
Abstract
Keywords
As neuroscientists involved in fundamental research, we are often asked how our animal behavioural work can advance knowledge about complex human mental illnesses such as schizophrenia. There appears to be a misconception that an animal model can and should reproduce the human disease in all its aspects. Rats and mice are obviously very different from humans and the simpler brain cortical structure of rodents [1] wouldmake it impossible to display the same kind of complex symptoms as humans. This paper will emphasize, however, that animal research is very important for our understanding of human psychiatric illness. We will focus here on schizophrenia, but the same principles also apply to other psychiatric illnesses. We will describe a number of animal behavioural models with relevance to schizophrenia and illustrate their usefulness with recent results from our own studies.
Animal experimentation ethics
When using experimental animals in behavioural research, ethical considerations as well as scientific considerations should be extremely important. There is concern in the general public and media about the use of animals, particularly primates, for scientific research. It is the responsibility of the scientists to explain exactly why animals are needed, what they are used for and how the results may benefit human clinical practice. If using animals for research, the scientist should use as few animals as possible, use the best possible methods and pay special attention to minimizing pain or discomfort in the animals. Animal research in Australia is done within the guidelines of the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes [2]. Although these guidelines have provided a workable environment, even without such guidelines, ethically responsible use of experimental animals should be of the utmost importance for any researcher. In addition, it is clear that animals that endure pain or distress may provide erroneous data, particularly where subtle changes in behaviour are studied.
Animal models
Geyer and Markou discussed the validity of animal models of psychiatric disorders [3]. They state that ‘The primary purpose of animal models is to enhance our understanding of a human phenomenon’ [3] and recognize that it is very difficult to reproduce a disease as complex as schizophrenia in an animal. Other possibilities are then to only use animals to study the effect of new treatments. This is essentially a rather restricted and circular bioassay approach. For example, amphetamine-induced locomotor hyperactivity in rats has been widely used to test new compounds for antipsychotic activity. However, as will be discussed in more detail later, amphetamine-induced hyperactivity in rodents is mediated by enhanced subcortical dopamine release. Compounds such as haloperidol, may reduce this activity by blocking dopamine receptors, but compounds with weak dopamine receptor affinity, such as clozapine and other atypical antipsychotics, will have little effect. Modern advances in receptor binding technology and computer modelling in chemistry have largely superseded the use of animals purely as bioassay systems.
Although it may be impossible tomodel the entire complex symptomatic spectrum of a psychiatric illness in an animal, selected symptoms may be successfully mimicked and have validity. Behavioural models should at minimum have predictive validity and be reliable [3–5]. Predictive validity refers to the predictive value that observations made in animals will have for the human condition. Reliability refers to the accuracy with which both the experimental and clinical observations are made [3],[4]. These concepts are particularly relevant for assay models, such as those used in the development of new antipsychotic drugs. However, face validity relates to phenomenological similarity between the behaviour exhibited by the animal and the specific symptoms of the human condition [3]. Therefore, it would be unrealistic to expect similar behaviours in rodents as in humans and it is more important to search for relevant equivalents based upon the brain areas and transmitter systems assumed to be involved. Construct validity is then more relevant, as it refers to similarity in the underlying mechanisms that are involved in particular behaviours, even though the precise expression of behavioursmay differ between humans and experimental animals. An extension of contrast validity is aetiological validity, which refers to the degree of similarity of aetiology between behavioural changes in humans and the experimental animal model studied. For complex human illnesses, such as schizophrenia, aetiological validity is difficult to assess, because so little is known about the aetiology of the illness. However, the animal model can be used to test hypotheses about the possible aetiology of the illness. Any animal model, where either the entire illness, aspects of it, or treatments for it are modelled, should be discussed as to their predictive validity, face validity, construct validity or aetiological validity.
Two of the most widely used animal models of psychosis have been the measurement of the extent of locomotor hyperactivity induced by amphetamine or phencyclidine treatment in rats [3]. Prepulse inhibition (PPI) has been widely used to assess sensory information processing problems (sensorimotor gating) that may underlie some symptoms of the illness. However, other aspects of schizophrenia, such as negative symptoms, appear to be very difficult to model in rodents [5].
Psychotropic drug-induced hyperactivity
For more than 25 years, the effect of psychotropic drugs, such as amphetamine, on locomotor activity in rodents has been used to model positive symptoms of schizophrenia, particularly psychosis. Early studies in rats showed that prior depletion of dopamine in the ventral striatum (nucleus accumbens) could completely prevent amphetamine-induced hyperactivity, suggesting the importance of subcortical dopamine release in the effects of amphetamine and similar drugs [6],[7]. It was only recently that similar conclusions were drawn in human research. Using imaging techniques in normal human subjects, Drevets et al. found that amphetamine, at a dose very similar to that used in rats, caused a much greater dopamine release in the ventral striatum than in the dorsal striatum [8],[9]. Furthermore, the euphoric effect of amphetamine treatment correlated with its neurochemical effect in the ventral striatum, but not in the dorsal striatum [8]. These observations are relevant for schizophrenia. Amphetamine-induced dopamine release in the striatum was found to be markedly increased in patients with schizophrenia compared to controls [10] and this enhancement was seen at onset of illness and in never-before treated patients during psychotic episodes and relapse, but not during periods of remission [11]. Taken together, these studies suggest that enhanced subcortical dopamine release is a prominent feature of psychosis in patients with schizophrenia, a comparable enhancement of dopamine release can be obtained with amphetamine treatment, and that such an effect of amphetamine is similar in humans and experimental rodents.
This comparison of the effects of amphetamine also emphasizes that, when studying animal behaviour, one has to look for behavioural effects that are relevant for that species: in humans, amphetaminemay induce euphoria and psychosis in addition to hyperactivity, whereas the same treatment in rodents induces mostly locomotor hyperactivity [6–9]. Although it is difficult to reliably assess other effects of amphetamine in rats and mice, locomotor activity can be easily quantitated by several means. Techniques range from human observations and rating scales, which are labour-intensive and prone to subjectivity and human error, to automated photocell set-ups, which allow objective measurements and high throughput.
We have used the amphetamine-induced locomotor hyperactivity model to study the role of central serotonergic projections in the brain in the modulation of dopaminergic activity. Interest in the possible role of serotonin in the pathophysiology of schizophrenia has originated from the observation that many atypical antipsychotics display significant serotonin receptor subtype binding affinity. Furthermore, neurochemical measurements in postmortem samples have revealed marked changes in indices of serotonergic activity in patients with schizophrenia. For example, affinity of the serotonin transporter was significantly reduced in the ventral hippocampus, whereas the density of 5-HT2A receptors was reduced in the frontal cortex [12].
We assessed the effect of selective serotonin depletion in the brain of rats on locomotor hyperactivity induced by amphetamine or phencyclidine in these animals [13]. Microinjection of the serotonergic neurotoxin 5,7-dihydroxytryptamine into the midbrain dorsal raphe nucleus (DRN) caused marked depletion of serotonin in the striatum and hypothalamus, but not hippocampus, whereas this microinjection into the median raphe nucleus (MRN) depleted serotonin in the hypothalamus and hippocampus, but not striatum [14]. Studies in humans could never induce similarly selective depletions of serotonin. Current strategies, such as dietary amino acid depletion [15] induce serotonin depletion throughout the brain, a global effect that makes it difficult to ascribe function to any brain regions or serotonergic projection system in particular. In our studies, rats with lesions of the DRN showed no changes in the extent of locomotor hyperactivity induced by either amphetamine or phencyclidine. However, in rats with selective MRN lesions, locomotor hyperactivity to phencyclidine, but not amphetamine, was significantly enhanced (Fig. 1) [13]. Further studies determined that serotonin depletion in the dorsal hippocampus, to which the MRN projects, also leads to marked enhancement of the locomotor hyperactivity response to phencyclidine treatment [16].
Effect of lesions of the median raphe nucleus (MRN) or dorsal raphe nucleus (DRN) on the change in locomotor activity induced by treatment with amphetamine (0.5mg kg−1) or phencyclidine (PCP, 2.5mg kg−1). Lesions were induced by microinjection of 5,7-dihydroxytryptamine into either one of the raphe nuclei 2 weeks before the behavioural experiments. In MRN-lesioned rats, the effect of PCP was markedlly enhanced, supporting a role of brain serotonergic projections from this nucleus in symptoms of schizophrenia. For further details see [13].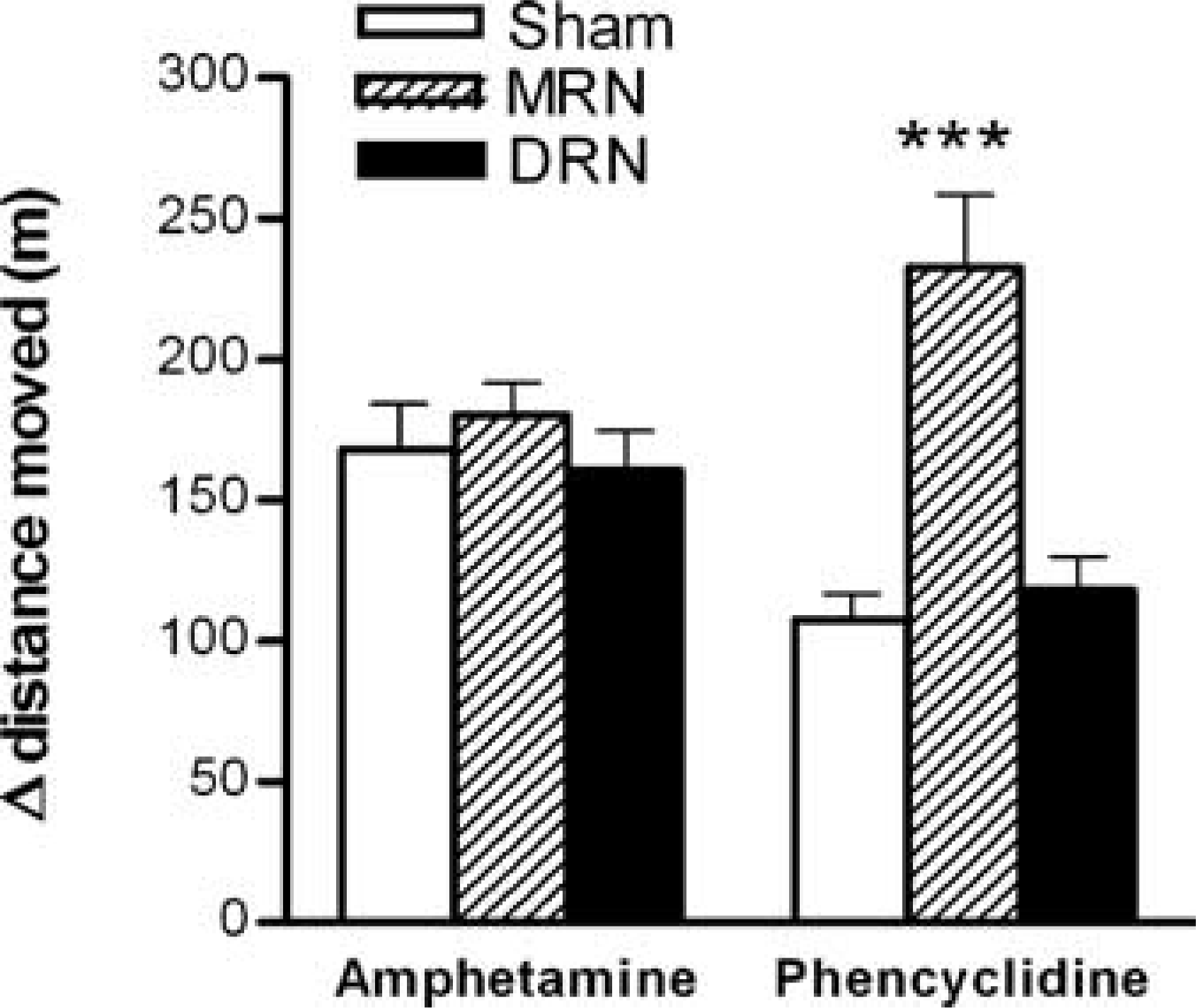
Although these effects were studied relatively early after the lesions (2–3 weeks), we are now studying the effect of chronic interventions on the neurodevelopment of behaviour. For example, the effect of neonatal stress, combined with later stress or treatment with stress hormones, may provide a way to study the synergistic effects of early and late developmental insults on the pathophysiology of schizophrenia [17]. Such studies are of great importance in order to test the ‘two-hit’ hypothesis of schizophrenia [18],[19] and would be very difficult to carry out in humans.
Prepulse inhibition (PPI)
Another example of behavioural observations with close similarity between humans and experimental animals, is PPI. As extensively reviewed by Geyer, PPI is an operational measure of sensorimotor gating, essentially a shielding mechanism against sensory information overload or ‘inundation’ [3],[17],[20–22]. Prepulse inhibition of the acoustic startle uses a very simple behavioural response to study this gating mechanism. Startle is mediated by a simple neuronal circuit in the brainstem, the activity of which ismodulated by several limbic and cortical regions. The latter brain regions include medial prefrontal cortex, nucleus accumbens, hippocampus and amygdala.
Prepulse inhibition is similar between different species (17],[20],[23]. It should be noted, however, that in rats as well as mice, differences have been observed in the sensitivity to the disruption of PPI by drugs such as apomorphine and amphetamine [20],[23–26]. Strain-related differences in baseline PPI and sensitivity to drug treatments become particularly important in studies using genetically modified mice, where the background strain of mice could determine or contribute to the extent of any deficiency in PPI [23],[27].
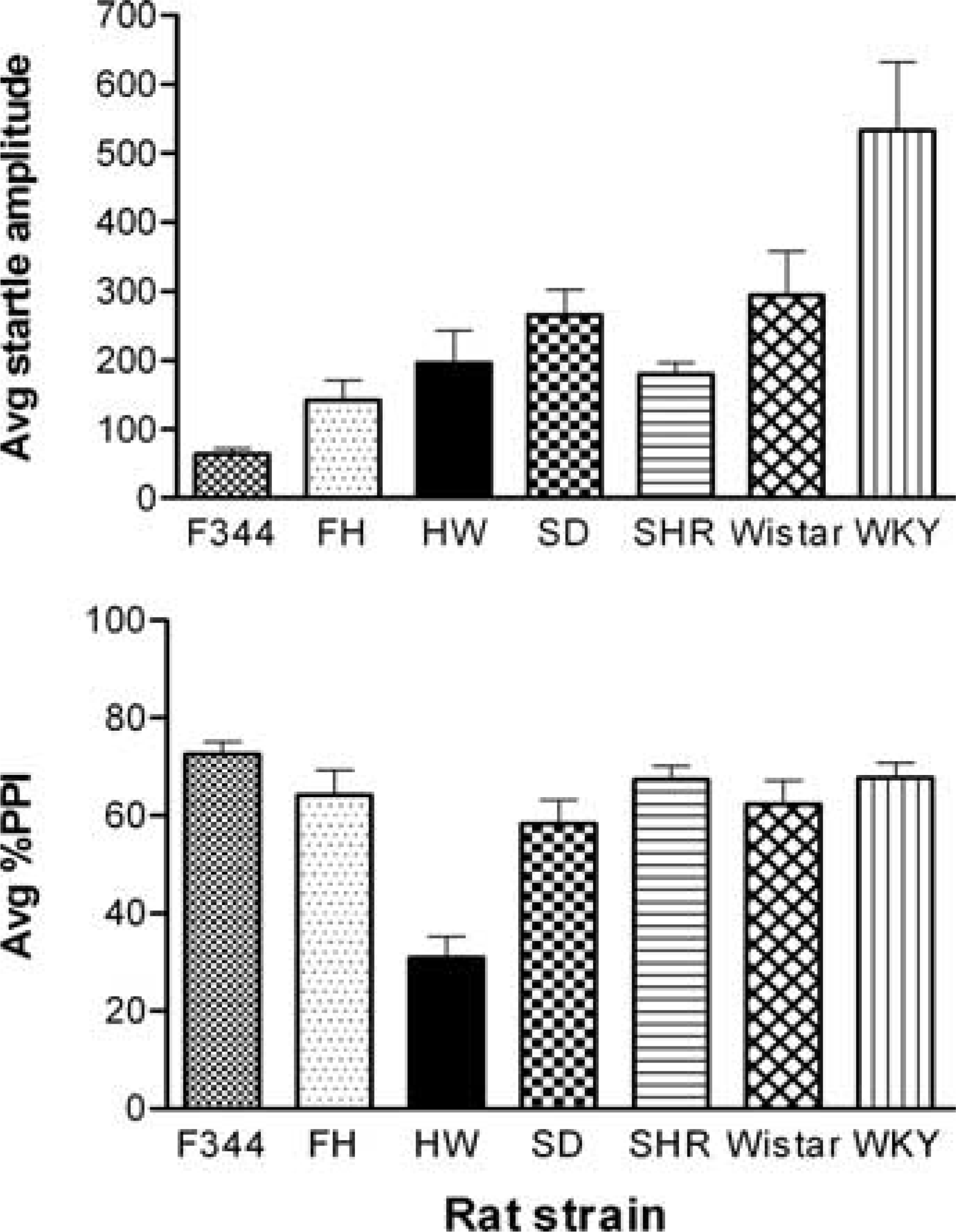
Comparable prepulse inhibition (PPI) of acoustic startle in male C57BL6/129J mice, Sprague–Dawley rats and Dunkin–Hartley guinea pigs. PP2 to PP16 refers to prepulse intensity levels of 2–16 dB above the 70 dB background noise used in the experiments. % inhibition refers to the percentage inhibition of the startle response by prepulses 100 msec before the 115 dB acoustic pulses [24],[48],[51],[52]. There were 6–12 animals per group. Comparison of acoustic startle amplitude (top panel) and prepulse inhibition (PPI) of acoustic startle (bottom panel) of seven rat strains. Startle amplitude was calculated as the average amplitude of 20 startle trials during a PPI session; PPI is expressed as the average percentage inhibition obtained from a range of prepulse intensities (see 17],[24–26]. Although marked differences were found between rat strains with respect to startle amplitudes, PPI tended to be similar, except in Hooded–Wistar rats (HW) that displayed significantly impaired responses [24]. F344, Fischer 344 rats; FH, Fawn–Hooded rats; HW, Hooded–Wistar rats; SD, Sprague–Dawley rats; SHR, spontaneously hypertensive rats; WKY, Wistar–Kyoto rats.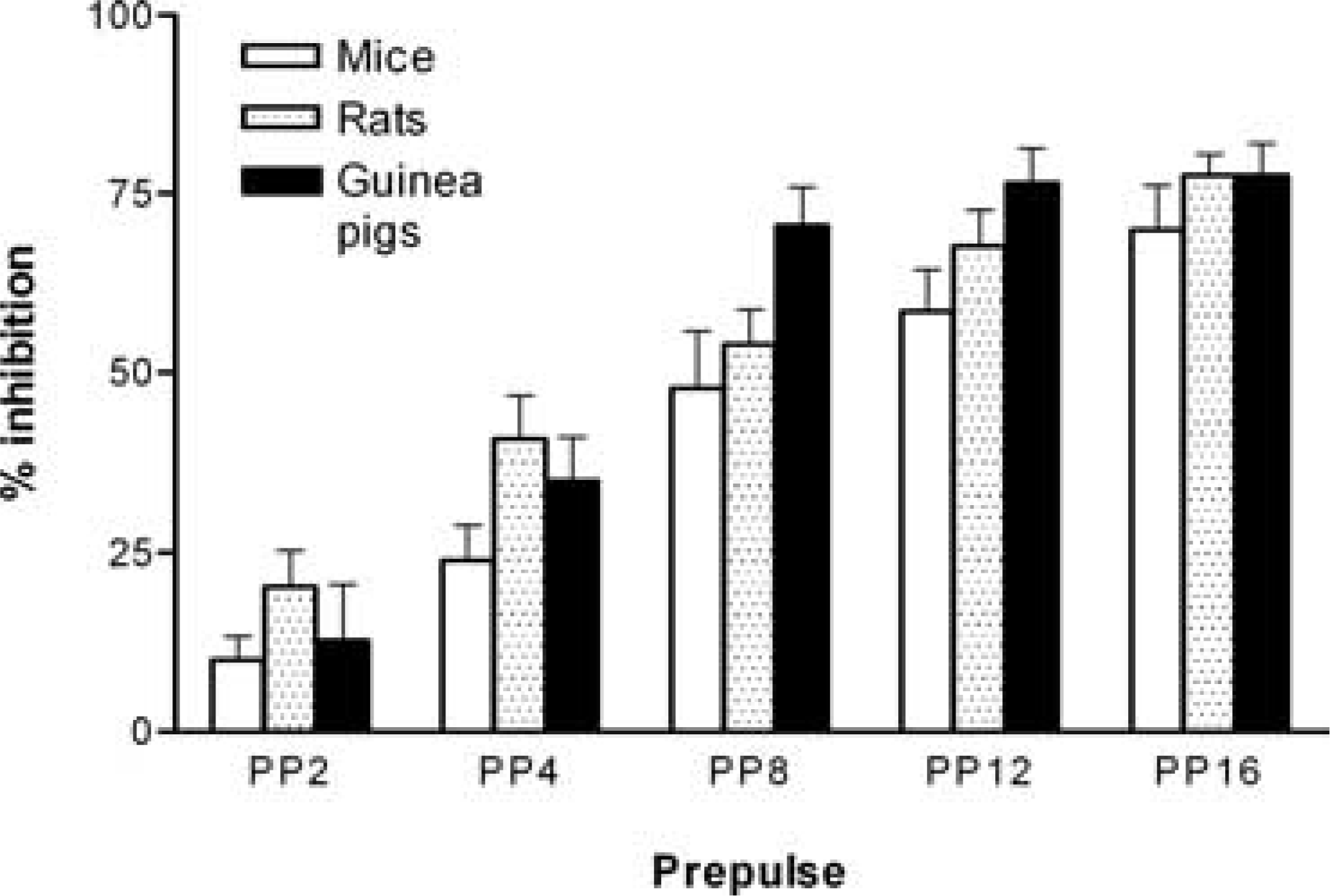
Several serotonergic treatments also cause disruption of PPI [20],[23]. Recently, we observed that selective serotonergic lesions of the MRN, but not DRN, cause a marked reduction of PPI [13]. This indicates that such lesions have effects in more than one model of the aspects of schizophrenia.
In other studies, we observed that administration of the serotonin-1A receptor agonist 8-hydroxy-di-propylaminotetralin (8-OH-DPAT) also disrupts PPI and that this effect could be modulated by sex steroids. In male rats, castration prevented the effect of 8-OH-DPAT on PPI, whereas testosterone treatment restored it [28]. Interestingly, in female rats, ovariectomy did not alter the effect of 8-OH-DPAT on PPI; however, treatment of ovariectomized rats with either a high dose of oestrogen or a combination of a low dose of oestrogen with progesterone, prevented the disruption [29]. These findings show that sex steroids potently, and in a sex-specific way, modulate the effect of serotonin receptor stimulation on PPI, with the effect of testosterone being essentially permissive in male rats and oestrogen and progesterone being protective in female rats. Such an effect of sex steroids could be important for our understanding of how hormones could be involved in sex differences in the first occurrence and symptomatology of schizophrenia [30]. It has been shown by Kulkarni et al. that addition of oestrogen treatment could accelerate and enhance the effect of antipsychotic treatment in acutely psychotic women [31],[32]. Our findings in rats may help to explain the mechanism of this interaction.
Negative symptoms of schizophrenia
Ellenbroek and Cools recently reviewed the published work on animal models for the negative symptoms of schizophrenia [5]. They suggested that ‘few of the negative symptoms lend themselves to modelling in animals’. For example, alogia and affective flattening are particularly difficult to measure reliably in rats and mice [5]. Only social withdrawal is relatively easy to measure in such animals and in monkeys. In rats, social interaction can be quantified and this process has, in some studies, been automated to facilitate reliability of measurements and increase throughput [33],[34]. Rats that are treated with phencyclidine showa syndrome of stereotyped behaviour and reduced social interaction when observed in a test environment with another untreated rat [34]. Treatment with antipsychotic drugs can inhibit these behaviours [34].
There are several learning and memory tests in rodents, focusing on different aspects ofmemory. For example, the classic Morris water maze determines long-term spatial memory, a behaviour particularly dependent on intact hippocampal function [35]. The Y-maze is a simple test for short-term spatial memory [36], whereas the T-maze test has been used to assess working memory in rodents [37]. These tests could be used to assess cognitive changes in experimentally treated animals, providing an insight into central mechanisms involved in cognitive changes in schizophrenia.
New genes and gene products
Schizophrenia and other mental illnesses have a strong genetic component [38] and molecular and genetic studies have revealed several candidate genes that could be involved in the development of the illness [39]. An important role for animal research in psychiatry will be fundamental in vivo research into the role of such new genes, gene products and basic brain mechanisms, preferably before drug development. Traditionally, this has been attempted by combining research on different rat strains with genetic linkage analysis [40],[41]. More recently, particularly work on genetically modified mouse models has become the method of choice to provide greater understanding of the role of genes in brain mechanisms in schizophrenia. Using molecular biological tools, transgenic mice have had genes added, leading to overexpression of the gene's products, whereas knockout mice have had genes removed or inactivated, leading to a shortage of the products of that gene. These models have been very valuable to assess gene function and in the future we will see this even more so for genetic modifications that are brain-region-specific and can be induced by external biochemical means at specific ages (for a discussion and references on the technical aspects of such modifications, see [42]). A recent review on in vivo pharmacology pointed out that ‘molecular biology, combinatorial chemistry and computer modelling cannot predict the integrated response in the whole animal (or human), particularly for novel genes. The human genome project will identify more than 30 000 new gene products and a crucial component of the identification and characterization of these gene products will be whole-animal studies’ [43]. Many of these genes, including ones implicated in schizophrenia [39], have not been ‘traditionally’ linked to psychiatric illnesses and a challenge for in vivo neuroscientists will be to identify their role in animal models of psychiatry, using behavioural tests with relevance to the illness of interest.
It is important to realize that transgenic or knockout mouse models, with their marked overexpression or deletion of gene function, almost never replicate the subtle changes in gene function seen in complex mental illnesses such as schizophrenia. Moreover, it is unlikely that mutation of a single gene can ever fully replicate the multigenetic mechanisms that are involved in schizophrenia [38]. These genetically modified mice can therefore only be used to obtain global information about the function of the gene on brain function. This new insight then has to be used as the basis for further studies, for example, on the effect of more subtle alterations in the function of this gene, or administration of the gene product or inhibition of its synthesis.
One consideration in the use of animal behavioural models in research on new gene products is which behavioural models to use. Also questions as to whether to study male and female animals, what age to choose and which neurotransmitter systems to focus on, are not simple to answer. As Crawley and Paylor stated: ‘If called by a molecular geneticist to test a knockout for a newly discovered gene expressed in the brain, with no a priori hypothesis about the function of the gene, the behavioural neuroscientist may be tempted not to answer. One wonders where to start and how far to go’ [44]. It is important always to test new knockout models in a battery of behavioural tests. The specificity of behavioural responses needs to be assessed: if there are problems with the general health of the animals or neurological complications, this will influence the outcome of many behavioural tests which often rely on movements by the animals. A wide range of tests may also reveal other unexpected negative findings, ‘side-effects’ that could complicate interpretation of the behaviour of primary interest. However, there could be unexpected positive findings, that is, a new role of the targeted gene could come to light [44]. In deciding which tests to use, there are important practical issues. In a recent review, Crawley listed several different behavioural categories and almost 40 specific behavioural paradigms that could be used to assess behavioural changes in new genetically modified mouse models (Table 1, see [44]). More tests means more animals required and larger numbers of animals may cause problems with adequate housing. A wide range of tests requires expertise. All this may make a phenotyping facility a very expensive place to run. One way to meet these issues is for several specialized laboratories to collaborate. Another possibility is to establish specialized phenotyping facilities that have a range of ‘standard’ behavioural tests available and collaborate with specialized research groups if less common tests are needed.

We have conducted some behavioural screening in a number of ‘knockout'mice. For example, because several studies have shown sex differences in the occurrence of schizophrenia [28],[29],[45], we studied aromatase knockout (ArKO) mice. These animals have a mutation in the gene encoding for the enzyme that mediates the conversion of the sex steroid testosterone to oestrogen [46]. These mice display all the physiological and endocrine hallmarks of loss of oestrogen production, such as infertility and morphological changes in the gonads [46],[47]. As such, this knockout does not replicate a known deficit associated with schizophrenia, but the mice were used to better understand the role of oestrogen (or rather the lack of) in behaviour. The mutation did not appear to have any effect of gross motor control, as measured with the rotarod, or anxiety, as measured on the increased plusmaze [48]. However, in these mice, we found an agedependent reduction of PPI that was present in males, but not in females [48]. This decline in function was suggested to be mediated by an age-dependent reduction of dopaminergic activity in these animals, rendering them in a functional hyperdopaminergic state similar to that seen after treatment with dopaminergic drugs such as amphetamine or apomorphine. Consistent with this hypothesis, male, but not female ArKO mice also showed an age-dependent increase in the locomotor hyperactivity induced by amphetamine [48]. These studies are important for schizophrenia in that they show that loss of oestrogen production may induce accelerated age- and sex-dependent alterations in dopaminergic function that results in changes in behaviour in animal models with relevance to the disease. This work in ArKO mice may shed light on the mechanisms behind sex differences in the occurrence and symptoms of schizophrenia. Several other studies in knockout mouse models have similarly provided new insight into possible pathophysiological mechanisms involved in this illness [23],[42],[49].
Conclusion
Studies in experimental animals can provide potentially important new insight into a range of brain mechanisms with relevance to schizophrenia, such as detailed investigations on the role of certain brain areas in behaviour, the mechanism of action of psychoactive and antipsychotic drugs, the interaction of classical and ‘novel’ neurotransmitters and genes in brain function, and neurodevelopmental mechanisms. Several of these studies would be very difficult to carry out in humans, both from a technical and an ethical point of view. Clearly, complex psychiatric illnesses, such as schizophrenia, cannot be exactly reproduced in species such as rats and mice. Nevertheless, animal ‘models’ are an important tool in studying the symptoms and development of such illnesses, alongside approaches such as post-mortem studies, psychophysiological studies, imaging and epidemiology.