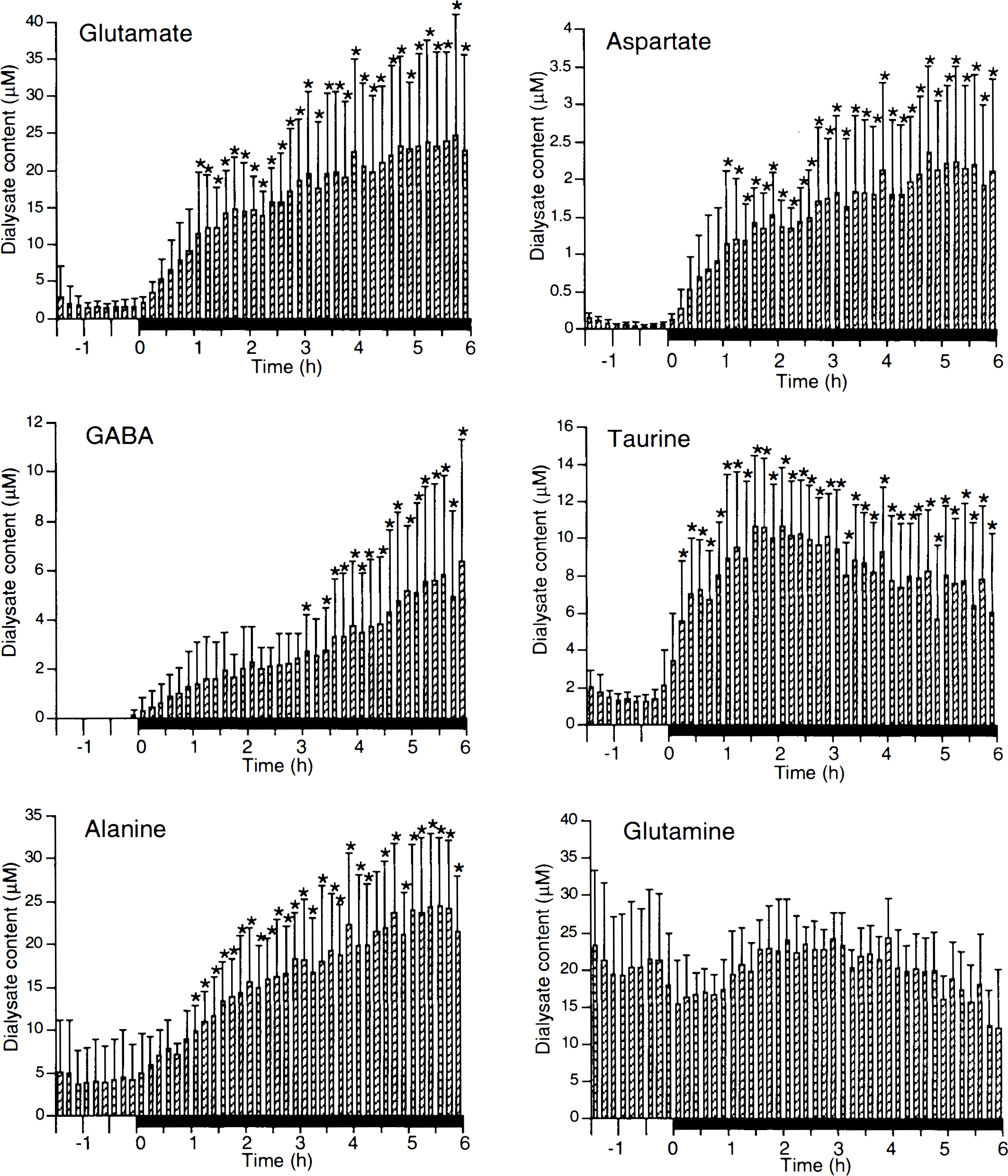
Amino acid concentration in dialysate (mean ± SD) in 6-h permanent occlusion. Solid bars designate occlusion periods. *p < 0.05 vs. preocclusion basal levels using multifactor analysis of variance with Dunnett's t test for multiple comparison. GABA, γ-aminobutyric acid.
Correlations between primary and secondary amino acid release
Linear regression analysis showed that in animals subjected to transient (2-h) ischemia–reperfusion, secondary (reperfusion) glutamate, aspartate, and GABA effluxes were significantly correlated with primary (ischemic) effluxes (r = 0.964–0.984) (Fig. 4). However, there were no significant correlations in levels of taurine and glutamine during ischemia and reperfusion.
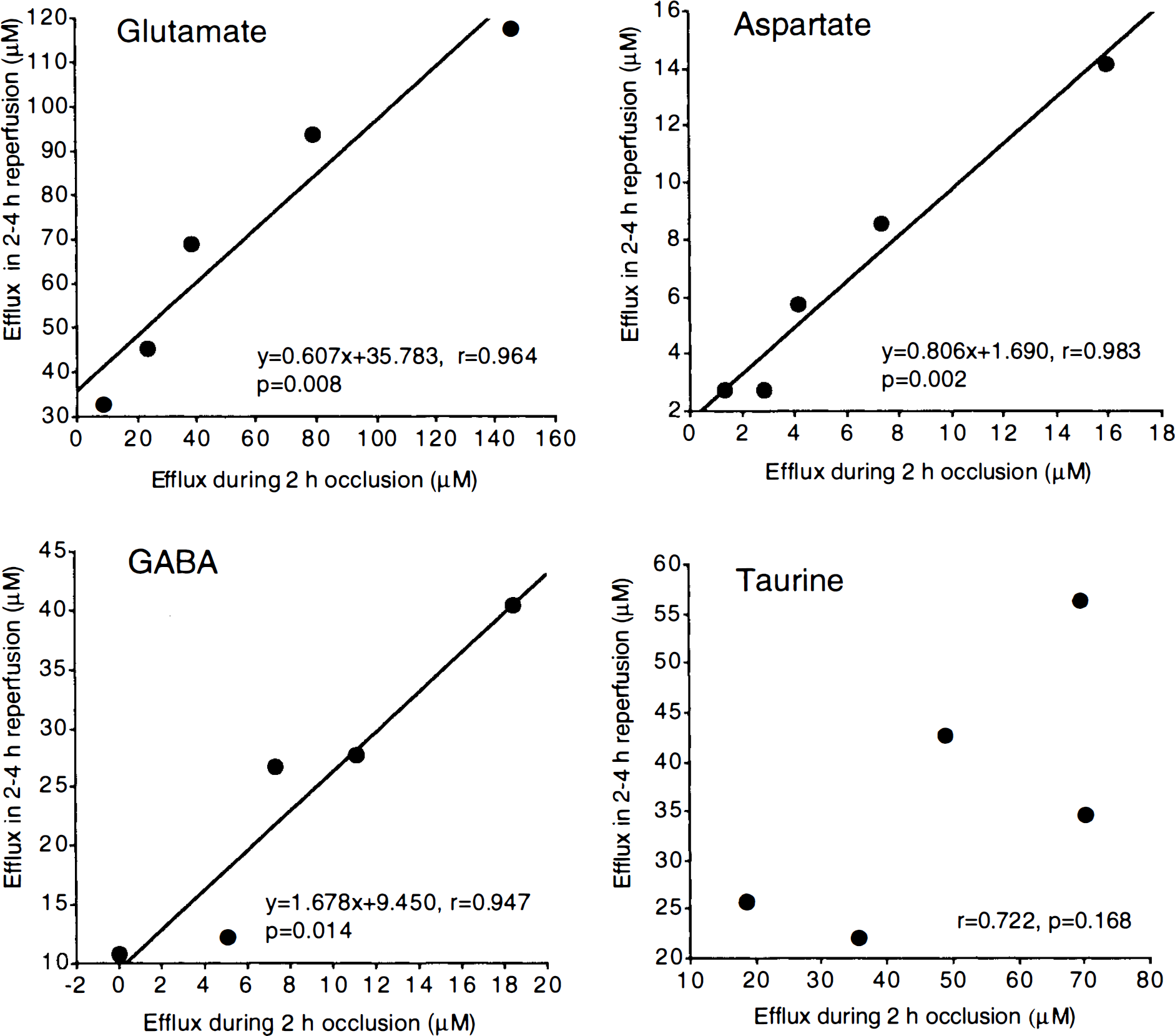
Correlations between amino acid efflux during 2-h occlusion and amino acid efflux in 2- to 4-h reperfusion following 2-h occlusion. Total amino acid efflux in each period was calculated as a sum of dialysate content during the period. GABA, γ-aminobutyric acid.
Amino acid efflux ratios
To compare the efflux profiles between the various amino acids, especially with respect to glutamate, ratios of total amino acid release for each hour in permanent ischemia and transient (2-h) ischemia were calculated (Fig. 5). GABA/glutamate ratios increased slowly over time; i.e., by the end of the experiment, relatively more GABA was released than glutamate. However, a significant difference was noted between transient versus permanent occlusions. By the late reperfusion phase, GABA/glutamate efflux ratios in transient ischemia were significantly higher than ratios for comparable timepoints in permanently occluded animals (Fig. 5). Taurine/glutamate and alanine/glutamate efflux ratios decreased postischemia. In the ischemia–reperfusion group, a transient increase in taurine/glutamate and alanine/glutamate occurred immediately after unocclusion, reflecting the brief normalization of glutamate but not taurine or alanine during reperfusion (see Fig. 2). Glutamine/glutamate ratios dropped immediately after occlusion and remained stable throughout the rest of the experiment.
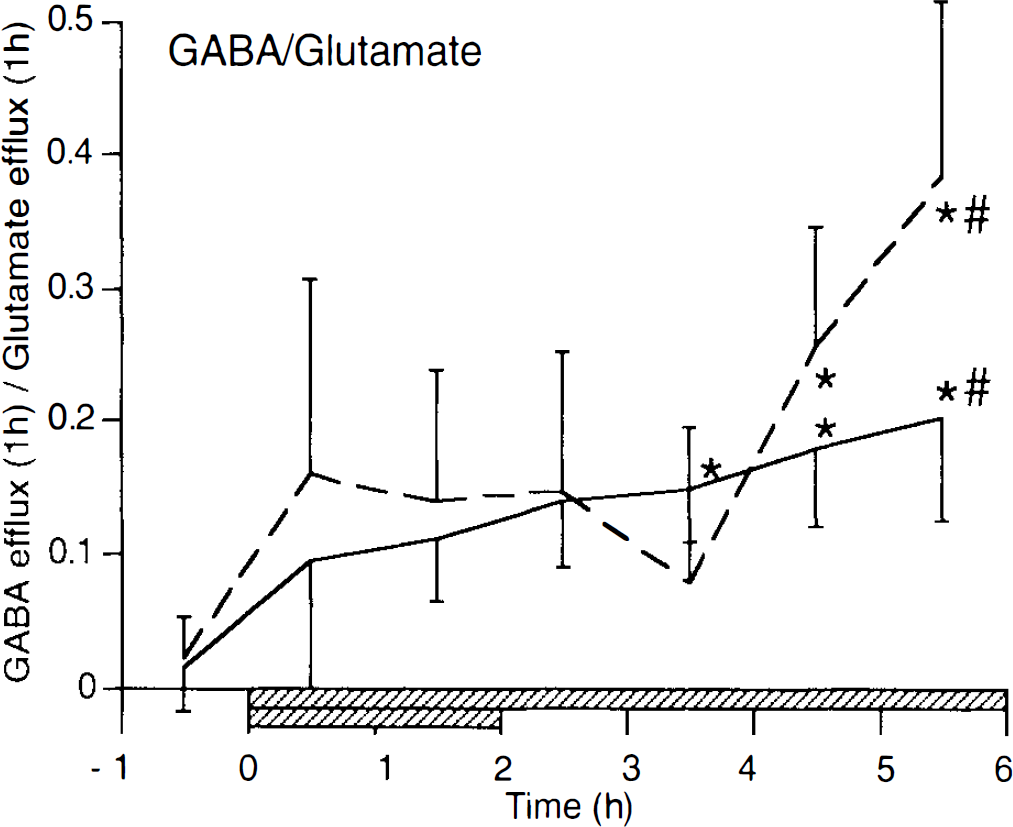
Temporal changes of the ratio of γ-aminobutyric acid (GABA) vs. glutamate efflux in 1-h intervals (mean ± SD). Solid line shows permanent occlusion group and dashed line shows 2-h occlusion–recirculation group. Hatched bars indicate the two different occlusion periods. *p < 0.05 from preocclusion values using one-factor repeated measures analysis of variance with Scheffé F test for multiple comparison; #p < 0.05 between 2-h occlusion–recirculation group and permanent occlusion group using two-factor repeated measures analysis of variance with unpaired t test.
Amino acid compositions in CSF, plasma, and brain tissue
To examine plasma leakage and nonspecific cell lysis as potential sources of amino acid efflux during ischemia–reperfusion, CSF, arterial blood plasma, and brain tissue homogenates were analyzed. Figure 6 shows that amino acid changes in CSF may reflect changes in extracellular space (dialysate) but not in plasma since amino acid compositions in dialysate and CSF were different from those in plasma during ischemia and reperfusion. For example, GABA is undetectable in plasma, whereas large amounts of extracellular GABA accumulate during ischemia–reperfusion. Amino acid composition in normal tissue homogenates is also different from those in dialysate and CSF (Fig. 6). Although glutamate levels are much higher than alanine in tissue, relatively larger elevations of extracellular alanine occur during reperfusion.
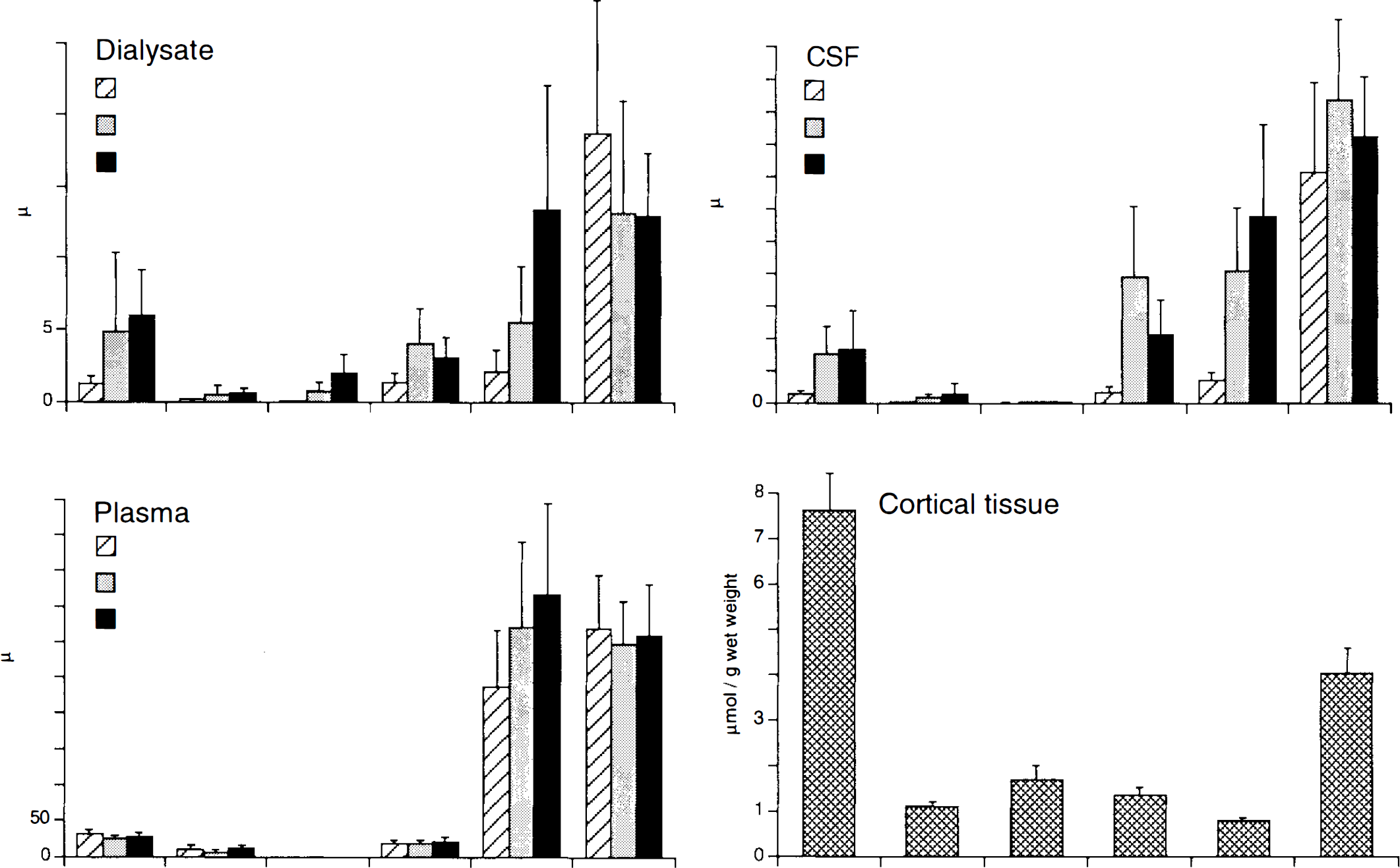
Amino acid compositions in dialysate (n = 5), CSF (n = 5), plasma (n = 3), and cortical tissue (n = 6). Dialysate concentrations are averaged from 6 samples (1 h) for preischemia, 12 samples during 2-h ischemia, and 12 samples during 2-to 4-h reperfusion. CSF and plasma samples were collected preischemia, during 2-h occlusion, and during 4-h reperfusion. Plasma γ-aminobutyric acid (GABA) levels were undetectable (n.d.). Cortical tissues were taken from contralateral normal cortex.
Lesion volumes
Ischemic lesions defined using tetrazolium staining showed the expected distribution of injury, encompassing cortex and basal ganglia. In all cases, probe tracks were visually identified and confirmed to reside within the ischemic lesion area. Lesion volumes in the 2-h occlusion group (26.81 ± 11.79% hemispheric volume) and permanent (6-h) occlusion group (33.06 ± 9.79% hemispheric volume) were significantly larger than lesion volumes in the 30-min transient ischemia group (5.51 ± 4.87% hemispheric volume) (Fig. 7). When all animals were analyzed as a single group, there were moderate but significant correlations between the volume of ischemic damage and the total amounts of released glutamate (r = 0.656, p = 0.008), aspartate (r = 0.644, p = 0.010), GABA (r = 0.686, p = 0.005), taurine (r = 0.799, p = 0.0004), and alanine (r = 0.595, p = 0.019).
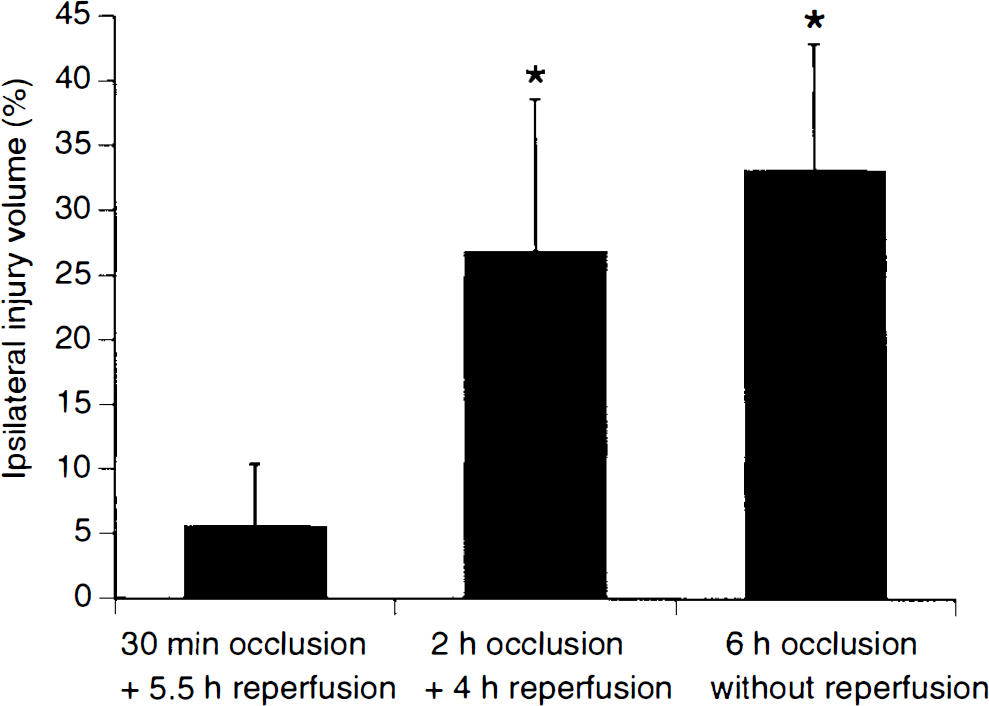
Lesion volumes on tetrazolium staining in three different ischemia–reperfusion groups (mean ± SD). *p < 0.05, from 30-min occlusion + 5.5-h reperfusion group using one-factor analysis of variance with Scheffé F test for multiple comparison.
DISCUSSION
Large elevations of excitatory amino acid transmitters during cerebral ischemia have been demonstrated in various animal models (Benveniste et al., 1984; Hillered et al., 1989). However, neurotransmitter profiles during recirculation after transient ischemia have not been extensively investigated. Most studies report a rapid return of transmitters to baseline levels upon unocclusion (Benveniste et al., 1984; Hagberg et al., 1985; Globus et al., 1988; Andine et al., 1990; Sciotti et al., 1992; Torp et al., 1993). However, recent studies have suggested that additional perturbations in neurotransmitter profiles may occur during the late reperfusion phase. Following transient forebrain ischemia in rats, transmitter levels initially returned to normal, but small delayed increases in excitatory amino acids and acetylcholine have been observed after 6–8 and 1–3 h of recirculation (Andine et al., 1991; Bertrand et al., 1993). Lin et al. (1992) showed larger secondary amino acid elevations during reperfusion after multiple ischemic insults that were hypothesized to reflect an increased excitotoxic index after global ischemia. In a rat model of transient focal ischemia, no secondary elevations in amino acids were seen during reperfusion, although it may be important to note that microdialysis measurements in this study were performed in the ischemic penumbra (Takagi et al., 1993). In a cat focal ischemia model, late elevation in glutamate levels was noted at 15 h after reflow following 4-h occlusion (Taguchi et al., 1993). Finally, in a retinal preparation, large increases in glutamate were noted during reperfusion after 45 min of transient ischemia (Louzada-Junior et al., 1992).
In this study, we measured extracellular amino acid levels for up to 6 h following different periods of ischemia–reperfusion in a rabbit focal occlusion model. As a result, we found large secondary elevations in transmitter amino acids in the reperfusion phase following 2-h transient focal ischemia. There were highly significant correlations between primary (ischemic) amino acid transmitter efflux and secondary (reperfusion) transmitter efflux, suggesting that secondary elevations are related to the severity of the primary insult. No secondary elevations were evident when a milder 30-min occlusion period was used. In permanent occlusion, amino acid levels remained elevated throughout the entire measurement period. Reperfusion is clearly beneficial when induced within 30 min to 1 h postocclusion (Selman et al., 1991). However, beyond certain periods of occlusion, ischemia–reperfusion may lead to more damage than permanent ischemia alone (Selman et al., 1991; Yang and Betz, 1994). Our data may provide an excitotoxic basis for these observations.
There are several possible mechanisms that may underlie these secondary amino acid transmitter elevations. These mechanisms include nonspecific amino acid leakage from infarcted cells, increased accumulation of plasma amino acids from a disrupted blood–brain barrier, delayed hypoperfusion leading to secondary ischemic events during reperfusion, and perturbations in transmitter release–reuptake systems caused by reperfusion injury. Some of these are more plausible than others.
First, nonspecific leakage from lysed brain cells is not likely to be a major cause since we have previously shown that infarction with generalized cellular lysis does not occur in this acute model of stroke (Steinberg et al., 1988, 1989, 1991). Ischemic injury is limited to acute neuronal changes such as nuclear basophilia and pyknosis. Secondary release was also specific for certain amino acids, and the proportion of released amino acids was quite different from tissue homogenate compositions. In tissue homogenates, there is approximately eight times more glutamate than alanine, but in dialysate, relatively equal amounts of glutamate and alanine are released during ischemia, and more alanine is released during reperfusion. It has been previously shown that total tissue alanine increases rapidly after cerebral ischemia but normalizes after reperfusion (Erecinska et al., 1984). In the absence of measurements of intra- versus extracellular gradients and absolute concentrations, these comparisons provide further indirect evidence against the role of nonspecific cell lysis. Finally, preliminary studies (unpublished data) from our laboratory showed potentiated transmitter responses to high potassium stimulation during reperfusion, further suggesting that viable neurons are still present during this phase.
Second, plasma leakage via a disrupted blood–brain barrier is also an unlikely cause since the proportion of secondary release was again quite different from plasma composition. Furthermore, plasma GABA levels were undetectable throughout all experiments, whereas there were significant accumulations in extracellular GABA levels during reperfusion following 2-h ischemia. Hence, large secondary elevations of transmitter amino acids in extracellular space are not likely to be due to leakage through a damaged blood–brain barrier.
A third possibility involves the phenomenon of delayed hypoperfusion that has been demonstrated to occur in various models of transient cerebral ischemia (Hossmann et al., 1973; Nemoto et al., 1975; Miller et al., 1980; Pulsinelli et al., 1982; Van den Kerckhoff et al., 1983; Grogaard et al., 1989). Transmitter release during reperfusion would then be mediated by a secondary decrease in cerebral blood flow that falls below the ischemic threshold. The temporal profile of cerebral blood flow in this model of rabbit focal ischemia has been previously established (Steinberg et al., 1991). Although delayed hypoperfusion does indeed occur and approaches ischemic thresholds (Shimada et al., 1989), time of onset is 3–4 h after reperfusion, whereas secondary amino acid release in this study clearly began at 1.5–2 h following recirculation. We did not directly measure blood flow in this study; this remains a caveat and limitation of these experiments. Further detailed correlations between blood flow and amino acid efflux during reperfusion are required to investigate the precise nature of these relationships.
The final possibility involves perturbation of neurotransmitter release–reuptake systems by reperfusion injury. It has been suggested that reperfusion injury is mediated via free radical formation and lipid peroxidation mechanisms (Gaudet et al., 1980; Yoshida et al., 1980; Sakamoto et al., 1991; Tegtmeier et al., 1990; Traystman et al., 1991). Free radicals and arachidonic acid generated during reperfusion are potent inhibitors of amino acid reuptake in glia and neurons (Yu et al., 1986; Barbour et al., 1989; Pellegrini-Giampetro et al., 1990) as well as enhancers of amino acid release in neurons (Rhoads et al., 1983; Gilman et al., 1994). Under normal conditions, extracellular concentrations of amino acid neurotransmitters are strictly controlled by glial and neural reuptake systems (Nicholls and Attwell, 1990). In the reperfusion phase following transient ischemia, perturbations in these release–reuptake systems may account for the secondary elevations documented in this study.
Analysis of the ratios of released amino acids may provide indirect information on the potential mechanisms involved. Our data showed that there were significant differences between amino acid ratios in permanent versus transient ischemia. In particular, when recirculation was performed, GABA/glutamate ratios were significantly higher during the late reperfusion phase than in permanently occluded animals. This suggests that a more severe deterioration of GABA release–reuptake systems occurred during reperfusion injury. The difference between GABA versus glutamate reuptake is that GABA may be primarily removed from extracellular space via neuronal uptake (Schousboe et al., 1977; Larsson et al., 1981), whereas glutamate is removed mainly via glial uptake (Hertz, 1979; Schousboe et al., 1983). Therefore, our data might suggest that reperfusion injury affected neuronal reuptake systems more severely than glial reuptake systems. GABA reuptake has been shown to be more vulnerable to ionic disturbance than those for glutamate or aspartate (Balcar and Johnston, 1972; Martin and Smith, 1972; Erecinska, 1987). On the other hand, with in vitro cell culture preparations, both neuronal and glial reuptake systems are vulnerable to arachidonic-acid-induced damage (Yu et al., 1986). GABA/glutamate ratios may also reflect differential effects on transmitter pools for GABA versus glutamate. It has been shown that in ischemia–reperfusion, activation of glutamic acid decarboxylase leads to rapid conversion of cellular glutamate into GABA (Erecinska et al., 1984), which may also contribute to increased GABA/glutamate ratios.
2,3,5-Triphenyltetrazolium chloride lesion volumes were significantly larger in 2-h transient and 6-h permanent occlusion animals than in the 30-min occlusion group. It has been shown that ischemic injury is proportional to the amount of amino acids released during ischemia (Butcher et al., 1990). In the present study, significant (albeit slightly lower) correlations were also noted between ischemic injury volume and the amount of released amino acids. Interestingly, however, for the putative transmitter amino acids (glutamate, aspartate, GABA), higher correlation coefficients were found between lesion area at the slice of probe location and secondary (reperfusion) efflux (r = 0.842, 0.699, 0.804) than primary (ischemic) efflux (r = 0.677, 0.621, 0.730). These data suggest that secondary amino acid release during reperfusion may play a role in the evolution of cerebral damage after transient ischemia. Our results may help explain the various published studies that show effective neuroprotection by glutamate antagonists even when infused postischemia, i.e., during the onset of recirculation (Steinberg et al., 1988, 1989; Nellgard and Wieloch, 1992; Nishikawa et al., 1994). Furthermore, it has been demonstrated that, for this rabbit model of transient ischemia, load (intraischemic) plus maintenance (reperfusion) therapy protocols are needed for successful neuroprotection (Kunis et al., 1991).
In summary, we evaluated the temporal profile of extracellular amino acids in various paradigms of ischemia–reperfusion and found significant secondary elevations in glutamate, aspartate, and GABA in the reperfusion phase following 2 h of transient ischemia. Abnormal excitatory transmitter dynamics during reperfusion may lead to additional metabolic stresses. These results suggest that an excitotoxic component of reperfusion injury may further contribute to the ischemic cascade.
Footnotes
Acknowledgment:
This work was supported by Sterling-Winthrop Pharmaceutical Research Division and grant NS32806 to EHL from NINDS. R.N. was supported by Neurex. The authors thank Drs. Gerald Wolf and Michael Moskowitz for valuable discussions.