
Abstract
Radiotherapy as a primary treatment for thoracic malignancies induces deleterious effects, such as acute or subacute radiation-induced lung injury (RILI). Although the molecular etiology of RILI is controversial and likely multifactorial, a potentially important cellular target is the lung endothelial cytoskeleton that regulates paracellular gap formation and the influx of macromolecules and fluid to the alveolar space. Here we investigate the central role of a key endothelial cytoskeletal regulatory protein, the nonmuscle isoform of myosin light chain kinase (nmMLCK), in an established murine RILI model. Our results indicate that thoracic irradiation significantly augmented nmMLCK protein expression and enzymatic activity in murine lungs. Furthermore, genetically engineered mice harboring a deletion of the nmMLCK gene (nmMLCK−/− mice) exhibited protection from RILI, as assessed by attenuated vascular leakage and leukocyte infiltration. In addition, irradiated wild-type mice treated with two distinct MLCK enzymatic inhibitors, ML-7 and PIK (peptide inhibitor of kinase), also demonstrated attenuated RILI. Taken together, these data suggests a key role for nmMLCK in vascular barrier regulation in RILI and warrants further examination of RILI strategies that target nmMLCK.
Radiotherapy, a primary treatment for thoracic malignancies, is often limited by deleterious dose-limiting side effects that manifest themselves as acute or subacute lung injury 6–24 weeks after radiation exposure. 1 Although the exact molecular mechanisms of radiation-induced lung injury (RILI) are controversial and likely multifactorial, a potentially important cellular target in RILI is the lung endothelium, a functionally dynamic tissue that regulates influx of macromolecules and fluid between the vascular compartment and the lung interstitial space. 2 Disruption of endothelial cell (EC) barrier integrity is considered a key feature of pulmonary inflammation in general and RILI in particular, resulting in increased vascular permeability and alveolar flooding. Thus, stabilization or enhanced restoration of lung vascular endothelial integrity is a critical step in preventing or reducing acute or subacute RILI, as we have previously demonstrated. 2
We have also reported the role of the nonmuscle myosin light chain kinase isoform (nmMLCK) as an essential element of the inflammatory responses, with variants in the MYLK gene contributing to acute lung injury (ALI), ventilation-induced lung injury (VILI) susceptibility, and asthma susceptibility.3,4 Nonmuscle MLCK is a calcium/calmodulin-dependent kinase critically involved in catalyzing the phosphorylation of myosin light chain (MLC), leading to cell contraction and vascular EC barrier disruption, which indicates a critical role for nmMLCK in regulating vascular permeability. 5
We have also reported that genetic variants (single-nucleotide polymorphisms) in MYLK confer significant susceptibility to sepsis- 6 or trauma-induced 7 ALI, in addition to contributing to the risk of severe asthma, another inflammatory lung disorder, in African Americans.3,4,8 Nonmuscle MLCK is activated by various permeability-enhancing factors, such as inflammatory cytokines and chemokines mediating cell contraction, formation of cell-cell and cell-matrix gaps, and vascular permeability. 9 We studied the role of nmMLCK in RILI development and evaluated the therapeutic potential of MLCK inhibitors in alleviating RILI, utilizing our previously characterized murine model of RILI.2,10 Our results indicate that single-fraction thoracic radiation (20 Gy) significantly augments nmMLCK protein expression and MLC phosphorylation in wild-type (WT) mice lungs, which peaked 3 days after radiation. Furthermore, mice with nmMLCK deletion were protected from RILI, as assessed by vascular leak, leukocyte infiltration, and histological evidence of inflammation and injury. Correspondingly, radiated WT mice treated with MLCK inhibitors ML-7 or PIK (peptide inhibitor of kinase, an MLCK inhibitory oligopeptide) also demonstrated attenuated RILI. Taken together, these data suggests a key role for nmMLCK in vascular barrier regulation in RILI and warrant further examination of RILI strategies that target nmMLCK.
METHODS
Reagents
Antibodies against nmMLCK, MLC, and phosphorylated MLC (p-MLC) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). ML-7 and β-actin antibody were purchased from Sigma (St. Louis, MO). Horseradish peroxidase–conjugated anti-mouse and anti-rabbit secondary antibodies were purchased from Cell Signaling (Danvers, MA). PIK (or MLCK inhibitor peptide 18) was purchased from Tocris Bioscience (Bristol, United Kingdom).
Murine model of RILI
All experiments and animal care procedures were approved by the Institutional Animal Care and Use Committee (IACUC). C57BL/6 (20–25-g) mice 8–10 weeks old were purchased from Jackson Laboratory (Bar Harbor, ME), and nmMLCK−/− mice were provided as gifts from D. M. Watterson 11 and bred according to approved IACUC protocol. Mice were anesthetized with ketamine (100 mg/kg) and acepromazine (1.5 mg/kg) and administered radiation (20 Gy) to the thorax, as we described previously. 2 Briefly, a 5-mm-thick lead block was used to shield the rest of the animal while the thorax, between the clavicles and below the sternum, was irradiated with a 250-kV x-ray beam at a dose rate of 2 Gy/minute using an orthovoltage animal irradiator. Body weight was measured for individual mice. Select mice were treated with MLCK inhibitor ML-7 (15 μg/mouse, every other day) or PIK (0.25 mg/mouse, every other day) or vehicle control via intraperitoneal injection, beginning 1 week before irradiation and continuing for up to 6 weeks afterward. The mice were then killed, and indices of lung vascular leak and inflammation were assessed via bronchoalveolar lavage (BAL) fluid protein levels and cell counts, as previously described. 2 Lungs were harvested for further evaluation.
BAL fluid analysis
BAL was performed after irradiation, as we have previously described. 2 Briefly, at the termination of each experiment, mice were euthanized by exsanguination under anesthesia according to the IACUC protocol. Both lungs were lavaged with 1 mL of cold Hank's buffered saline solution (HBSS). The BAL fluid collected was centrifuged at 500 g for 20 minutes at 4°C, and the supernatant was removed and recentrifuged at 12,000 g for 10 minutes for supernatant collection. The cell pellet was resuspended in cold HBSS for determination of total cell count and cell differential on slides prepared by cytocentrifugation (Cytospin 3; Shandon Instruments, Pittsburgh, PA) and Diff Quik staining (Dade Behring, Dugen, Switzerland). The protein concentration in BAL supernatant was determined by a protein assay (Bio-Rad, Hercules, CA).
Lung histology and immunohistochemistry
Lungs collected from radiated mice were fixed in 10% formalin for at least 48 hours, and lung paraffin sections were used for immunohistochemistry and stained with nmMLCK primary antibody (catalog no. sc-25428, Santa Cruz Biotechnology) using the avidinbiotin-peroxidase method. Briefly, lung sections (4–5 μm thick) were deparaffinized and rehydrated with graded ethanol. After 30 minutes incubation at room temperature with Tris-buffered saline (TBS) containing 3% normal serum or casein for blocking, the slides were incubated with nmMLCK antibody overnight at 4°C, followed by 3 washes in TBS and further incubation with a streptavidin-conjugated secondary antibody, followed by reaction with substrate and chromagen (diaminobenzidine [DAB]). Slides were then counterstained with hematoxylin.
Western blotting of nmMLCK, total-MLC, and p-MLC protein in lung tissue
Lung tissues were homogenized in a polytron with a buffer containing 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 2 mM EGTA (ethylene glycol tetraacetic acid), 5 mM β-glycerophosphate, 1 mM MgCl2, 1% Triton X-100, 1 mM sodium orthovanadate, 10 μg/mL protease inhibitors, 1 μg/mL aprotinin, 1 μg/mL leupeptin, and 1 μg/mL pepstatin. Lysates were centrifuged at 500 g for 5 minutes at 4°C, equal amounts of protein (40 μg) were loaded onto 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels, and Western blotting was performed according to standard protocols. 12 Nonmuscle MLCK (catalog no. sc-365352, Santa Cruz Biotechnology), and p-MLC (catalog no. 3675, Cell Signaling) were purchased.
Quantitative real-time polymerase chain reaction (RT-PCR) confirmation of gene change
Quantification of the selected transcripts was performed by relative quantitative RT-PCR with SYBR Green RT-PCR assays and the CFX384 Real-time PCR system (Bio-Rad). The complementary DNAs (cDNAs) were generated from 1 μg of total RNA with a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). The resulting cDNA was subjected to a 40-cycle PCR amplification using manufacturer's protocol. Three replicates were run for each gene for each sample in a 384-well plate. The relative quantitation method (ΔΔCt) was used (against internal control GAPDH), with the relative level of the messenger RNA (mRNA) for the gene of interest calculated. The significance of the SYBR Green data was calculated; results were considered significant when P < 0.05.
Statistical analysis
Two-way analysis of variance (ANOVA) was used to compare the means of data from two or more different experimental groups. If significant difference was present by ANOVA (P < 0.05), a least significant differences test was performed post hoc. Subsequently, differences between groups were considered statistically significant when P values were <0.05.
RESULTS
Lung radiation induces nmMLCK expression and MLC phosphorylation
Nonmuscle MLCK is crucial in driving cytoskeletal rearrangement via phosphorylating MLCs, leading to cell contraction and EC barrier dysfunction. To examine radiation-induced alterations in MLCK expression and activity, C57BL/6J (WT) mice were radiated with a 20-Gy single-fraction thoracic radiation dose, and expression levels of nmMLCK protein and mRNA levels were examined 6 weeks after radiation. Both immunohistochemistry and Western blotting data demonstrated significantly increased nmMLCK expression levels 6 weeks after radiation in mouse lungs compared to lungs of nonradiated WT controls, suggesting that nmMLCK is influenced by RILI exposure (Fig. 1A, 1B). Similarly, nmMLCK activity was rapidly increased after radiation (1 day; Fig. 1C), reflected by increased levels of MLC phosphorylation, beginning 1 day after radiation, peaking at 3 days, and dissipating over 6 weeks after radiation. Correspondingly, mRNA expression of nmMLCK was upregulated significantly in radiated lungs 6 weeks after radiation (Figs. 1D, S1; figs. S1, S2 available online), findings also observed in lung microarray data collected from a Gene Expression Omnibus (GEO) data set (GEO data set ID: GSE41789; Fig. S2). 13
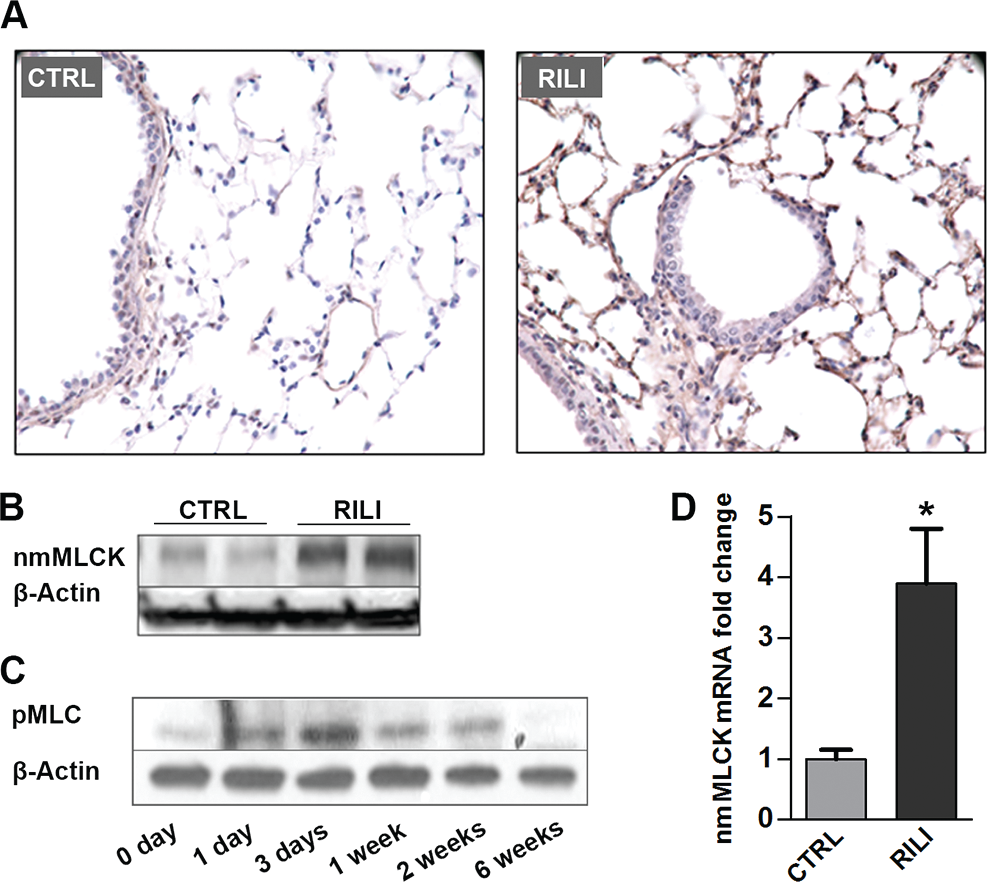
Nonmuscle myosin light chain kinase (nmMLCK) is upregulated in murine radiation-induced lung injury (RILI) lungs. C57BL/6J mice were exposed to a single-fraction thoracic radiation (20 Gy), and lung tissues were examined for nmMLCK expression and activity. A, Lung immunohistochemistry staining for nmMLCK; B, lung protein Western blot analysis for nmMLCK; C, lung protein phosphorylated MLC (pMLC) Western blot analysis (with different time points after radiation); D, quantitative polymerase chain reaction analysis for nmMLCK messenger RNA (mRNA). *P < 0.05 compared to nonradiated samples. n = 5–6. CTRL: control.
Mice with targeted deletion of nmMLCK are protected from RILI
Utilizing a previously characterized preclinical model of RILI, 2 C57BL/6 (WT) and nmMLCK−/− mice were exposed to a single dose of whole-thoracic radiation (20 Gy) to assess RILI in terms of BAL protein (reflecting vascular leak) and inflammatory cell influx (reflecting inflammation). Radiation induced significant increases in the levels of protein and total cells in BAL fluid of WT mice, compared to nonradiated respective controls. In contrast, nmMLCK−/mice were protected from RILI in terms of vascular leak, inflammation, and percentage body weight loss, compared to age-matched radiated WT mice (Fig. 2).
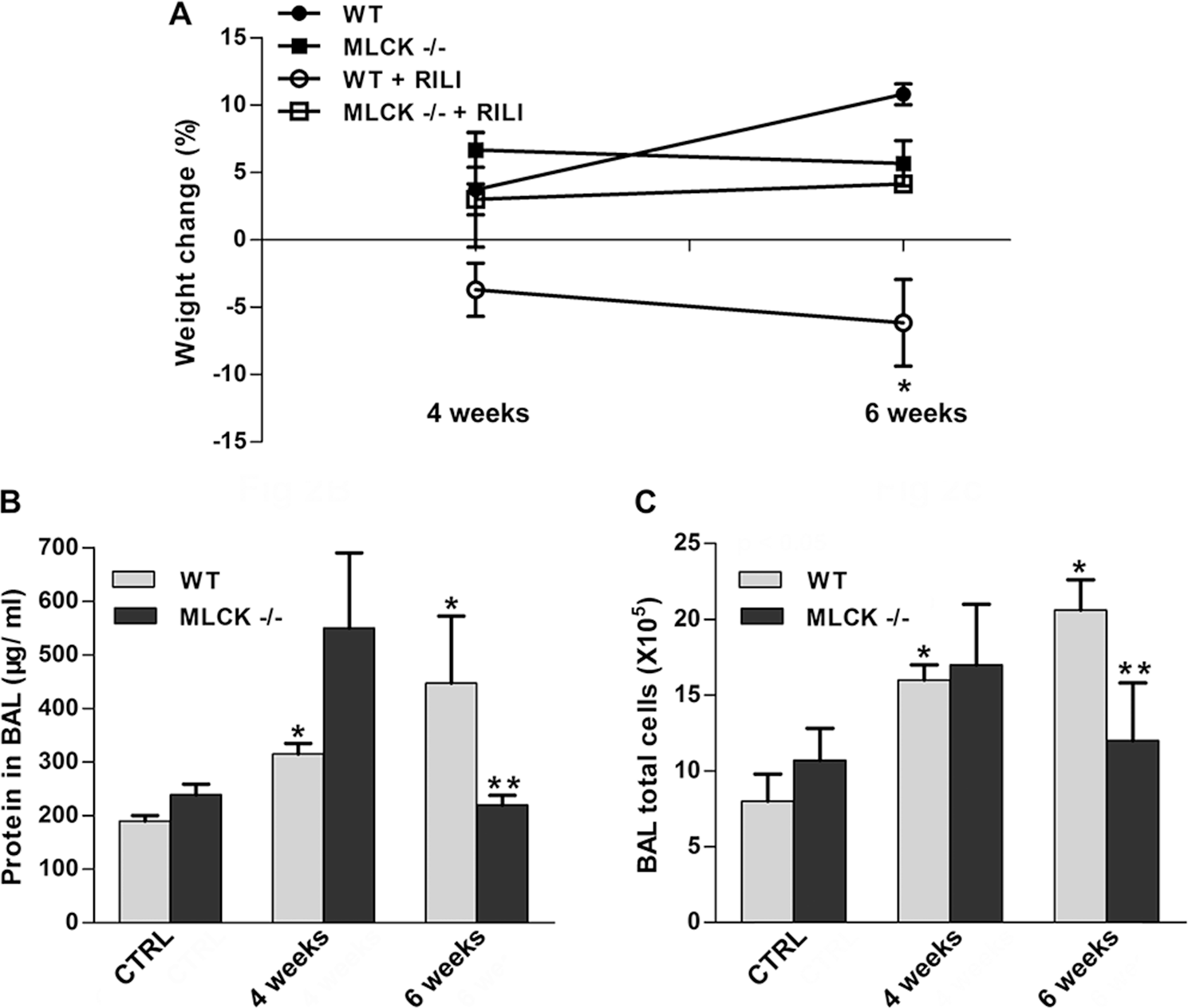
The nmMLCK−/− mice are protected from radiation-induced lung injury (RILI). nmMLCK−/− mice and wild-type (WT) mice received thoracic radiation (20 Gy), and bronchoalveolar lavage (BAL) fluid was extracted/examined for inflammatory profile. A, Body weight of the corresponding mice was examined 2 weeks, 4 weeks, or 6 weeks after radiation. *P < 0.05 between WT+RILI and other groups. n = 5–6. B, C, BAL protein levels (B) and total cell counts (C) were analyzed. *P < 0.05 compared to WT-control (CTRL) group. ***P < 0.05 compared to the WT–6-week group. n = 5–6.
Protective effects of inhibitors of MLC kinase activity on RILI
Our previous in vitro and in vivo studies demonstrated beneficial effects of the membrane-permeant inhibitor of MLC kinase activity (PIK, a highly specific peptide inhibitor of MLCK) 14 by inhibiting MLC kinase activity and MLC phosphorylation. 15 ML-7, an nmMLCK chemical inhibitor, 16 was also adapted to define nmMLCK function. Radiated mice were administered PIK (0.25 mg/mouse) or ML-7 (15 μg/mouse) every other day for 6 weeks in this RILI model. Both PIK- and ML-7-treated mice were protected from RILI, compared to untreated radiated controls, as evidenced by BAL protein content and total BAL cellularity (Fig. 3A–3C).
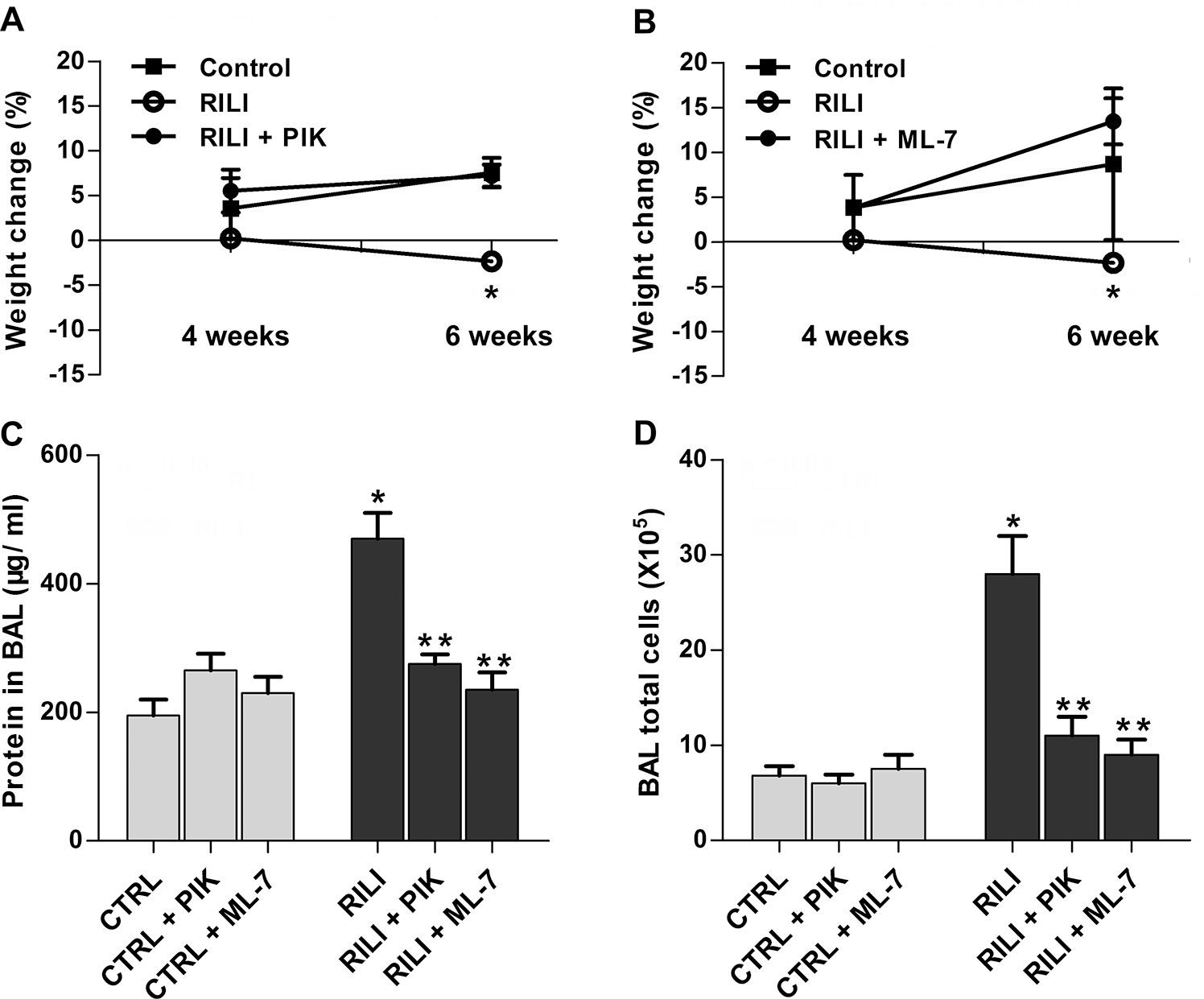
Protective effects of nonmuscle myosin light chain kinase (nmMLCK) inhibitors ML-7 and PIK (peptide inhibitor of kinase) on radiation-induced lung injury (RILI). Mice were treated with MLCK inhibitor ML-7 (15 μg/mouse, every other day), PIK (0.25 mg/mouse, every other day), or vehicle control (CTRL) via intraperitoneal injection beginning 1 week before irradiation and continuing for a period of up to 6 weeks. A, B, Body weight of the corresponding mice was examined 2 weeks, 4 weeks, or 6 weeks after radiation. A, *P < 0.05 between RILI and RILI+PIK groups. n = 5–6. B, *P < 0.05 between RILI and RILI+ML-7 groups. n = 5–6. C, D, Bronchoalveolar lavage (BAL) protein levels (C) and total cell counts (D). *P < 0.05 between CTRL and RILI groups. **P < 0.05 compared to RILI group. n = 5–6.
DISCUSSION
The molecular mechanisms underlying the development of RILI remain enigmatic and controversial, impeding the identification of novel therapeutic targets. To date, therapeutic strategies have largely been designed to ameliorate the acute effects of radiation via neutralizing proinflammatory cytokines or attenuating inflammatory cell infiltration. As we have shown that endothelial and epithelial barrier integrity is perturbed in murine RILI,2,10 we hypothesized that preservation of vascular barrier integrity is an effective therapeutic strategy in RILI as well as in other acute lung injury syndromes.15,17 By facilitating formation of a paracellular gap, nmMLCK plays a central role in EC cytoskeletal rearrangement during lung injury. 18 We have previously reported that nmMLCK knockout mice, as well as mice treated with an inhibitory peptide to reduce MLCK activity, were protected against VILI. 15 Furthermore, our studies identified MYLK gene variants (single-nucleotide polymorphisms) that confer susceptibility to sepsis, sepsis- and trauma-induced ALI, and increased risk of severe asthma in African Americans.3,4 In this study, we report that nmMLCK contributes to RILI by modulating MLC phosphorylation induced in radiated mouse lungs.
The striking paucity of mechanistic information that addresses RILI pathogenesis magnifies the importance of appropriate preclinical models of human RILI. We employed our established RILI model to define the role of nmMLCK in RILI pathogenesis. Murine RILI, followed by a single clinically relevant radiation dose (18–25 Gy) over a clinically relevant time (∼12 weeks), exhibits increased vascular leak (BAL protein) and leukocyte infiltration (BAL cell count), two critical parameters to assess acute lung inflammation.
Nonmuscle MLCK is upregulated in lung tissue by radiation consistently (Figs. 1D, S2), from a combination of sources: endothelium/epithelium (Fig. 1A) and infiltrated inflammatory leukocytes. 19 Furthermore, we also tested the mRNA level of two major nmMLCK splice variants, nmMLCK1 and nmMLCK2, which were induced after 6 weeks of radiation treatment (Fig. S1). Regulation of nmMLCK transcription is possibly mediated by the increased generation of reactive oxygen species (ROSs) evoked by radiation-induced injury. We have identified multiple antioxidant response elements located in nmMLCK core promoter (−2.5 kb to transcription start site), suggesting the activation of nmMLCK expression by ROS-dependent pathways. In addition, we have found that Nrf2 knockout mice exhibit a lower expression of nmMLCK (data not shown), which also supports that nmMLCK levels are maintained by Nrf2 positively. Although inhibitors of nmMLCK and nmMLCK knockout mice exhibited similar RILI protection effects, we believe that the MLC phosphorylation is biphasic. Nonmuscle MLCK is activated via a calmodulin-dependent mechanism in the acute phase (minutes to hours), 20 and nmMLCK is kept upregulated in an ROS-Nrf2-dependent manner in the delayed phase (days to weeks). This biphasic mechanism makes nmMLCK inhibitors consistent and reliable for RILI.
Nonmuscle MLCK knockout mice were significantly protected from RILI (Fig. 2), similar to our previous findings in acute respiratory distress syndrome and VILI murine models, suggesting a highly conserved function of nmMLCK in regulation of EC barrier integrity during inflammatory stimuli. Interestingly, this protection effect in nmMLCK knockout mice was not present at an early time point (4 weeks in Fig. 2B, 2C), which suggests a complex role of nmMLCK in barrier disruption 15 and restoration. 21 Importantly, pharmacological inhibitors of nmMLCK (ML-7 and PIK)22,23 effectively attenuated RILI, similar to the effects observed in nmMLCK knockout mice at a delayed time point, suggesting that regardless of its dual role in endothelial barrier regulation, nmMLCK is an effective therapeutic target of RILI, a devastating and life-threatening syndrome.
Besides playing the central role in endothelial barrier regulation in response to stimuli, 5 nmMLCK is also essential for neutrophil transmigration and epithelial barrier dysfunction during lung injury response.19,22 It would be interesting to characterize nmMLCK function in neutrophil-epithelial interaction with RILI in our future work. Remarkably, it is demonstrated that p21-activated kinase and extracellular signal-regulated kinase are critical activators of MLC involved in vascular permeability regulation during inflammation development. 24 Those results provided another avenue for identifying the therapy targets of RILI.
In summary, this concise study characterizes the upregulation of nmMLCK expression and activity in RILI and suggests a key role for nmMLCK in vascular barrier regulation in RILI. Further examination of RILI strategies that target nmMLCK is warranted.