
Abstract
Depression is accompanied by an increase in activity in the amygdala and a decrease in the rostral anterior cingulate cortex (rACC), with the former attributed to a failure of the latter to exert its normal inhibitory influence. This failure is likely due to regression of synaptic connections between the rACC and the amygdala, a process reversed in part by selective serotonin reuptake inhibitors (SSRIs). The present work presents a hypothesis as to how SSRIs might bring about this process and hence normalization of activity, at least in patients that are responsive to SSRIs. Serotonin receptors of the excitatory 5-HT2AR class increase N-methyl-D-aspartate receptor (NMDAR) efficacy, while those of the inhibitory 5-HT1AR class decrease NMDAR efficacy. A decrease of 5-HT transporter (5-HTT) efficacy, either during human development through functional polymorphisms, or in animals through 5-HTT transgenic knockouts, is accompanied by a decrease in 5-HT1AR and hence an increase in excitability and NMDAR efficacy which drives an increase in synaptic spines in the amygdala. As the limbic region of the brain normally possesses high levels of 5-HT1AR the effect of loss of these is to increase excitation in this region, as is observed. Changes in the level of extracellular 5-HT in adult animals also modulates the density of synaptic spines, with these increasing with an increase in 5-HT, possibly as a consequence of increases in 5-HT2AR activity over that of 5-HT1AR. Increasing extracellular levels of 5-HT with SSRIs would then lead to an increase in excitability and in synaptic spines for afferents in the dorsal rostral anterior cingulate cortex but not in the ventral regions such as the amygdala that have few 5-HT2AR. This allows dorsal regions to once more exert their inhibitory influence over ventral regions. In this way, SSRIs may exert their effect in normalizing dorsal hypometabolism and ventral hypermetabolism in those suffering from depression.
Keywords
Mood disorders such as depression are accompanied by changes in the structure and function of the prefrontal cortex, cingulate cortex and amygdala [1]. In particular, specific connections between these areas are lost [2] and animal studies suggest that this probably occurs as a consequence of the disappearance of spines on neuronal dendrites leading to the regression of synapses [3]. Selective serotonin reuptake inhibitors (SSRIs) are known to at least partly reverse this loss of synapses but no consistent account of how this might occur has been given [4]. N-methyl-D-aspartate receptor (NMDAR) activation is a principal means of ensuring the integrity of synaptic spines [5]. The present work considers a means by which changes in SSRIs modulate NMDA receptors and so determine the formation and regression of synaptic spines and hence the viability of synaptic connections between cortical regions and the amygdala that accompany depression.
Synaptic connections of the anterior cingulate cortex and the amygdala
The anterior cingulate cortex (ACC) consists of four regions, delineated in Figures 1a and 1b: dorsal ACC (BA 24b’, 24c’ and 32’; compare Figure 1b with Figure 1c); rostral ACC (BA 24a, 24b and 24c; compare Figure 1b with Figure 1c), and a subgenual ACC which can be subdivided into anterior (continuation of rostral BA 24a and 24b; compare Figure 1b with Figure 1c) and a posterior component (BA25; compare Figure 1b with Figure 1c). Rostral ACC is anatomically connected with the dorsal prefrontal cortex (BA9, 10,46) (Figure 2a), while subgenual ACC is primarily connected to the orbitofrontal cortex (BA11) as well as the hippocampus and amygdala [6–8] (Figure 2a).
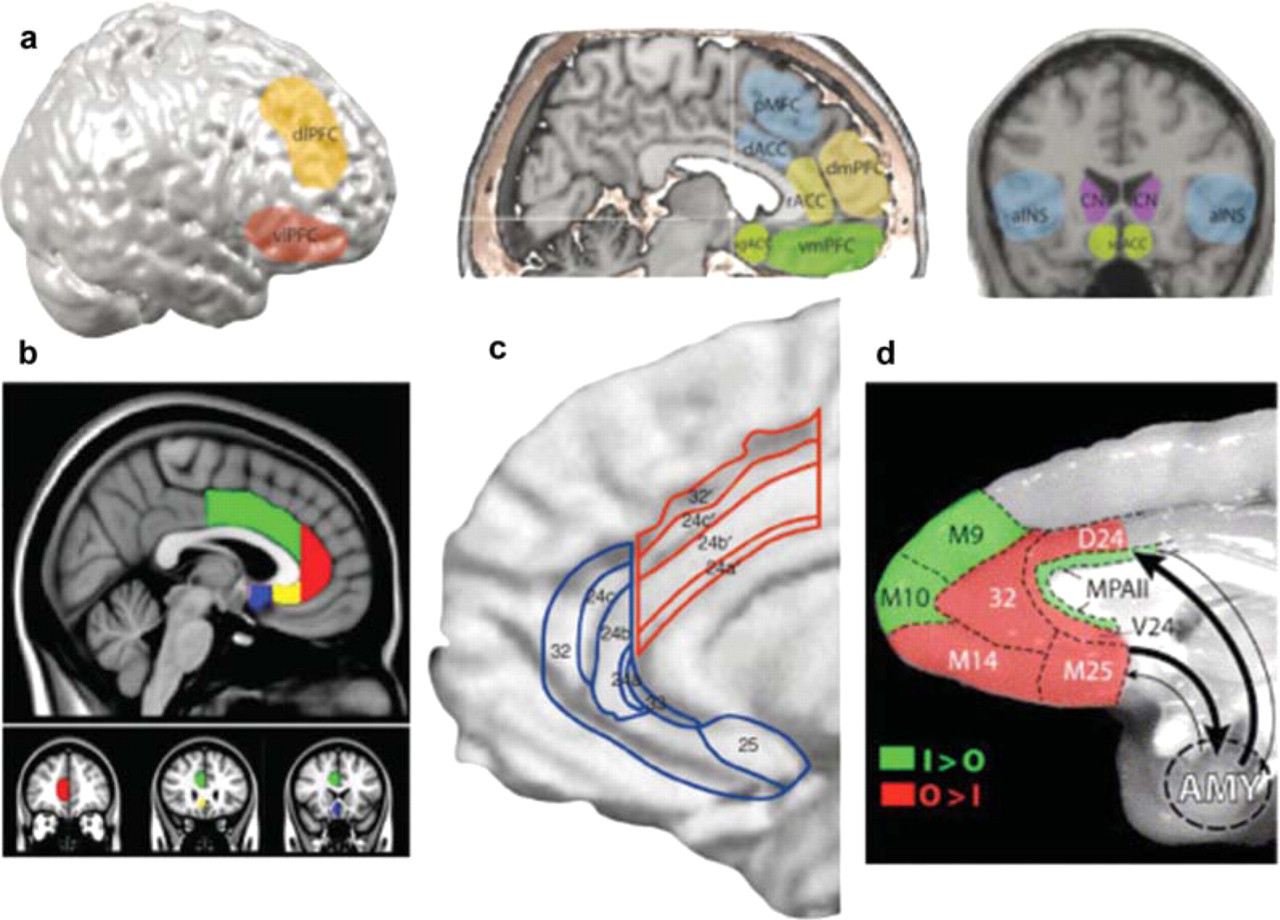
Anatomy of the anterior cingulate cortex, dorsal and ventral prefrontal cortex and the amygdala in humans. (a) The delineations of anterior insula (a INS); CN, caudate nucleus; dACC, dorsal anterior cingulate cortex; dlPFC, dorso-lateral prefrontal cortex, dmPFC, dorsomedial prefrontal cortex; vmPFC, ventromedial prefrontal cortex (includes the medial aspect of orbito-frontal cortex); pMFC, posterior medial cortex; rACC, rostral anterior cingulate cortex; sgACC, subgenual anterior cingulated cortex; vmPFC, ventromedial prefrontal cortex (after Figure 1 in [11]). (b) Parcellation scheme for subregions of the anterior cingulate cortex (ACC)). The green region indicates the dorsal ACC (consisting of BA 24b', 24c' and 32′ (see Figure 1c)); the red region indicates rostral ACC (BA 24a, 24b and 24c; sometimes referred to as pregenual ACC, anterior to the genu of the corpus callosum); the yellow region indicates anterior subgenual ACC (continuation of the rostral cingulate gyrus (BA 24a and 24b), which wrap around the corpus callosum and generally corresponds to the subgenual region (originally identified by [29] as ACC ventral to the genu of the corpus callosum). The blue region indicates posterior subgenual ACC (BA 25; sometimes referred to as the subclossal gyrus) (after Figure 1 in [59]). (c) Detailed anatomy of the anterior cingulate cortex in humans. The cortical surface has been partially inflated to allow simultaneous viewing of gyri and sulci, with a single cingulate gyrus lying between the cingulate sulcus and the corpus callosum. The numbered cytoarchitectural areas of the ACC delineate what are taken to be the cognitive division (areas 24’, 24b’, 24c‘and 32’) and the affective division (24a, 24b, 24c, 25, 32 and 33) (after Figure 1 in [9]). (d) Relative proportion of input and output connections in prefrontal cortex with projections involving the amygdala in the monkey. Shown is the medial surface of the prefrontal cortex. Prefrontal areas with input from the amygdala greater than output to the amygdala are shown (after Figure 8a in [60]). The densest connections of the monkey prefrontal cortex with the amygdala are similar to those in the human, and involve the posterior medial cortex (M25 and D24 and MPA11) and the posterior orbitofrontal cortex (OPA11, OPro). Of these cortices, cingulate areas D24 and M25 project more heavily to the amygdala than they receive projections from the amygdala, in contrast to the caudal orbital areas that receive more projections than they send. Figures 1a, 1c and 1d has been reproduced with permission from Elsevier. Figure 1b has been reproduced with permission from John Wiley & Sons, Inc.
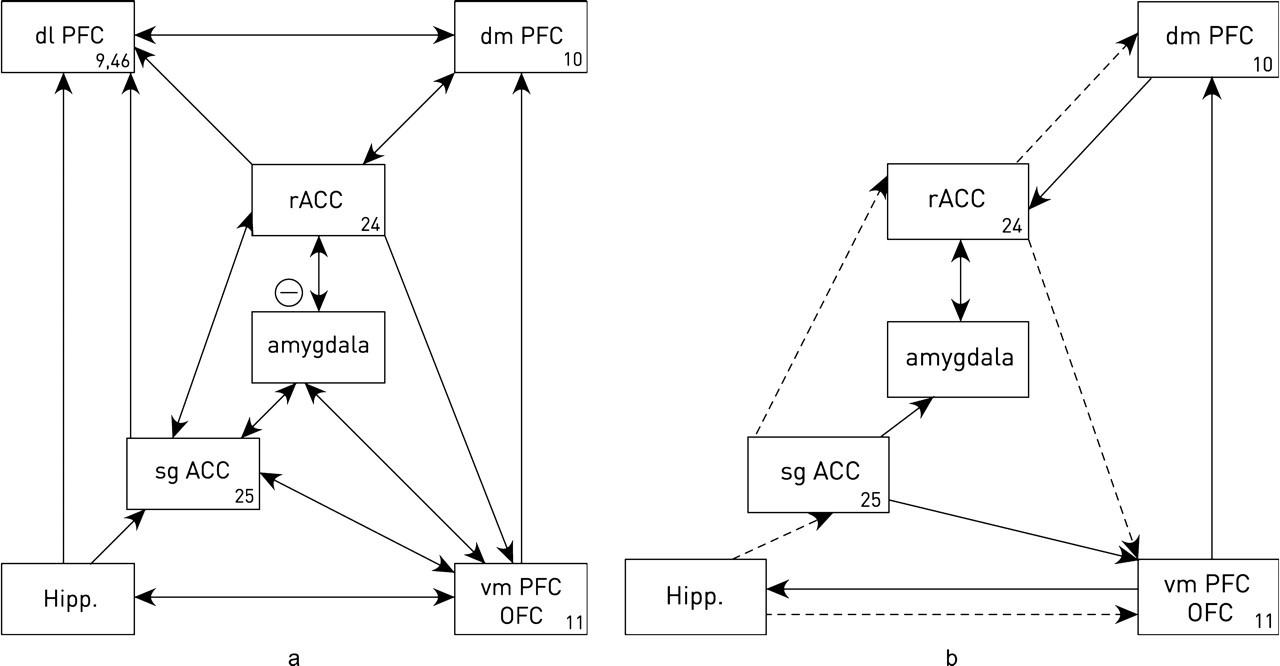
Connection of the anterior cingulate cortex, dorsal and ventral prefrontal cortex and the amygdala in humans. (a) Control connections based on anatomical determinations. (b) Connections determined for depressed patients. Definitions of the acronyms are given in the legend to Figure 1a (after Figure 2 in [61]).
Anatomical connectivity determinations indicate that these regions can be compartmentalized into dorsal ACC (comprising BA24a’, 24b’, 24c’ and 32’), with dense connections to subgenual ACC, medial prefrontal (B Al 0), orbitofrontal cortex (BA11) and amygdala; and a rostral and subgenual region (comprising areas BA24a, 24b, 24c and 25), with dense connections to the amygdala and orbitofrontal cortex [9,10]. It has been suggested that dorsal areas are concerned with cognition and exert control over ventral areas such as the subgenual ACC that are concerned with the affections (Figure 1a; described and criticized in [11]). This claim may be reflected in the fact that in healthy subjects there is a relative deactivation in dorsal ACC during emotional paradigms and a relative deactivation in ventral ACC during cognitive tasks [6].
Changes in synaptic connections of the anterior cingulate cortex and the amygdala in depression
During psychological tests with fearful stimuli, functional magnetic resonance imaging (fMRI) shows that there is tight functional coupling of a feedback pathway involving synaptic connections formed by amygdala axons projecting to the subgenual ACC and synaptic connections formed by the subgenual axons projecting to the rostral (pregenual) ACC, which has synaptic connections formed by axons projecting to the caudal amygdala which lead to its inhibition (Figure 2a; [7]). Thus when the rostral (pregenual) ACC is stimulated it causes inhibition in the amygdala [12,13].
Of great interest is the fact that synaptic connections are partially lost in depression, primarily those subserving the projection from the amygdala to subgenual ACC and less along the synaptic pathway from rostral (pregenual) ACC that causes inhibition in the amygdala (Figure 2b). There is a decrease in the rostral (pregenual) ACC synaptic connectivity to the amygdala in both unipolar and bipolar depressed patients [14,15]. Reduced synaptic projections between subgenual ACC and the amygdala have been observed in a patient with bipolar disorder post mortem [16]. Different regions in the medial prefrontal cortex and orbito-frontal cortex exert an inhibitory influence over activity in the amgdala, and fail to do so in mood disorders (compare Figures 2a and 2b; [1,15,17]).
Mayberg and her colleagues have used structural equation modeling to investigate the synaptic connectivity, determined by fMRI, of the cortical dorsal (cognitive) area with the limbic ventral (affective) area of normal subjects compared with those suffering from depression [18]. First they confirmed in normal subjects the synaptic projection from the rostral (pregenual) ACC to the sub-genual ACC, as well as that of the subgenual ACC with the orbitofrontal cortex and the dorsolateral prefrontal cortex (Figure 2a). In addition, they showed strong synaptic input from the hippocampus-amygdala to the sub-genual ACC (Figure 2). Of further interest was the finding that in depressed patients there is a lack of functional synaptic connections between the subgenual ACC and the rostral (pregenual) ACC, as well as of the hippocampal input to the subgenual ACC and all functional synapses with the dorsolateral prefrontal cortex (Figure 2b). This amounts to failure of most of the regulatory synaptic inputs to the subgenual ACC. Such observations may help explain the enhanced metabolic and fMRI activity in subgenual ACC of depressed patients [2].
Use of multivariate techniques combined with structural equation modeling, applied to resting-state positron emission tomography scans of acutely depressed patients, show differences in known anatomical and physiological pathways. An estimate has been made of the strength and direction of ‘effective synaptic connections’ between these areas [19–21]. Changes are observed between subgenual ACC, pregenual ACC, orbitofrontal cortex, hippocampus (amygdala), medial pre-frontal cortex, dorsolateral prefrontal cortex and the thalamus (Figure 2b).
The decrease in rostral (pregenual) ACC to amygdala functional synaptic connectivity in bipolar disorder, even detected in patients in different mood states, could reflect a reduction in the perigenual ACC's inhibitory control over the amygdala [22]. Disrupted structural integrity of white matter bundles that subserve rostral (pregenual) ACC to amygdala synaptic connections might contribute to these perigenual ACC to amygdala functional synaptic connectivity deficits [22].
Changes in synaptic connections of the anterior cingulate cortex and the amygdala of depressed patients following serotonergic uptake blockers
In major depressive disorder there is increased blood flow and metabolism in the subgenual ACC [15,23–25]. There is a decrease in metabolic and fMRI measured activity in the dorsolateral prefrontal cortex (BA9, 46) concomitant with the increased metabolic activity and fMRI signal in the subgenual ACC (BA25) in patients with depression, leading to the conjecture that it is failure of control from the dorsal areas to the ventral areas that leads to increased activity in the ventral areas (Figures 1 and 2; [11]). In general, SSRI medication-responding patients show normalization of dorsal hypometabolism and ventral hypermetabolism [26–28] so that effective anti-depressant action is associated with reduced subgenual ACC activity [15,23,29]. The question arises as to how SSRIs bring about such normalization of metabolism.
The mechanism by which changes in serotonin transporters bring about changes in synaptic activity in anterior cingulate cortex and amygdala
5-HT receptor distributions on neurons and their interaction with NMDA receptors
Serotonergic 1A receptors (5-HTIAR) are localized at relatively high density on the axon hillock of pyramidal neurons in the cerebral cortex, and more diffusely over dendrites, synaptic spines and the perikaryon (Figure 3; [30–32]). In contrast to this distribution the 5-HT2A/c receptors are found localized to the proximal dendritic shafts, and more diffusely on synaptic spines of pyramidal neurons, in association with the NR1 subunit of NMDA receptors, as well as on the perikaryon of inhibitory interneurons (Figure 3; [31–34]). It is likely that the different pools of serotonergic neurons in the Raphe nucleus project to the 5-HT1AR on the axon hillocks of neurons, to the 5-HT2AR on the proximal dendritic shafts and to the 5-HT2AR on inhibitory interneurones [32].
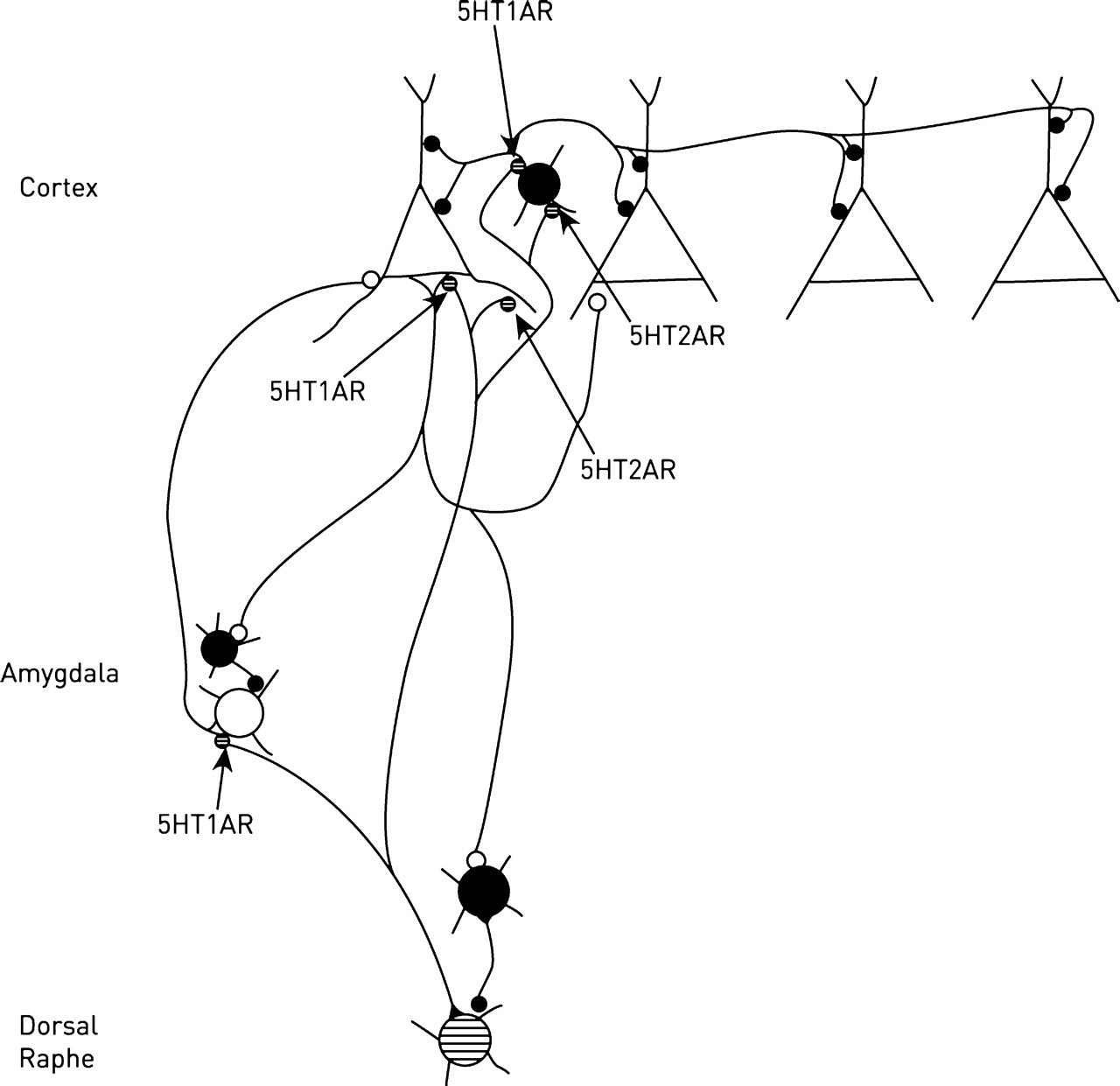
Synaptic connections of serotonin (5-HT) containing neurons in the Raphe nucleus (hatched) with neurons in the amygdala, pyramidal neurons in the cortex and inhibitory GAB A-containing neurons (filled) in both. Shown are the 5-HT synapses formed on the axon hillock of neurons, with localized 5-HT1A receptors, and on dendrites, with localized 5-HT2A receptors (after 5-HT receptor distributions given in Figure 9 of [31]; Figure 3 in [32]).
5-HTIAR activation decreases NMDA receptor-mediated currents through inhibition of protein kinase A, which leads to a decrease in microtubules involved in clustering the NR2B subunit of NMDAR [35,36]. In contrast to this, 5HT2AR activation increases NMDAR-mediated currents by increasing protein kinase C and activating ERK via the beta-arrestin-dependant pathway, thus counteracting the effects of 5HTIAR activation in decreasing NMDAR-mediated currents (see above and [35,36]). In summary, the inhibitory 5-HT1AR both decrease excitability at the axon hillock of pyramidal neurons and also indirectly reduce excitability through down-regulation of NMDA receptors. In contrast, the opposite is the case for the 5-HT2AR.
The mechanism by which changes in serotonin transporter genes alter the level of excitability in anterior cingulate cortex and amygdala
Transgenic mice knockouts for the 5-HT transporter (5-HTT) show a marked decrease in 5HTIAR in the amygdala [37–40]. It would then be expected, following consideration of the serotonergic circuit in Figure 3, that the 5HT1AR-mediated inhibition of pyramidal neurons at their axon hillock would be lost. This should lead to a net loss of inhibition of these neurons, an enhancement of NMDAR currents and therefore an increase in synaptic spines. Such an increase is observed in the amygdala, a site of normally high 5HT1AR density (Figure 4b; [41]).
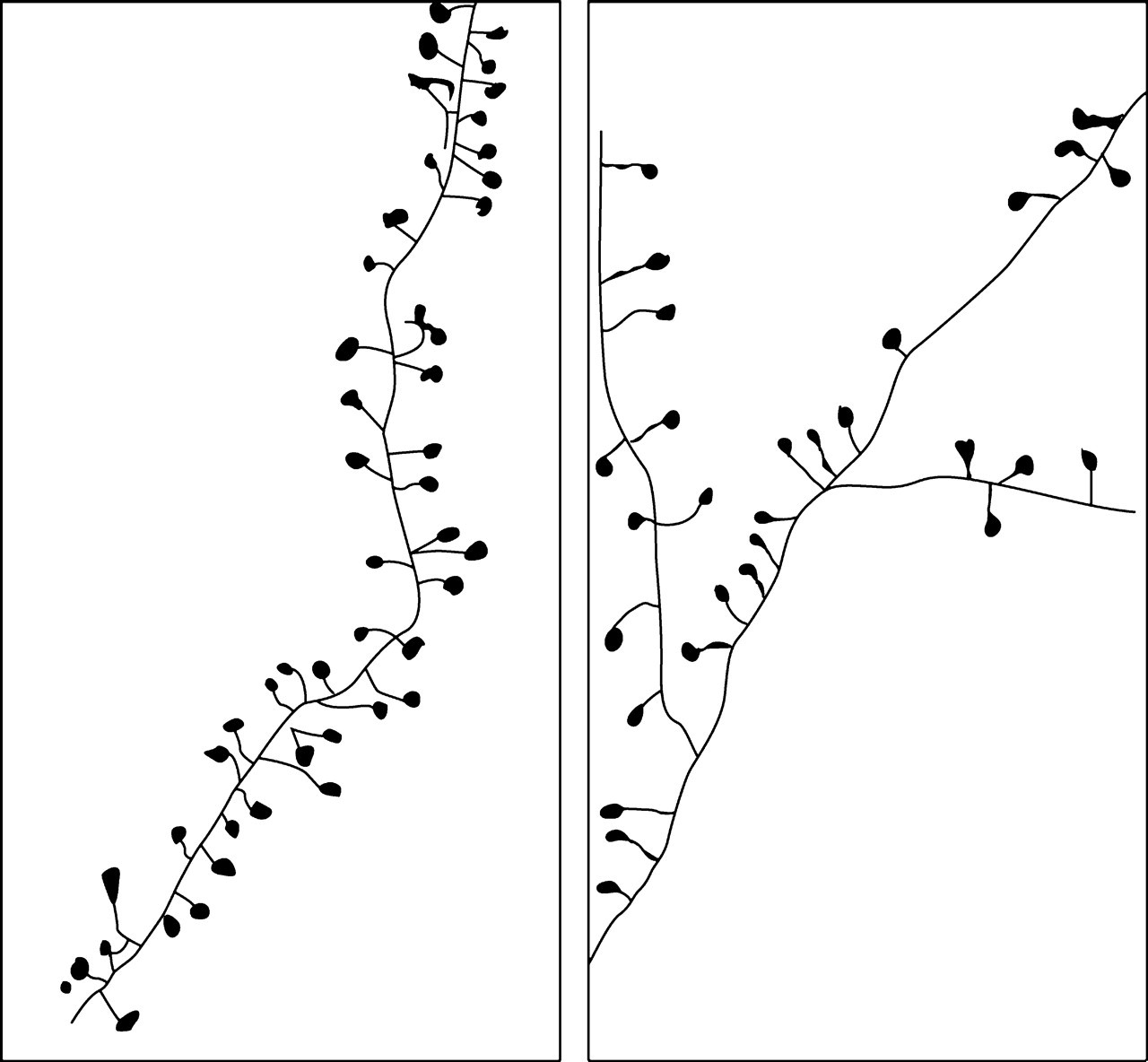
Abnormal synaptic-spine density on the dendrites of pyramidal neurons in the amygdala of 5-HTT knockout mice. The left panel shows a sketch of dendritic synaptic-spines on a fourth-order branch of a Golgi-stained pyramidal neuron in a wild-type mouse. Right panel shows the distribution of synaptic-spines on such branches in a 5-HTT knockout mouse (after Figure 7a in [41])
Increases in excitability of pyramidal neurons are also expected to occur in humans with functional polymorphisms in their 5-HTT genes (specifically 5-HTTLPR short genotype), as this results in lower 5-11:32 PMHTIAR [42]. Given that 5HTIAR are at highest density in the limbic areas of the brain, it might be that the enhanced activity and hypermetabolism in the amygdala observed in depressed patients arise in part as a consequence of down-regulation of 5-HT1AR there. It is interesting to note that such down-regulation also occurs following release of corticotrophin releasing factor during stress [43].
The subgenual ACC is the major region of gray matter volume loss in carriers of the short allele of the serotonin transporter gene 5-HTTLPR that have increased amygdala activity and elevated risks of depression [7]. Decreases in gray matter in the subgenual ACC have been observed in patients with mood disorders [23], including bipolar disorder [44] and major depressive disorder [15]. If these changes in gray matter are primarily due to a decrease in the neuropile, consisting of the processes of synapses and astrocytes, then the increase in NMDAR contingent on the loss of 5-HT1AR would be anticipated to give an increase in synapses, the opposite of what is observed. On the other hand, the use of 5-HT2AR antagonists as antidepressants leads to a down-regulation of the receptor and so an expected increase in NMDAR activity is accompanied by synaptic-spine growth [43,45]. However another observation that cannot be easily accommodated with the idea that it is ultimately the activity of the NMDAR that determines the growth and stability of synaptic spines is that in some cases the NMDAR antagonist ketamine is an effective anti-depressant, unless is it claimed that this antagonist is principally acting on NMDAR in the amygdala [46].
The mechanism by which serotonin transporter antagonists alter the level of excitability in anterior cingulate cortex and amygdala
Depletion of 5-HT in neonatal and adult mice leads to a 20% decrease in density of spines and the size of dendrites in the dorsomedial prefrontal cortex [47] as well as of both dentate granule cells and CA1 pyramidal cells in the hippocampus [48–50]. In contrast to this, blocking the 5-HTT in developing mammals with antagonists such as fluoxetene increases the ambient level of 5-HT [51] and synaptic spine density [4]. Fluoxetine also restores the density of synaptic spines on hippocampal pyramidal cells of ovariectomized female rats [52] and increases synaptophysin-labeled synapses in normal rats [53]. These are the results to be expected if a decrease in 5-HT leads to an enhancement of inhibitory over excitatory modulation of excitability of these neurons through 5HT1AR and 5HT2AR (see Figure 3), giving rise to a decrease in NMDA receptor activity and hence spine loss, with an increase in 5-HT leading to an enhancement of excitatory over inhibitory modulation and hence producing a gain of spines. SSRIs such as fluoxetine have been reported to change the sensitivity of 5HT1AR, at least in the Raphe nucleus where they can act as autoreceptors, but the matter is controversial (for a review see [54]).
The means by which blocking the 5-HTT leads to the return of dorsal hypometabolism and of ventral hypermetabolism towards normal levels might then be as follows. Increases in 5-HT following the block of 5-HTT enhances excitability through 5-HT2AR over that through inhibitory 5-HT1AR giving rise to increases in NMDA receptor activity at afferent synapses in the dorsal areas which is further enhanced by an increase in spines contingent on this increase in NMDA activity (Figure 3; [5]). As a consequence dorsal areas are excited and able to exert enhanced inhibitory control over the subgenual anterior cingulate cortex and the amygdala (see Figure 2a), giving a normalization of levels of metabolism in these structures. It might be argued that increasing 5-HT after blocking 5-HTT should also increase excitability in the ventral regions as it is claimed to do in the dorsal regions. However, there is a relative paucity of 5-HT2AR receptors on principal neurons in the amygdala compared with 5-HT1AR. Dorsal regions then produce an enhanced inhibitory control over the ventral regions (Figure 3).
Whether these mechanisms involving 5-HTT also operate in bipolar disorder is unknown, but it should be noted that the transporter might undergo a conformational change in patients with this disorder [55–57], although no changes in the density of either 5-HT2AR or 5-HT1AR have been detected in either the frontal cortex [58] or hippocampus [55].
Conclusion
During development, decreased 5-HTT activity in humans as a consequence of functional polymorphisms or complete loss of such 5-HTT activity in knockout animals, leads to a loss of inhibitory 5-HT1AR and so an increase in NMDAR activity with resulting increase in density of synaptic spines. In either case it is suggested that this contributes to ventral hyperexcitability in the subgenual anterior cingulate cortex and the amygdala as these areas are normally rich in 5-HT1 AR Changes in the level of extracellular 5-HT in adult animals also modulates the density of synaptic spines, with these increasing with an increase in 5-HT, probably as a consequence of increases in 5-HT2AR activity over that of 5-HT1AR Increasing the extracellular levels of 5-HT with SSRIs leads to an increase in excitability and of synaptic spines for afferents in the dorsal rostral anterior cingulate cortex but not in the ventral regions such as the amygdala that have few 5-HT2AR allowing dorsal regions to once more exert their inhibitory influence over ventral regions. It is suggested that it is in this way that SSRIs exert their effect in reversing dorsal hypometabolism and ventral hypermetabolism in those suffering from depression.
Footnotes
Acknowledgements