
Abstract
Increased exposure to ambient particulate matter (PM) is associated with elevated morbidity and mortality in patients with cardiopulmonary diseases and cancer. We and others have shown that PM induces lung microvascular barrier dysfunction which potentially enhances the systemic toxicity of PM. However, the mechanisms by which PM disrupts vascular endothelial integrity remain incompletely explored. We hypothesize that PM induces endothelial cell (EC) cytoskeleton rearrangement via Rho GTPase-dependent pathways to facilitate vascular hyperpermeability. Fine PM induced time-dependent activation of cytoskeletal machinery with increases in myosin light chain (MLC) phosphorylation and EC barrier disruption measured by transendothelial electrical resistance (TER), events attenuated by the Rho-dependent kinase (ROCK) inhibitor Y-27632 or the reactive oxygen species (ROS) scavenger, N-acetylcysteine (NAC). Both Y-27632 and NAC prevented PM-induced stress fiber formation and phospho-MLC accumulation in human lung ECs. PM promotes rapid accumulation of Rho-GTP. This event is attenuated by NAC or knockdown of RhoA (siRNA). Consistent with ROCK activation, PM induced phosphorylation of myosin light chain phosphatase (MYPT) at Thr850, a post-translational modification known to inhibit phosphatase activity. Furthermore, PM activates the guanine nucleotide exchange factor (GEF) for Rho, p115, with p115 translocation to the cell periphery, in a ROS-dependent manner. Together these results demonstrate that fine PM induces EC cytoskeleton rearrangement via Rho-dependent pathways that are dependent upon the generation of oxidative stress. As the disruption of vascular integrity further contributes to cardiopulmonary physiologic derangements, these findings provide pharmacologic targets for prevention of PM-induced cardiopulmonary toxicity.
Introduction
Air pollution, with increased exposure to ambient particulate matter (PM), remains a persistent, worldwide risk factor for morbidity and mortality in patients with cardiopulmonary diseases and cancer.1–6 PM exerts chronic and oxidative injury in biological systems, via variable and complex chemical components, including inorganic transitional metals, elemental and organic carbons, and biological residuals such as endotoxin and pollen. 7 Importantly, transitional metals, 7 as well as polycyclic aromatic hydrocarbons (PAH) 8 in PM, generate a burst of reactive oxygen species (ROS) and exert pro-inflammatory impact on the respiratory system. The oxidative stress induced by PM active components leads to further systemic injurious effects, such as endothelial barrier disruption. Recently, we reported that direct challenge with PM produces vascular barrier dysfunction in vitro9,10 and in vivo.10,11 Mice exposed to PM (intratracheal instillation) lead to increased protein leakage into the bronchoalveolar space,10,11 a marker for loss of pulmonary endothelial integrity. 12 Increased lung endothelial cell (EC) monolayer permeability not only exacerbates pulmonary inflammatory responses, but contributes to amplified systemic responses via increased translocation of pollutant particles into the circulation. 12
We have previously demonstrated the critical role of myosin light chain (MLC) phosphorylation in endothelial barrier regulation, and its regulation by key proteins myosin light chain kinase (MLCK) and myosin light chain phosphatase (MYPT),13,14 in various pulmonary inflammatory diseases.15,16 MLC phosphorylation, triggered by edemagenic agonists such as thrombin, TNFα, or LPS, facilitates actin stress fiber formation, cell contraction, and creation of paracellular gaps between ECs, thereby increasing lung fluid imbalance and leukocyte infiltration, as well as transport of pollutant particles to the systemic circulation. 17
In the present study, we demonstrate that PM induces EC barrier disruption via cytoskeleton rearrangement. We also show that PM activates ROCK to increase lung EC permeability. We further demonstrate that activation of RhoA/ROCK promotes phosphorylation and inactivation of MYPT, thereby increasing actomyosin contraction generated by the accumulation of phosphorylated MLC. Together these results demonstrate that fine PM induces EC cytoskeleton rearrangement via Rho-dependent pathways that are dependent upon the generation of oxidative stress. These findings provide pharmacologic targets for prevention of PM-induced cardiopulmonary toxicity.
Materials and methods
Reagents and chemicals
Polyacrylamide gels, buffers, and protein assay reagents were purchased from Bio-Rad (Hercules, CA, USA). Texas Red phalloidin- and Alexa 488-conjugated secondary antibodies were purchased from Molecular Probes, Inc. (Eugene, OR, USA). MYPT1 polyclonal antibody was purchased from Covance Inc. (Berkeley, CA, USA). Site-specific phospho-MYPT1 (MYPT1) antibody and p115-RhoGEF antibody were purchased from Upstate Biotechnology (Lake Placid, NY, USA). Cell lysis buffer and diphospho-MLC antibody were obtained from Cell Signaling (Beverly, MA, USA). Rho-kinase specific inhibitor Y-27632 was obtained from Calbiochem (La Jolla, CA, USA). All other chemicals and reagents were obtained from Sigma-Aldrich Co. (St. Louis, MO, USA) unless stated otherwise. Fine PM was collected (April of 2005) from the Fort McHenry Tunnel, Baltimore, MD, USA using a high-volume cyclone collector, with chemical components previously described. 9 This PM sample has been used in related in vitro and in vivo studies.9,10 This particular PM sample dose- and time-dependently disrupts endothelial integrity, with the ideal experimental concentration of 100 µg/mL identified by previous studies. 9 At this concentration, the maximum effects on endothelial barrier disruption and ROS generation were observed without cytotoxicity (within 24 h). 9 The physiological relevance of this particular concentration has also been justified, 9 approximately equivalent to a short-term exposure in the particular collection location with high ambient PM levels. We chose 100 µg/mL as the experimental concentration for this study.
Cell culture
Human pulmonary artery ECs obtained from Lonza (Basel, Switzerland) were cultured as previously described 18 in EGM-2 complete medium (Lonza) with 10% fetal bovine serum at 37℃ in a humidified atmosphere of 5% CO2 and 95% air, with passages 6–8 used for experimentation. Twenty-four hours before the PM challenge, EC media was changed to EGM-2 with 2% fetal bovine serum (Lonza). Human lung ECs were transfected with siRNA (Darmacon RNA Technologies, Inc., Lafayette, CO, USA) using siPORT Amine (Ambion, Austin, TX, USA) as the transfection reagent according to the manufacturer’s protocol. ECs (∼40% confluent) were serum-starved for 1 h, followed by incubation with 100 ng/mL total target siRNA (or control siRNA) for 6 h in serum-free medium. Serum-containing medium was then added (10% serum final concentration) for 42 h before further assays were conducted.
Transendothelial electrical resistance (TER)
ECs were grown to 90–95% confluence in polycarbonate wells containing evaporated gold microelectrodes (Applied Biophysics, Troy, NY, USA) and TER measurements were made by using an electrical cell-substrate impedance sensing system (ECIS) (Applied Biophysics, Troy, NY, USA) as previously described in detail. 19 PM samples were added into the culture chamber to achieve designated concentration while the TER were being continuously measured. 9
Western blot
EC cell lysate was prepared with cell lysis buffer with adequate protease and phosphatase inhibitors (Cell Signaling, Beverly, MA, USA) after indicated treatment as previously described. 9 Equal amounts of protein samples were separated on SDS-PAGE, transferred to nitrocellulose membranes, blocked with 5% BSA in TBS-Tween20 (0.5%, TBST, 1 h), and incubated with primary antibodies in 5% BSA in TBST (overnight at 4℃). The primary antibody was detected by incubation with horseradish peroxidase–coupled second antibody (1:2000 in TBST with 5% BSA) at room temperature for 2 h. The protein blots detection was performed by using chemiluminescence reagent from cell signaling using its standard protocol.
Endothelial cell imaging
ECs were grown on gelatin-coated glass coverslips before exposure to various conditions as described for individual experiments. EC were then fixed in 3.7% formaldehyde, washed with cold TBS, and permeabilized with 0.25% Triton X-100 for 5 min. Cells were then washed in TBS, blocked with 2% bovine serum albumin in PBS for 30 min, and then incubated for 60 min at room temperature with primary antibody (diphospho-MLC or p115-GEF, 1:500 dilution) prepared in TBS with 2% bovine serum albumin. After washing three times with cold TBS, ECs were incubated for 60 min with Alexa 488-conjugated secondary antibody prepared in TBS with 2% bovine serum albumin. After washing three times with cold TBS, ECs were incubated with Texas Red–conjugated phalloidin for 60 min. Coverslips were mounted using Slow Fade (Molecular Probes, Inc.), followed by washing three times with cold TBS. Confocal microscopy was performed using the Radiance Laser scanning 2100 system (Bio-Rad). Images were recorded and stored.
Rho activation assay
Rho activation in EC culture was analyzed using a Rho assay kit available from Upstate Biotechnology. Briefly, pulmonary ECs grown in 100-mm petri dishes were treated with PM for the indicated periods of time. At the end of the experiment, cells were rinsed with ice-cold PBS, and cell lysate was collected with cell lysis buffer to be incubated with rhotekin Rho-binding peptide immobilized on agarose overnight, and activated GTP-Rho bound to rhotekin-agarose was detected by western blot with anti-Rho antibody.
Statistical analysis
Data are presented as means ± SE for each group, and analyzed by two-way ANOVA for more than two groups or by an unpaired Student’s t-test for two groups. Significance in all cases is defined as P < 0.05. Each experiment was repeated three to five times if not specifically mentioned.
Results
PM induces MLC phosphorylation
MLC phosphorylation at Thr18 and Ser19
20
upon edemagenic agonist stimulation facilitates the formation of F-actin stress fibers. This process induces cell contraction and leads to paracellular gap formation that results in barrier disruption.
13
In order to examine the mechanisms of PM-induced EC barrier disruption, we initially determined levels of MLC phosphorylation. PM challenge (100 µg/mL) of human lung ECs produced approximately a twofold increase in MLC phosphorylation levels in a time-dependent manner peaking at 30 min post PM stimulation (Fig. 1a). In addition, we also observed a slight increase of MLC total protein. We previously demonstrated that PM stimulation in human lung EC increases ROS production, with ROS cellular effects prevented by the ROS scavenger, N-acetylcysteine (NAC).
9
PM-induced MLC phosphorylation was also attenuated by NAC (5 mM) in human lung ECs, and it was also prevented by the ROCK inhibitor Y-27632 (10 μM) (Fig. 1b). Consistently, NAC and Y-27632 each significantly attenuated the PM-induced endothelial barrier disruption (Fig. 1c), reflected by transendothelial resistance measurements. These results confirm PM-induced increases in EC permeability are mediated by ROCK and ROS. The Y-27632 and NAC-mediated attenuations in MLC phosphorylation or TER were not additive, which suggests ROCK activation is downstream of ROS.
PM induces MLC phosphorylation and endothelial barrier disruption, both of which are attenuated by a ROS scavenger or a ROCK inhibitor. (a) PM mediated time-dependent MLC phosphorylation. (b) PM-induced MLC phosphorylation was reduced by Y-27632 (Y, 10 μM) or NAC (5 mM). (c) PM-induced endothelial barrier disruption (by TER measurements) was significantly inhibited by Y compound, NAC or a combination of Y and NAC. n = 4–5. *P < 0.05 compared with control. **P < 0.05 compared with PM only group.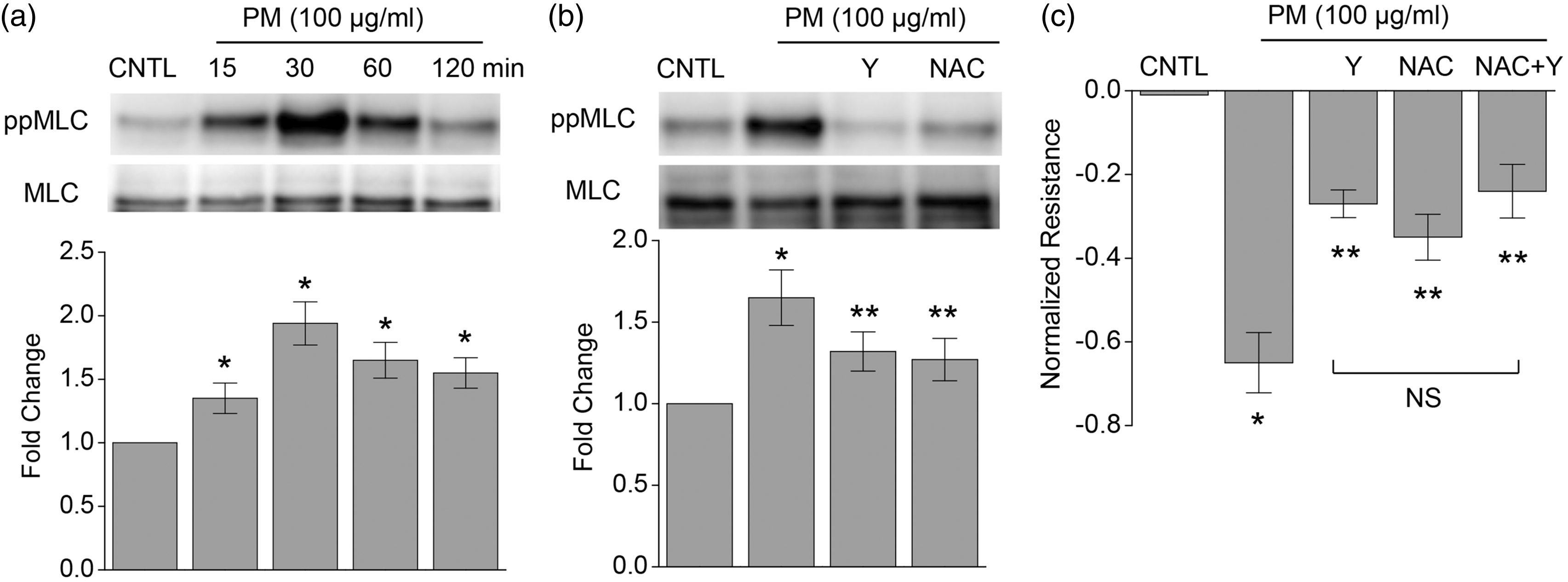
PM induces lung EC stress fiber formation
Decreased TER upon PM challenge is associated with actin rearrangement with stress fiber formation, characteristic of cytoskeleton rearrangement mediated EC barrier disruption.
13
Under basal conditions, lung EC actin fibers are primarily localized at the cell periphery and are organized into cortical bands. The PM challenge promoted formation of F-actin stress fibers that span the cytoplasm in a spindle-like pattern (Fig. 2) with phosphorylated MLC co-localized in these bands of polymerized actin. ROCK inhibition by Y-27632 and/or anti-oxidant treatment with NAC abolished this PM-induced F-actin rearrangement, as well as PM-induced MLC phosphorylation, consistent with mediation of PM-induced MLC phosphorylation and stress fiber formation by ROS and ROCK.
PM-induced phospho-MLC co-localizes with stress fibers. PM challenge (100 µg/mL) stimulated stress fiber (F-actin) synthesis and MLC phosphorylation. Phospho-MLC was co-localized with stress fibers. Y (10 μM, 1 h pretreatment) or NAC (5 mM, 1 h pretreatment) abolished PM-induced stress fiber formation and MLC phosphorylation in ECs. These exhibiting images are representative images from three independent repeats.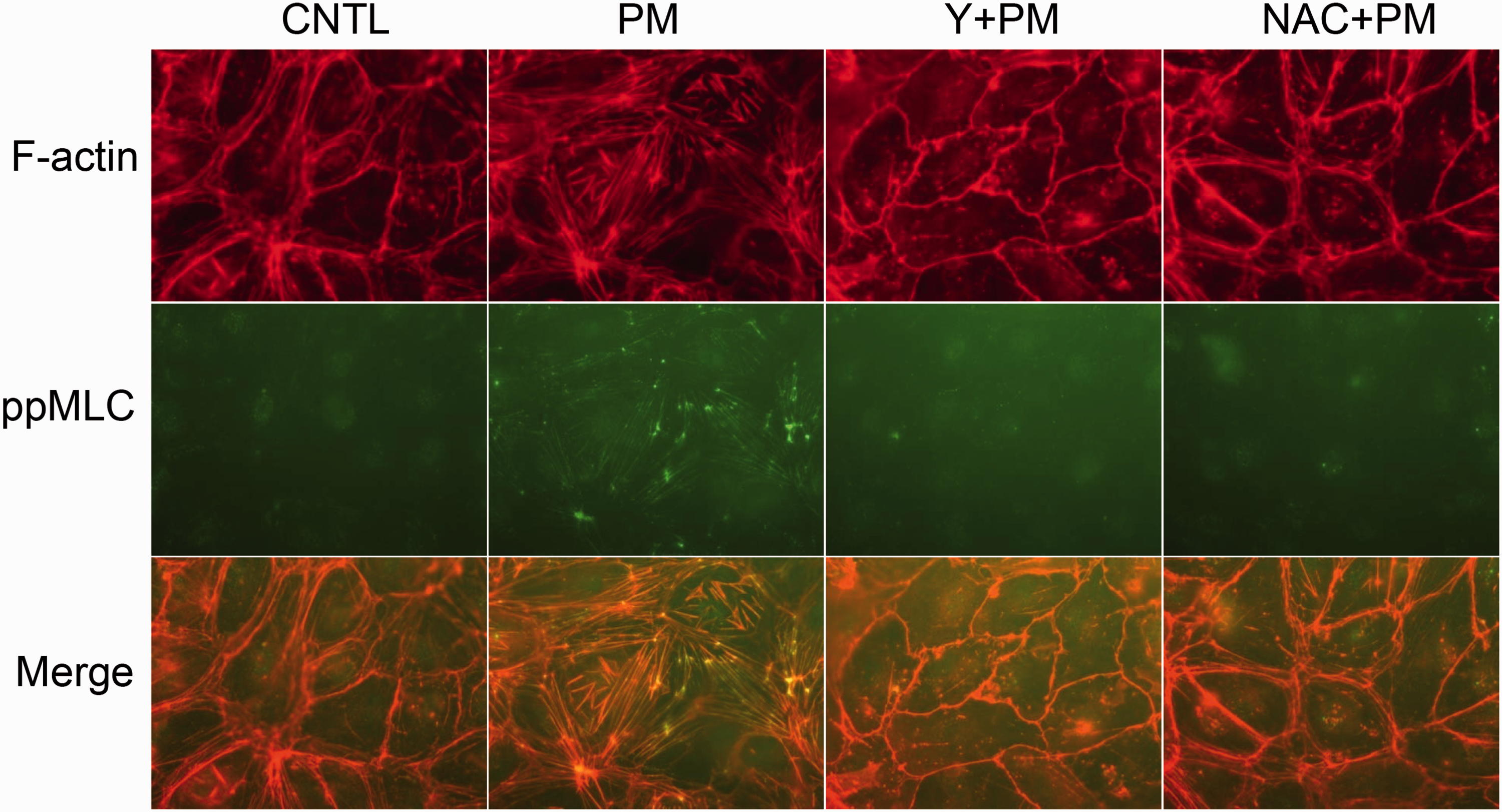
PM activates Rho in human lung EC
RhoA is a member of the family of Ras small GTPases that activates ROCK and regulates the actin cytoskeleton.
13
Fig. 3a depicts the dramatic increase in Rho-GTP complex (the active form of Rho) formation in PM-treated human lung ECs compared with basal levels with these effects attenuated by NAC (Fig. 3a). Reduced RhoA expression (siRNA) abolished PM-induced phosphorylation of MLC (Fig. 3b), decreased PM-induced MLC phosphorylation, and prevented PM-induced EC barrier disruption (Fig. 3c). These data suggest that PM accelerates the exchange of GDP to GTP on Rho proteins, and the family member RhoA is a key protein responsible for PM-induced MLC phosphorylation.
PM activates RhoA in human lung EC. (a) PM challenge (100 µg/mL) enriched the active form of Rho (GTP bound Rho) in ECs, which was inhibited by NAC (5 mM). (b) RhoA siRNA reduced RhoA expression and inhibited PM-induced MLC phosphorylation. (c) RhoA siRNA significantly inhibited PM-induced endothelial barrier disruption. n = 4–5. *P < 0.05 compared with siControl.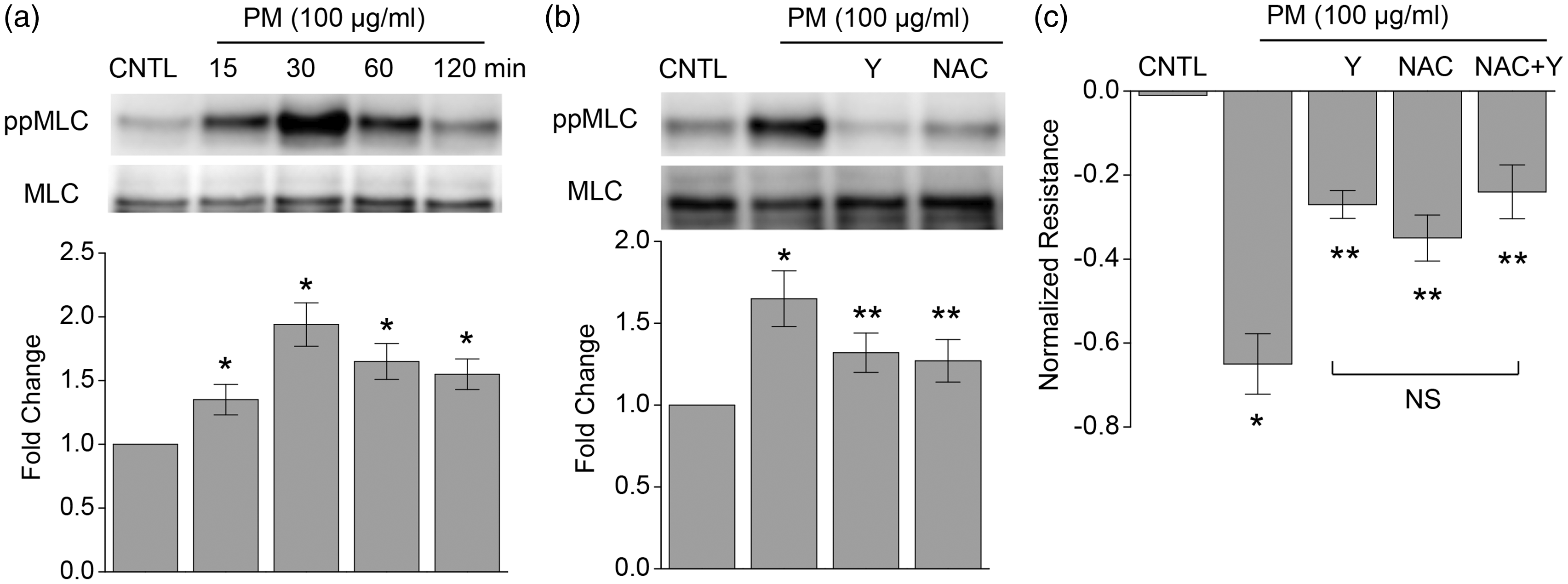
PM induces ROCK1/2-mediated MYPT1 phosphorylate
ROCK phosphorylates and inactivates myosin phosphatase (MYPT1), resulting in an increase in MLC phosphorylation.
13
The PM challenge of human lung ECs resulted in MYPT phosphorylation at Thr850 which was attenuated by either NAC or Y-27632 (Fig. 4a). ROCK exists as two major isoforms, ROCK1 and ROCK2, that exhibit similar biological function in ECs.
21
We utilized isoform-specific siRNAs to reduce ROCK1 and ROCK2 expression (Fig. 4b) with reduction of either isoform retaining the capacity to attenuate PM-induced MYPT phosphorylation (Fig. 4c). Finally, PM stimulated increases in EC permeability were ameliorated by reduced ROCK1 and ROCK2 expression (Fig. 4d), indicating PM-induced barrier disruption is ROCK dependent.
PM induces ROCK-mediated MYPT1 phosphorylation. (a) PM induced MYPT phosphorylation at Thr850, which was inhibited by Y (10 μM, 1 h pretreatment) or NAC (5 mM, 1 h pretreatment). (b) siRNAs of ROCK1 (R1) or ROCK2 (R2), or a combination of siRNAs of ROCK1 and ROCK2 (R12) successfully reduced protein expression of ROCK1 or ROCK2 or both after 48 h incubation, respectively. (c) PM-induced MYPT phosphorylation was inhibited by siRNAs of R1, R2, R12, or RhoA. (d) PM-induced endothelial barrier disruption was significantly inhibited by siRNAs of R1, R2, or R12. n = 4–5. *P < 0.05 compared to siControl (the first group).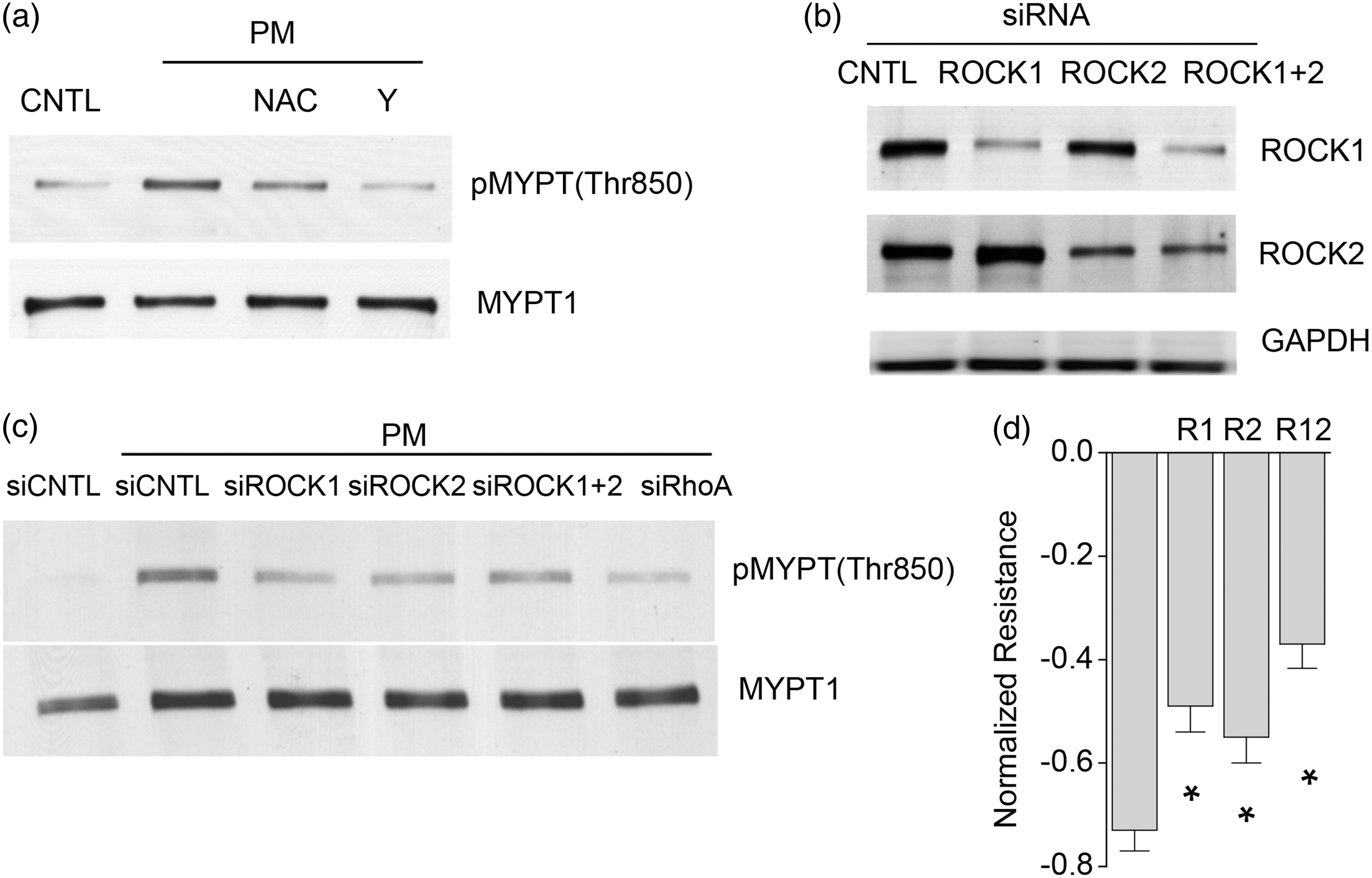
Discussion
Ambient PM is a significant environmental and health problem throughout the world, most notably in populated geographic and industrial regions. PM and environmental pollution exposures are linked to increases in mortality and morbidity in patients with pulmonary disorders such as asthma and chronic obstructive pulmonary disease (COPD), cardiovascular diseases such as cardiac arrhythmias and congestive heart failure (CHF), and cancer. 22 The complex composition of PM across various regions has hampered the understanding of the molecular mechanisms that produce these deleterious health risks. In this report, we have elucidated a molecular pathway by which PM exposure increases vascular permeability that implicates RhoGTPase activation, MYPT inactivation, and a contractile EC phenotype. As the Rho/ROCK/MYPT axis has been shown to be associated with inflammatory diseases in the lung and heart, 23 Rho/ROCK/MYPT axis constituents could be potentially useful targets for prevention of PM-induced cardiopulmonary dysfunctions.
PM samples commonly stimulate ROS generation, leading to cellular injury,9,24,25 which further contribute to PM-exacerbated pathobiology. Previously, we have shown that PM induces cytoskeleton rearrangement and stress fiber formation led by oxidative stress pathways, causing cells to contract and resulting in formation of paracellular gaps. 9 In ECs, PM stimulates the generation of ROS, which leads to p38beta MAPK mediated HSP27 phosphorylation and stress fiber stabilization. 9 However, PM-induced EC permeability is only partially blocked by p38-MAPK inhibition, suggesting additional mechanisms for increased permeability by PM exposure. In the present study, we demonstrated that Rho/ROCK/MYPT signaling is a contributing mechanism for PM-induced pulmonary EC barrier disruption. We demonstrated that PM increases phosphorylation of MLC (at Thr18 and Ser19), which is co-localized with newly formed stress fibers with these biochemical events attenuated by a ROS scavenger or a ROCK inhibitor. This is consistent with our previous reports of MLC signaling in ECs, 26 and agrees with previously described generation of ROS by PM in various cell types.27,28 Excessive ROS generation increases oxidative injury, such as DNA damage and mitochondria dysfunction, in target cells or tissues. We now demonstrate yet another deleterious consequence of PM and ROS that is relevant to inflammatory reactions in lung, vascular, and heart tissues.
Rho family GTPases are key regulators of the EC cytoskeleton and control cell–cell and cell–matrix adhesion, cell migration, and vascular integrity via the cytoskeleton. One of the Rho family GTPases, RhoA, in particular has been shown to be a critical regulator of actin cytoskeleton modulation. 26 In the present study, we demonstrate that PM exposure in ECs activates RhoA through production of ROS, and is a critical step in increasing paracellular permeability. As shown in Fig. 3, increased GTP binding to RhoA by PM is significantly attenuated in the presence of an antioxidant (NAC). RhoA could be directly activated by ROS,29,30 or activated via Rho guanine exchange factor (RhoGEF), which promotes exchange of GDP by GTP on Rho. 31 We have observed that ROS-dependent translocation of RhoGEF1 (p115-RhoGEF) occurs upon PM stimulation (Supplementary Fig. S1), suggesting RhoGEF is involved in PM-mediated MLC phosphorylation. In combination with our present findings, we strongly suggest that PM-induced RhoA activation is mediated by increased generation of ROS. Similarly, cigarette smoke has been shown to increase oxidative stress in lung vasculature that subsequently causes endothelial barrier dysfunction that is mediated by RhoA signaling, 32 further supporting the premise that oxidative stress is a factor for activating RhoA and subsequent disruption of endothelial barriers.
We further show the increase in phosphorylation of MYPT by PM in pulmonary ECs. Phosphatase activity of MYPT is inhibited by phosphorylation of MYPT at Thr850 by the kinase ROCK, leading to accumulation of phosphorylated MLC in contracting ECs. In our TER study (Fig. 4), inhibition of ROCK by the pharmacological inhibitor Y-27632 or siRNA knockdown of ROCK1 or 2 attenuated phosphorylation of MYPT Thr850 with the combination of ROCK1/2 silencing most significantly attenuating EC barrier disruption triggered by PM, suggesting both ROCK1 and ROCK2 play roles in inhibiting MYPT activity with the possibility of redundant functions of ROCK1 and ROCK2 in ECs. 21
In summary, we have delineated a molecular pathway by which PM increases vascular permeability via generation of ROS that activates RhoA/ROCK and inhibits myosin phosphatase (MYPT) with these events essential for the cytoskeleton rearrangement that produces paracellular gap formation and EC barrier dysfunction. These results demonstrate that fine PM dysregulates EC cytoskeleton rearrangement via Rho-dependent pathways that are dependent upon the generation of oxidative stress. As the disruption of vascular integrity further contributes to cardiopulmonary physiologic derangements, these findings provide pharmacologictargets for the prevention of PM-induced pulmonary and cardiovascular toxicity.
Footnotes
Acknowledgments
The authors appreciate the technical support from Lakshmi Natarajan on endothelial cell culture.
Conflict of interest
The authors declare that there are no conflicts of interest.
Funding
This study is supported by Parker B Francis Foundation, National Institutes of Health grants R01HL091899, P01HL126609, P30ES006694, and T32HL007249. This study is also supported by National Natural Science Foundation of China (grant no. 81570011), Anhui Provincial Natural Science Research Project of University (KJ2013A188), and International Science and Technology Cooperation Project – Key Research and Development Program of Anhui Province (1604b0602026).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.