
Abstract
Stress during childhood and adolescence has implications for the extent of depression and psychotic disorders in maturity. Stressful events lead to the regression of synapses with the loss of synaptic spines and in some cases whole dendrites of pyramidal neurons in the prefrontal cortex, a process that leads to the malfunctioning of neural networks in the neocortex. Such stress often shows concomitant increases in the activity of the hypothalamic–pituitary–adrenal system, with a consequent elevated release of glucocorticoids such as cortisol as well as of corticotropin-releasing hormone (CRH) from neurons. It is very likely that it is these hormones, acting on neuronal and astrocyte glucocorticoid receptors (GRs) and CRH receptors, respectively, that are responsible for the regression of synapses. The mechanism of such regression involves the loss of synaptic spines, the stability of which is under the direct control of the activity of N-methyl-
Stress and anxiety
A poor attachment and stressful relationship between mother and child in the first 18 months results in relative inhibition of the child in approaching novel objects or participating in novel events [1]. This maternal relationship during childhood is critical in determining depression behaviour in the adult [2], [3]. In the adolescent period, those at ultra-high risk of psychoses may be precipitated into such conditions by stressful events, such as loss of employment or shifting residence [4], [5] (for a review see [6], [7]). During this period an increase in the number of stressful events experienced in the 4 weeks immediately preceding a relapse into schizophrenia has been reported [8], [9] (for a critical review see [4]), and higher levels of familial risk for schizophrenia are associated with higher levels of emotional reaction to stressful events [10] (for a review see [11]). Anxiety following stressful events is known to precipitate major depressive disorder as well as schizophrenia in those so predisposed. Anxiety precedes the development of major depressive disorder in approximately 30% of first-episode cases and in approximately 75% of recurrent episodes, with a 50% comorbidity [12]. The number of stressful life events from age 21 years to 25 years is predictive of subsequent depression [13], [14].
This essay constitutes an enquiry into what the mechanisms are by which stress and anxiety in childhood have important effects on the development of adult depression and, in adolescence, promote psychoses.
Stress precipitates the regression of synapses in the brain
Decreasing general connectivity below a certain value in the neural networks of the brain is expected to lead to rapid loss of function [15]. A child or adolescent with fewer synapses than normal in the frontal regions following a stressful event might then be exceptionally vulnerable to depression or psychoses.
Synapse loss occurs at a high rate in the prefrontal cortex of adolescents, so that more than 30% of synapses found at the beginning of this period have normally regressed by the end of it [16], [17]. Approximately a further 30% of synapses are missing in schizophrenia [18], although it is not clear if they regressed during the adolescent period or were not formed in the first place during childhood (for a review see [19]). A more than 30% decrease in volume and activity of prefrontal cortex in major depression disorder (for reviews see [20–22]) probably also reflects a loss of synapses [22–26].
Stressful events precipitate the regression of synapses in animals. Mild stress of rats for 20 min during 7 consecutive days leads to decrease in synaptic spine density along apical tufts of layer V pyramidal cells of medial prefrontal cortex [27]. Daily restraint stress of rats, for approximately 3 h over 3 weeks, leads to a loss of approximately 30% of all axospinous synapses on the apical dendrites of layer II/III pyramidal neurons in medial prefrontal cortex, and this is accompanied by a loss of distal dendritic branches [28–31]. This loss of spines is almost certainly accompanied by a concomitant loss of terminals because nearly all spines have terminals on them in mature animals [32]. The loss of dendrites and spines is largely reversed if a period of rest from stress is allowed. Thus a 3 week recovery period following a 3 week regimen of daily stress results in the return of most of the apical dendrites lost during the stress period [33].
Stress enhances circulating glucocorticoids and the release of CRH in the brain
Stressful events, such as loss of employment or shifting one's house, are accompanied by activation of the limbic system (especially the amygdala) as well as the reticular formation, which engage the hypothalamus to secrete corticotropin-releasing hormone (CRH) [34]. This brings the adrenohypophysis of the pituitary into play so that it releases adrenocorticotropic hormone (ACTH) that acts on the adrenal cortex to in turn release the glucocorticoid cortisol, which functions as a negative regulator on both the pituitary ACTH and hypothalamic CRH, completing this hypothalamic–pituitary–adrenal (HPA) system.
Young children in an insecure relationship with a carer show elevated cortisol levels when experiencing depressing events, whereas those in secure relationships with a carer do not (for a review see [35]). Older children with such insecure relationships not only show enhanced cortisol levels in stressful situations but also a higher risk of behavioural and emotional problems in maturity [36], [37]. Indeed, adults suffering from clinical depression who have been abused as children show increased responsiveness of the HPA system, as compared with depressed adults who have not had such experiences when children [38], [39].
In adolescence, those at ultra-high risk of psychoses may be precipitated into such conditions by stressful events through elevation of cortisol in the hippocampus following activation of the HPA system [39]. Recent studies, however, show that plasma cortisol levels are not correlated with stressful events in patients at ultra-high risk of psychoses, but they are with the frequency of ‘hassles’, such as losing one's keys and being late for appointments, and with depression [40]. Glucocorticoid
CRH is also released from neurons in the brain, often as co-transmitters with glutamate and γ-aminobutyric acid [43], [44]. Genes for the CRH receptor CRHR1, possess single-nucleotide polymorphisms that modulate the effects, for better or worse, of child abuse on the risk of major adult depression [45]. CRH release in the hippocampus accompanies acute stress [46]. Of particular interest is that early-life administration of CRH can reproduce many of the long-term cognitive deficits found after chronic early-life stress [45].
CRH is modulated by stress during early development, when maternal care determines the level of central CRH expressions, and can alter the set point of CRH gene sensitivity into adulthood, through activation of the central-stress response [47]. Decreased maternal care gives rise to offspring that are more fearful when adults and possess higher brain levels of CRH [48–50]. Increased maternal care in rats, expressed as increased levels of pup licking/grooming and arched-back nursing (LG-ABN), as in high LG-ABN mothers, produces offspring with enhanced spatial learning abilities, with the opposite effect occurring for poorer levels of maternal care [51], [52]. Such increased maternal care during the first 10 days of life ensures that when they are adults, exposure to acute stress leads, among other things, to decreased levels of CRH messenger RNA and reduced plasma ACTH with a consequent reduction in the activation of the HPA systems responses to stress [53], [54]. These effects are possibly mediated by changes in the epigenome at a GR gene promoter through difference in DNA methylation [55]. Environmental enrichment, however, in the peripubertal period from 22 to 70 days of life, such as providing burrowing systems between large interconnected cages filled with regularly placed toys, can reverse the earlier adverse effects of poor maternal care [56].
Enhanced circulation of glucocorticoids and release of CRH in the brain couples stress to synapse regression
Are the high circulating glucocorticoid levels that accompany stress responsible for synaptic-spine regression? Chronic administration of glucocorticoids alone produces regression of spines [16]. Daily injections of the glucocorticoid corticosterone, which binds to GRs in the medial prefrontal cortex, leads to loss of pyramidal neuron spines and dendrites [57–59]. The release of CRH from neurons during acute stress, especially in the hippocampus [60], alters the growth of synapses, primes long-term potentiation [61], as well as engendering neuronal degeneration [61].
Mechanism of synapse regression: loss of synaptic spines
The synaptic-spine head grows as a consequence of the formation and elongation of actin filaments, namely F-actin that pushes against the spine-head plasmalemma (Figure 1). F-actin grows by incorporating monomeric actin subunits (G-actin) with bound ATP at the so-called barbed end of the F-actin. Older actin subunits are removed from the pointed end of F-actin in a treadmilling process. The older subunits are identified as a consequence of a process in which ATP-actin slowly hydrolyzes to ADP-actin. An Factin severing protein, ADF/cofilin, has high affinity for ADP-actin, so promoting disassembly of the older pointed ends of the actin filaments. Profilin catalyses the exchange of ADP for ATP on these ADP-actin monomers. In this way ATP-actin monomers are made available for addition to the barbed ends of Factin, so completing the treadmilling process. Filament growth is blocked by capping their barbed ends, for instance with gelsolin. The filament branching network is enabled by ARP2/3, which both caps the pointed end of F-actin, and acts as a template for the assembly of new filaments.
Synaptic-spine growth and regression are under direct control of the N-methyl- N-methyl-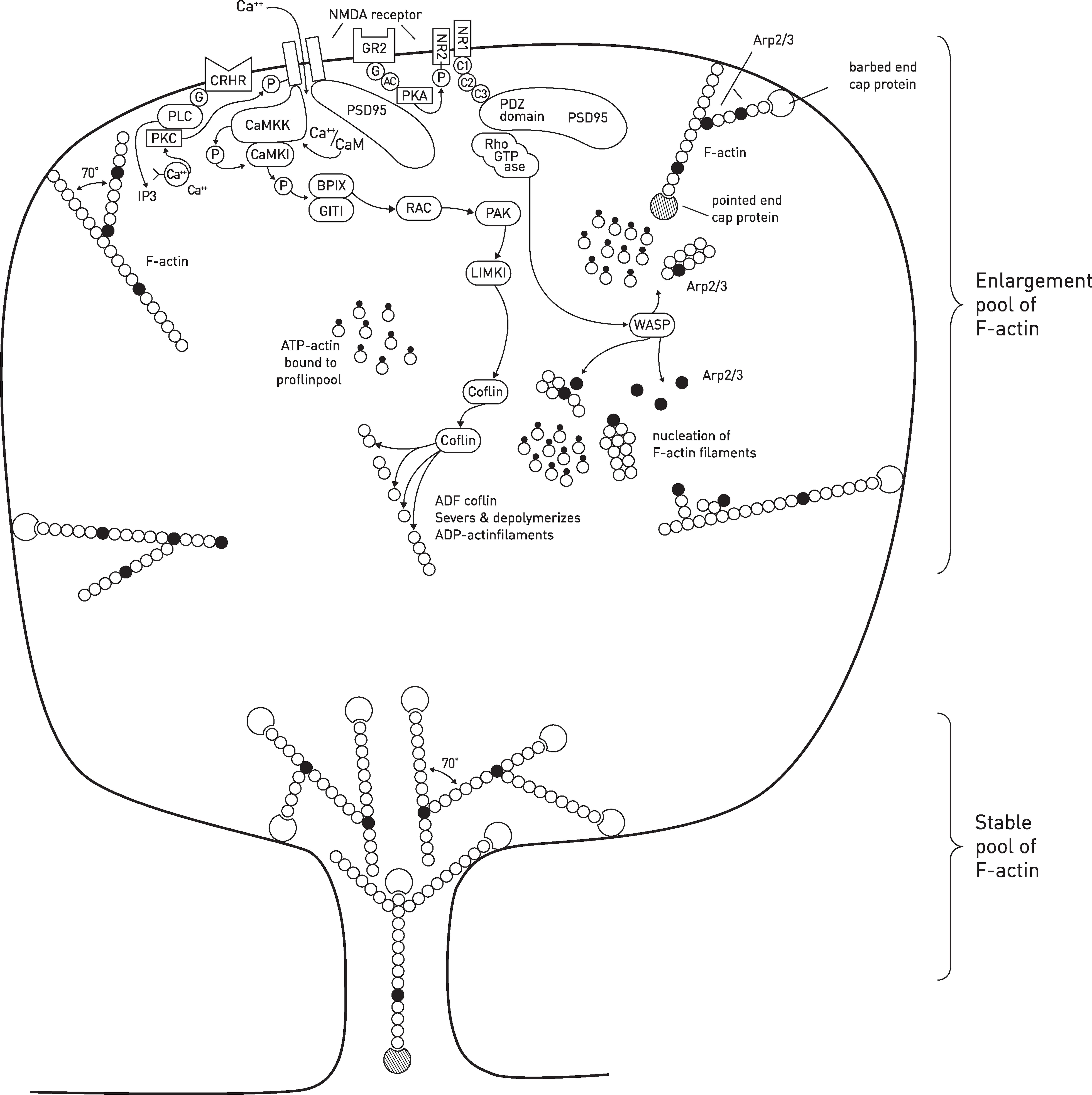
Regulation of NMDA receptors through activation of GRs and CRHRs may then have direct consequences for spine growth and regression through both the CaMKK and the PSD95/RhoGTPase pathways (Figure 1).
NMDA receptors are modulated by circulating glucocorticoids and CRH
Acute cortisone application (30 min) to GR2 on the plasma membrane or only intracellular to the membrane [68] decreases calcium influx through NMDA receptor channels of CA1 hippocampal pyramidal neurons [69], probably through a protein kinase A-mediated phosphorylation of the NR2 subunit of NMDA [70]. Acute CRH acting on CRHR2 also decreases calcium currents through the NMDA channels on amygdala neurons [71]. CRH acts on CRHR2 to activate protein lipase C that potentiates the release of calcium through IP3 channels of internal stores and protein kinase C, leading to phosphorylation of the NMDA channel and depression of its activity [72], [73]. Calcium through the NMDA channel activates calcium-calmodulin kinase and thereby the cascade of events that results in the upregulation of a Rho GTPase (RAC1) and hence coflin (Figure 1) [74], [75]. Coflin severs and depolymerizes ADP-actin filaments in the synaptic spine (Figure 1), thereby enlarging the pool of F-actin. If this pool is released into the dendritic shaft then the spines retract with eventual loss of the dendrite [76], [77]. Blocking CRHRs can block spine removal and stabilize them [78].
Glucocorticoid–cortisone–NMDA receptor hypothesis for synapse regression in stress and anxiety
The normal functioning of neural networks in the brain is dependent on adequate numbers of synapses for subserving the network. Synaptic spine regression or stabilization is dependent on the activity of NMDA receptors. Consideration is given here to a hypothesis concerning the determinants of this activity.
Acute application of a glucocorticoid such as cortisone to the brain elicits a transient high increase in extracellular glutamate for approximately 1 h, followed by a low and sustained increase in extracellular glutamate [79]. The locations of the GR2 in the neurons and astrocytes that mediate this increase in glutamate release are not known, although it is likely to be the GR found in nerve terminals at synapses and astrocyte processes rather than the nuclear GR, given the speed with which it occurs [80]. Although it is known that acute CRH facilitates the induction and stability of long-term potentiation in the hippocampus [81–83], whether it also increases extracellular glutamate is not known.
Exciting GR receptors on astrocytes greatly facilitates both the amplitude and propagation of calcium waves in astrocyte networks, with a concomitant increase in their release of ATP [84], [85]. Increased extracellular glutamate can act on alpha-ancino-3-hydroxy-5-methyl-4-isoxazo-lepropionic acid (AMPA) and NMDA receptors on astrocyte processes at the synapse to release ATP, and ATP itself releases ATP in an autocrine action on astrocyte purinergic receptors [86]. The large amounts of ATP generated through these mechanisms has the potential to act on presynaptic P2X7 receptors to enhance further glutamate release [87]. In this way the initial glutamate released by the action of glucocorticoids has the potential to set up a regenerative feedback mechanism that stabilizes synaptic glutamate release at a high level.
Increased extracellular levels of ATP act as proliferation agents and chemoattractants for microglia, the processes of which intercalate into the synaptic region [88] so that the increased levels of ATP afforded by the regenerative mechanisms described here have the potential to recruit microglia into the synaptic region. Such microglia release tumour necrosis factor-alpha (TNF-α) in response to glutamate, with this TNF-α acting in an autocrine manner on microglia TNFR-1 receptors to release further TNF-α [89]. High levels of this cytokine may then lead to further proliferation of microglia, so giving rise to a wave of TNF-α propagation over long distances accompanied by activation of microglia [90]. This proliferation of TNF-α-releasing microglia would be anticipated to elevate NMDA receptor activation [91–93].
In opposition to the aforementioned mechanisms for increasing NMDA receptor activity following enhanced glutamate release from nerve terminals by glucocorticoids is the fact that they act directly on post-synaptic GRs to depress currents through NMDA receptors (Figure 1) [94–96], as does CRH on CRHRs [72].
The present hypothesis proposes that circulating glucocorticoids, under the control of the HPA system, act on GRs to increase glutamate release from nerve terminals, which then engenders further glutamate release through an astrocyte-based P2X7 regenerative loop, so exciting post-synaptic NMDA receptors. In addition this glutamate engages the release of microglia to release TNF-α, which upregulates the post-synaptic NMDA receptors. In contrast, both glucocorticoids and neurally released CRH act on post-synaptic receptors to depress the activity of NMDA receptors. It is the balance between these modulators of NMDA receptor activity that determines spine loss or growth and hence synapse regression or formation and stabilization.
Conclusion
This speculative essay suggests that stress can engender the consequent appearance of major depressive disorder and schizophrenia in those so predisposed by triggering the regression of synaptic spines, especially during adolescence, below a critical level at which neural networks cannot adequately function. This process is initiated by the stress-induced release of corticosterone by the HPA system and of CRH from neurons. Several regenerative actions are then orchestrated by neurons, astrocytes and microglia, mediated by glutamate, ATP and TNF-α. These ultimately regulate the expression and activity of NMDA receptors on spines that directly control their growth, stabilization and regression. Such a hypothesis is open to direct experimental investigation.