
Abstract
Carlsson has put forward the hypothesis that the positive and negative symptoms of schizophrenia are due to failure of mesolimbic and mesocortical projections consequent on hypofunction of the glutamate N-methyl-d-aspartate (NMDA) receptor. The hypothesis has been recently emphasized in this Journal that the loss of synaptic spines with NMDA receptors, which can be precipitated by stress, can explain the emergence of positive symptoms such as hallucinations and that this synapse regression involves molecules such as neuregulin and its receptor ErbB4 that have been implicated in schizophrenia. In this essay these two hypotheses are brought together in a single scheme in which emphasis is placed on the molecular pathways from neuregulin/ErbB4, to modulation of the NMDA receptors, subsequent changes in the synaptic spine's cytoskeletal apparatus and so regression of the spines. It is suggested that identification of the molecular constituents of this pathway will allow synthesis of suitable substances for removing the hypofunction of NMDA receptors and so the phenotypic consequences that flow from this hypofunction.
Carlsson has put forward the hypothesis that the basic nervous system failure in schizophrenia is hypofunction of the glutamate receptor, N-methyl-d-aspartate (NMDA) [1, 2]. This hypothesis is attractive because it integrates failure of neural networks involving the prefrontal cortex, the ventral tegmental area, the nucleus accumbens and the thalamus with the positive and negative symptoms in schizophrenia, as well as with changes in dopamine and serotonin in these structures. One of the clearest concomitants of schizophrenia, however, namely the regression of synapses [3], with resulting positive symptoms such as hallucinations [4], has not yet been integrated into the NMDA hypofunction hypothesis. In the present essay the efficacy of NMDA receptors is shown to determine the integrity of synaptic spines within the neural networks. It is suggested that it is the loss of these synaptic spines that leads to the failure of the neural networks responsible for the positive and negative symptoms in schizophrenia. The cytoskeletal system within the spines determines their growth, stability and regression. This system is under the control of a range of membrane receptors, in particular NMDA receptors. Attention is given to those receptors that determine NMDA receptor efficacy, such as the ErbB4 receptors. Consideration is then given to how failure of the ErbB4 receptors is responsible for NMDA receptor decline, with concomitant regression of synaptic spines resulting in changes in neural network function that underlie the positive and negative symptoms of schizophrenia. It is suggested that fast through-put bioassays are now required to assess the effects of substances that will restore the functioning of the NMDA receptor and therefore re-establish synaptic spines.
NMDA receptor control of synaptic spine formation and regression
The main mechanisms of spine enlargement involve increasing F-actin polymerization at the apex of the spine head (Figure 1). The two principal ways of achieving this is to either increase Arp2/3 and so F-actin proliferation and branching, or to inhibit adenosine diphosphate (ADF)/cofilin and so prevent depolymerization of F-actin while allowing proliferation to proceed. Increases in profilin will also assist spine growth through accelerating adenosine diphosphate (ADP)-profilin to adenosine 5′-triphosphate (ATP)-profilin, so making the G-actin available for attachment at the ends of F-actin at the apex of the spine. The principal means of obtaining spine shortening is to activate myosin in the spine, so increasing the retrograde movement of F-actin from the apex of the spine head.
The actin nucleation model of growth and regression of synaptic spines. (1) Receptors activate signalling pathways that lead to Rho GTPases (2). (3) These activate WASP proteins, which, in turn, in (4), lead to activation of the Arp2/3 complex and the subsequent binding of adenosine-5′-triphosphate (ATP)-actin into the complex. (5) This leads to the formation of new actin filaments through attachment of the Wiskott–Aldrich syndrome protein (WASP)-Arp2/3-ATP-actin complex to an existing filament to form a side branch. This new filament grows as a consequence of being supplied from a high concentration of profilin bound actin (11), with the result that the spine membrane is pushed out (6). (7) Capping proteins bind to the growing barbed ends of the filaments, so terminating elongation or they can be protected from capping by plasmalemma-bound Vasodilator-stimulated phosphoprotein (VAMP). Actin-polymerizing factor (ADF)-cofilin severs and depolymerizes the adenosine diphosphate (ADP)-actin in the older regions of the filaments (8, 9). Profilin then promotes dissociation of ADP and binding of ATP to the dissociated subunits (10). (11) ATP-actin then binds profilin, making them available for assembly into actin filaments. (Diagram and description after Figure 1 in [5]).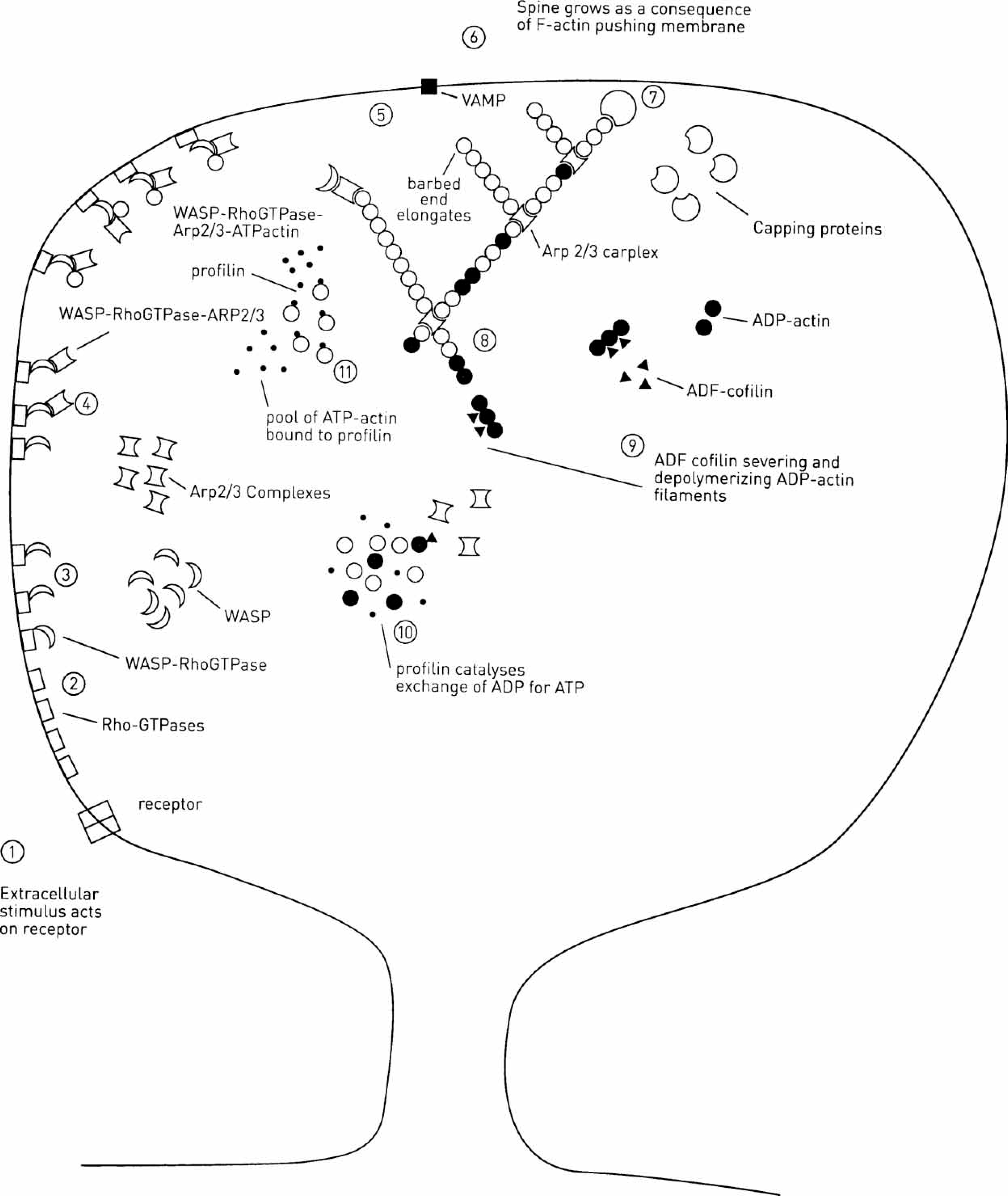
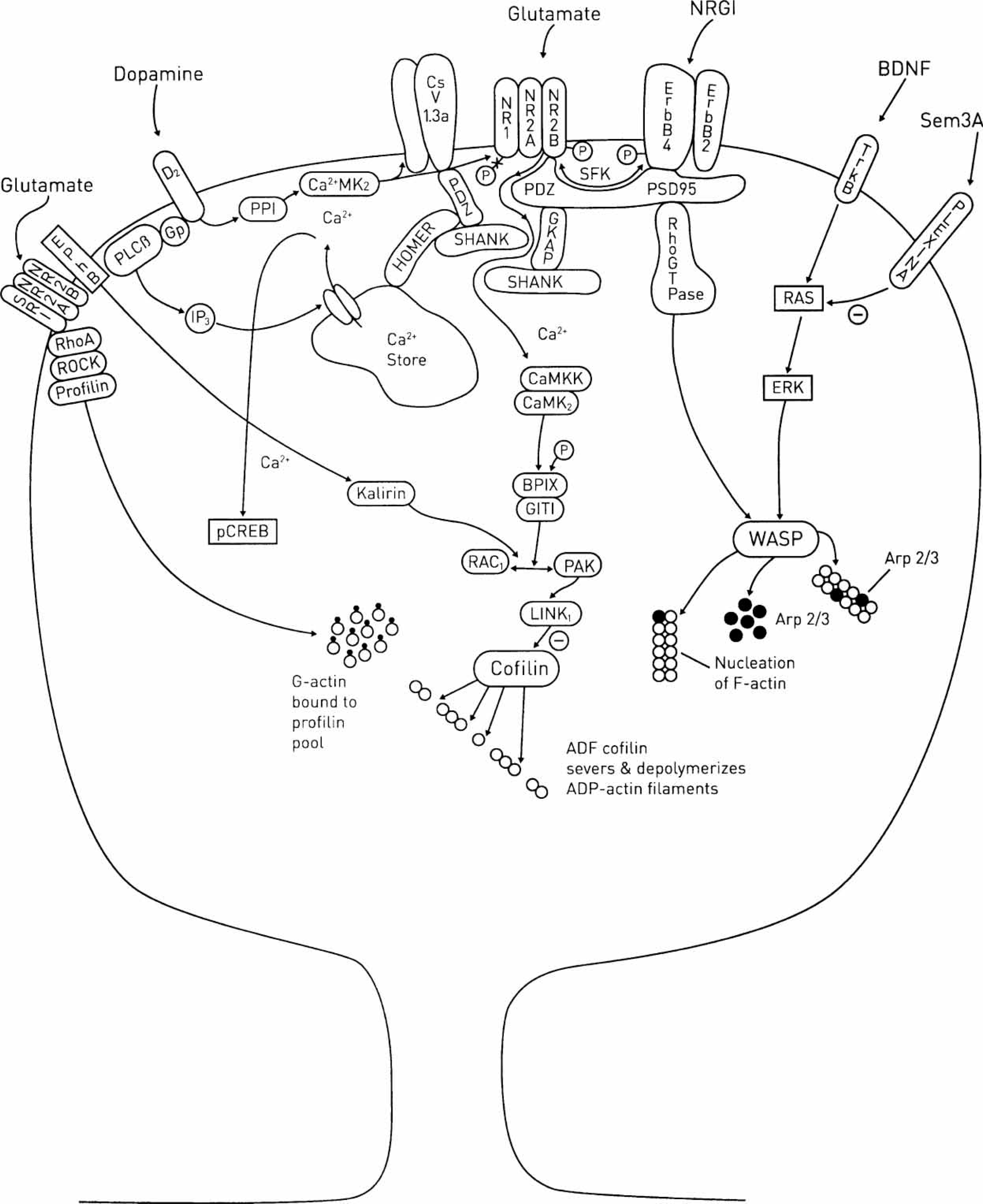
The principal receptors at the plasmalemma that activate signalling cascades that lead to changes in F-actin treadmilling and spine shape are neurotransmitter receptors as well as voltage-dependent calcium-ion channels on the one hand, and surface membrane receptors that activate intracellular signalling cascades that control activity of Rho GTPases on the other (Figure 2) [7]. The main transmitter-activated calcium-permeable channels are the NMDA receptors, which are modulated by ErbB4 receptors, and the main receptors controlling the RhoGTPases are ephrin [8], brain-derived neurotrophic factor (BDNF) [9] and plexin (Figure 3).
Upregulation of N-methyl-d-aspartate (NMDA) receptors by Src family kinase Fyn acting on the NR2B receptor subunit. Fyn, like Src, has domains UD (unique domain), SH3, SH2 and CD (catalytic domain) and also requires phosphorylation (P) in the CD loop in order to undergo a conformational change of an activation loop in order to produce a fully active Fyn kinase. Fyn is associated with psot-synaptic density (PSD)-95 through its protein PDZ3 domain and when the kinase is brought into close proximity with the NR2B subunit of NMDA it phosphorylates it at Y1472, Y1336 and Y1252 (Fig. 4), leading to increased efficacy of the NMDA receptor. (After Figure 1 in [6].) Membrane receptors and voltage-gated ionic channel control of synaptic spine actin cytoskeleton and so synapse formation and regression. The following receptor and ion channel pathways are indicated on the spines plasmalemma (read counter-clockwise around the spine). (1) Sem 3A acting on plexin A receptors to decrease RAS, ERK and so WASP nucleation of G-actin around Arp2/3. (2) BDNF acting on TrkB receptors to activate RAS, ERK and so WASP nucleation of G-actin around Arp2/3. (3) NRG-1 on ErbB4/ErbB2 receptors, anchored to the PSD-95 scaffolding protein and RhoGTPase to increase WASP as well as release of SFKs to act on the NR2B subunit of the NMDA receptor to phosphorylate it, so enhancing calcium influx through NMDA. (4) Glutamate acting on NMDA receptors to enhance calcium entry into the cytosol and via CaMKK, CaMK2, BPIX, GIT1, RAC, PAK, LIMK1 to inhibit cofilin depolymerization of ADP-actin in the F-actin filaments. (5) Voltage-dependent calcium channel, CAV1.3a, which is anchored by PDZ and SHANK in the cytosol and through HOMER releases calcium from calcium stores. (6) Dopamine acting on D2 dopamine receptors, which, through PP1, activates calcium calmodulin kinase 2 to modulate CAV1.3a and phosphorylates the NR1 subunit of NMDA. (7) Dopamine acting on D2 dopamine receptors, which through Gp and PLCβ, releases IP3 to activate calcium release from internal stores; this calcium activates pCREB. (8) Glutamate acting on NMDA receptors activates the RhoA, ROCK, profilin pathway to provide G-actin bound to profilin. (9) Ephrin binding to the EphB receptor activates Kalirin, which then acts on RAC1/PAK to excite LIMK1 and so inhibits cofilin depolymerization of F-actin. BDNF, brain derived neurotrophic factor; BPIX, guanine nucleotide exchange factor for RAC; CaMK2, calcium calmodulin-dependent kinase 2; CaMKK, calcium calmodulin-dependent protein kinase kinase; cofilin, severs and depolymerizes ADP-actin; D2, dopamine D2 receptor; EphB, ephrin receptor; ErbB2, receptors for neuregulin; ErbB4, receptors for neuregulin; ERK, extracellular signal-regulated kinases; GKAP, guanylate kinase-associated protein; Gp, G-protein; HOMER, scaffolding protein; IP3, inositol triphosphate; kalirin, Rho guanine nucleotide exchange protein (GEF); LIMK, LIM kinase, phosphorylates ADF/cofilin; NMDA, N-methyl-d-aspartate; NR1, NR2A, NR2B, subunits of the NMDA receptor; NRG-1, neuregulin 1; PAK, downstream effector of RAC (sometimes called P21-activated kinase); pCREB, phosphorylated cyclic AMP response element-binding protein; PDZ, protein domain; PLCβ, protein lipase Cβ; plexin A, receptor for Sema 3A; PP1, protein phosphatase 1; profilin, actin regulatory molecule; PSD-95, post-synaptic density 95, a scaffolding protein; RAC, Rho-GTPase; RAS, Rho-GTPase; RhoA, Rho-GTPase; Rho-GTPase, Rho-family GTPases, a subgroup of the superfamily of GTPases; ROCK, Rho-associated kinase; sema 3A, semaphorin 3A; SFK, src family kinase; SHANK, scaffolding molecule; TrkB, BDNF receptor; WASP, Wiskott–Aldrich syndrome protein that triggers actin polymerization via Arp 2/3 complex.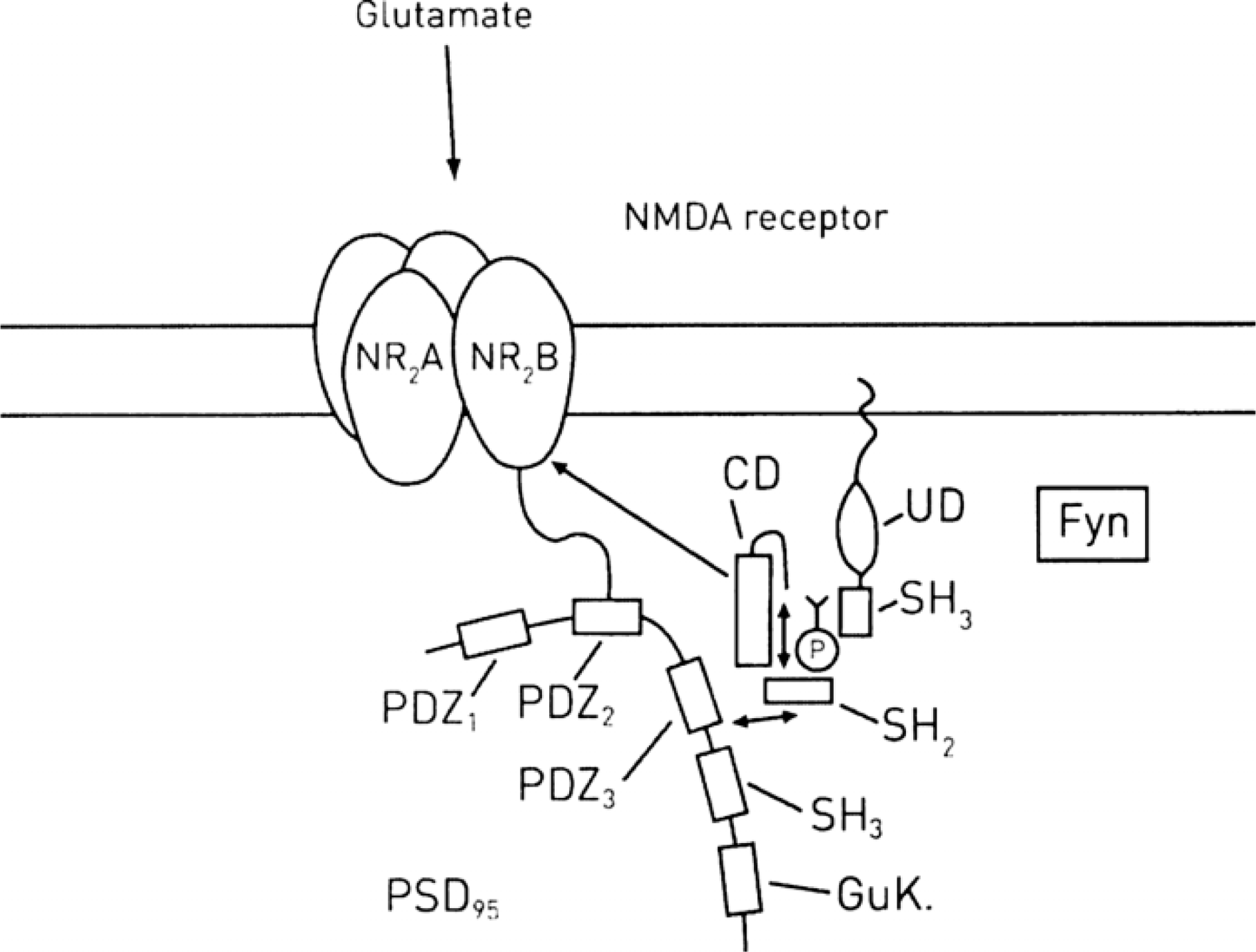
NMDA receptor activation by glutamate leads to the largest calcium influx that any receptor can generate. This calcium can in turn activate calcium-calmodulin kinase (CaMKII), which in turn phosphorylates and activates calmodulin-dependent kinase kinase (CaMKK–CaMK1). These kinases form a multimolecular complex with the guanine-nucleotide exchange factor βPIX, as shown in Figure 3, [10, 11]. Phosphorylation of this results in activation of Rac1 and Pak1. Pak1-mediated phosphorylation then activates LIM kinase (LIMK), which inactivates actin-depolymerizing protein ADP/cofilin, so promoting stabilization of F-actin and giving spine enlargement (Figure 3).
In hippocampal neurons an actin-regulated pathway is directed by Rho small GTPases, such as Rho A [12]. Rho A recruits and activates its specific Rho-associated kinase (ROCK), which in turn complexes with profilin Iia, so modifying actin stability through a RhoA/ROCK/profiling IIa pathway (Figure 3) [13]. It is known that RhoA associates with ionotropic glutamate receptors (iGluRs) and metabotropic glutamate receptors (mGluRs) at the plasma membrane of synaptic spines, with activation of the iGluRs leading to detachment of RhoA from these receptors and their recruitment to the mGluRs. This triggers local reduction of RhoA activity at the iGluRs, resulting in inactivation of RhoA-specific kinase ROCK there and so disruption of the ROCK-profilin IIa, producing rapid changes in the underlying actin cytoskeleton and remodelling of spine shape [14].
Activation of the NMDA receptors can also lead to increases in activity of LIMK, and hence inhibition of ADP/cofilin and spine enlargement, by the GTPase RhoA through RhoA-specific kinase (ROCK; (Figure 3). This pathway can also increase profilin and hence availability of G-ATP-actin for F-actin polymerization. ROCK can also activate myosin regulatory light chain (MLC) and simultaneously inhibit myosin light chain phosphatase (MLCP), so leading to myosin motor activity and increased retrograde flow of F-actin [15] with concomitant spine shortening. Collectively, glutamate stimulation of NMDA receptors can, through both CaKM11 and RhoA, lead to inhibition of ADF/cofilin (with perhaps a small enhancement from the phosphatase calcineurin) and increase profilin, which probably more than offsets in most instances the increased retrograde flow of F-actin due to enhanced myosin motor activity contingent on the increase in ROCK.
Neuregulin-ErbB4 receptor control of NMDA receptor efficacy
Neuregulin (NRG) and its ErbB4 receptor have been implicated in schizophrenia. It is then of special interest to inquire into how the ErbB4 receptor regulates the NMDA receptor leading to its hypofunction with subsequent spine loss (Figure 3).
Neuregulin transphosporylation of receptor kinase ErbB4 and recruitment of Src family kinases
The ErbB receptors are found in the plasmalemma (detergent-soluble) and membrane caveoli (detergent-insoluble), with translocation between these occurring on activation of the receptors with NRG [16]. Thus NRG promotes both the translocation of ErbB4 to the caveoli as well as its heterodimerization partner ErbB2, which is there tyrosine-phosphorylated to a level fourfold greater than ErbB2 in the non-caveoli regions of the membrane. NRG binds ErbB receptors to induce formation of ErbB4/ErbB2 heterodimers (Figure 4). This then activates intrinsic kinases, which leads to the phosphorylation of specific tyrosines located in the ErbB's cytoplasmic region. Such phosphorylated residues are docking sites for non-receptor protein kinases (Src) family signalling molecules, the recruitment of which stimulates the intracellular signalling cascades leading to upregulation of NMDA-receptor function [17] (Figure 4). The crystal structure of ErbB4 kinase has been described and shown, on activation by NRG, to adopt an asymmetric dimer confirmation [18].
ErbB-receptor tyrosine family members ErbB4 and ErbB2, after heterodimerization on activation by neuregulin, can phosphorylate N-methyl-d-aspartate (NMDA) receptor subunits NR2A and NR2B through mediation by interaction partners Src and Fyn of the ErBB-receptor tyrosine residues. These upregulate the function of NMDA receptors through Fyn acting at Y1252, Y1363 and Y1353 sites to phosphorylate NR2B and through Src acting at Y1492, Y1353 and Y1387 to phosphorylate NR2A. Src and Fyn are family kinases.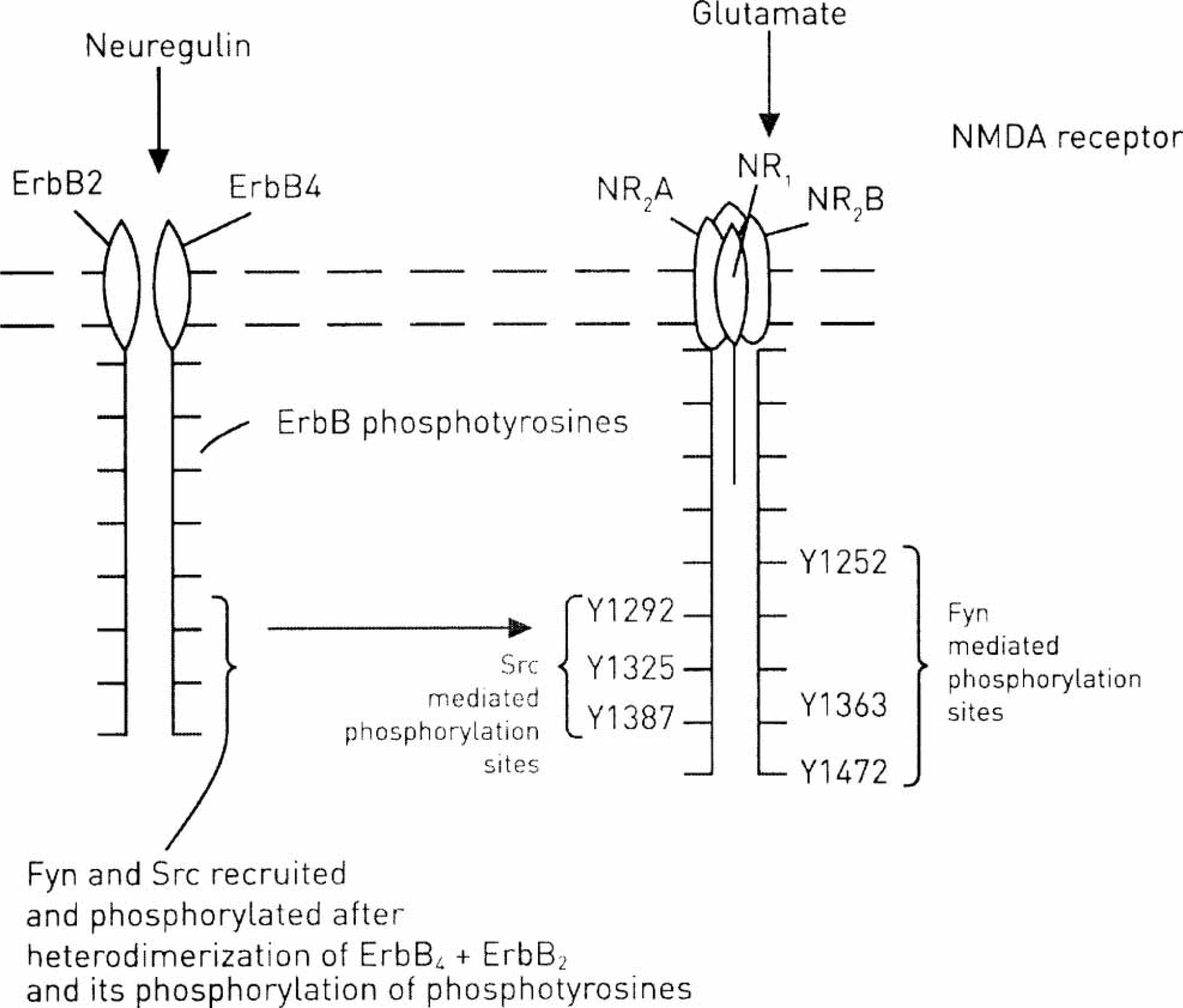
NRGs, encoded by four different genes, namely NRG-1, NRG-2, NRG-3 and NRG-4, are ligands for ErbB4 and promote its autophosphorylation [19]. Receptor dimerization occurs following formation of ligand–receptor complexes, so initiating signalling. ErbB4 most frequently forms a heterodimer with ErbB2. Autophosphorylation of ErbB receptors takes place at 18 tyrosine residues within the carboxy-terminal region [20, 21], of which approximately five are potential autophosphorylation sites (Figure 4). When phosphorylated, these conserved tyrosine residues can function as docking sites for downstream signalling molecules (Figure 4). Different downstream molecules are recruited, depending on the ErbB4 ligand, because this determines the pattern of its phosphorylation [22].
ErbB4 interacts directly with the first two PDZ domains of PSD-95 23–25, through its carboxy-terminal sequence TVV.
NMDA receptor modulation by phosphorylation through Src family kinases
Src and other Src family kinases, such as Fyn, regulate the activity of NMDA receptors, with Src and Fyn phosphorylating and thereby upregulating the function of NMDA receptors in long-term potentiation [26, 27], increasing currents through the receptor as well as intracellular calcium [28]. The Src adaptor protein NADH dehydrogenase subunit 2 (ND2) is required for Src to be anchored to the NMDA receptor (Figure 5) [6, 29].
Upregualtion of N-methyl-D-aspartate (NMDA) receptors by Src family kinase Src acting on NR2A receptor subunits. Src has domains UD (unique domain), SH3, SH2 and CD (catalytic domain). Phosphorylation (P) in the CD results in a conformational change of the activation loop, producing a fully active Src kinase. This can now act, via its tyrosine kinase activity, through an adaptor protein, to phosphorylate the NR2A subunit of the NMDA receptor in its carboxy (C)-terminal tail at least at sites Y1292, Y1325 and Y1387 (Fig. 4). Besides Src becoming a fully active kinase through its interaction with the ErbB receptor, it can also be activated by the non-receptor protein kinase cell adhesion kinase beta (CaKβ), which possesses three domains, BAND 4.1JEF (Janus kinase/ERM/FAK), CD (catalytic domain) and FAT (focal-adhesion targeting). CaKβ is activated by protein kinase C (PKC) and therefore indirectly by calcium and by G-protein coupled receptors (GPCRs). (After Figure 1 in [6]).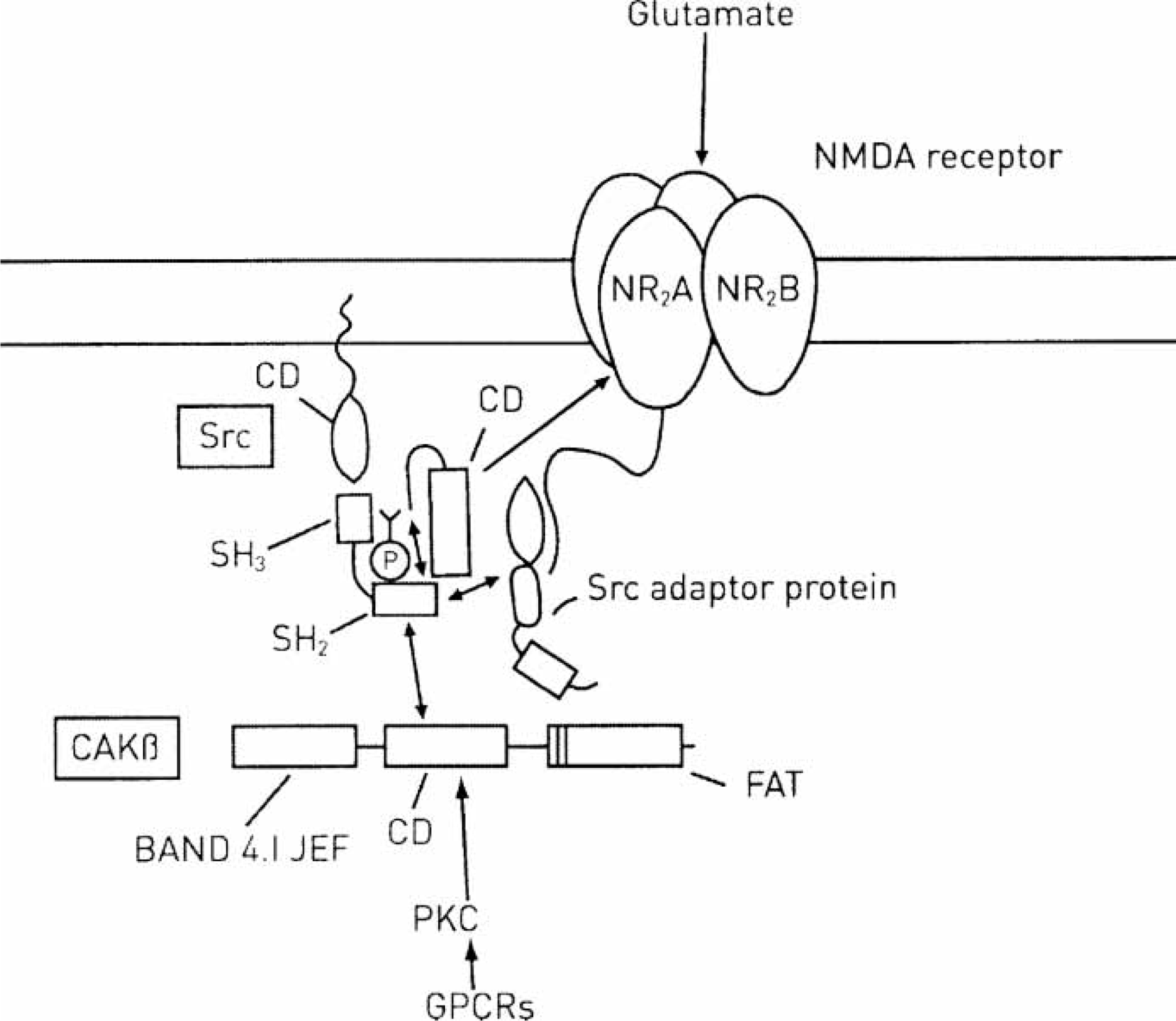
Activation of Src depends on the focal adhesion non-receptor protein kinase CaKβ (Figure 5) [30]. Activation of CaKβ is required for inducing long-term potentiation, and such activation is indirectly dependent on calcium, probably through stimulation of protein kinase C and perhaps calcium–calmodulin –dependent kinases protein (CaMKII) [31].
Activation of CaKβ leads to its autophosphorylation at Tyr-402 on the linker between the catalytic domain (CD) and the band 4.1, Janus kinase family, ERM protein family, focal adhesion kinase family domain (band 4.1-JEF domain) as well as on Tyr-579/580 in the CD (Figure 5) [6]. Phosphorylation on Tyr-402 creates an SH2 ligand through which CAKβ binds to the SH2 domain of Src and activates this kinase by relieving autoinhibition [32]. When this occurs, autophosphorylation of Y416 in the CD results in a conformational change of the activation loop, producing a fully active kinase. The SH2 domain binds to peptide motifs that contain phosphorylated tyrosine. The CD contains phosphorylated tyrosine. The CD contains the tyrosine kinase activity that phosphorylates the NR2A subunit of the NMDA receptor in its carboxy (C)-terminal tail at least at sites Y1292, Y1325 and Y1387 (Figure 5) [32].
Fyn is associated with PSD-95 that, like the adaptor protein for Src, brings the kinase into close proximity with the NMDA receptors (Figure 2) [6, 33]. This leads to phosphorylation of the NR2B subunit of N-methyl-
The kinetics of phosphorylation by the SH3 domain of Fyn have been described [37], as has the kinetics of phosphorylation by Src of the G protein-coupled receptor kinase GRK2 [38] as well as of dynamin [39].
It has been noted here that Src facilitates NMDA receptor function by increasing tyrosine phosphorylation of the NR2A subunit [40], whereas Fyn does so by phosphorylating the NR2B subunit (Figure 2) [32]. The Src-dependent regulation of NMDA receptor activity is regulated by an interaction between C-terminal Src kinase (Csk) and the NMDA receptor, probably through direct association of Csk with the Src-phosphorylated NR2 subunits [41]. In this way Csk maintains constant the excitatory synaptic transmission at impulse rates that do not evoke long-term potentiation by inhibiting calcium increases of Src kinase-dependent increases.
Kinetics of Erb (EGFR) transphosporylation and of subsequent Src family kinases
A detailed kinetic model of epidermal growth factor receptor (EGFR)/ErbB signalling pathways, describing the activation, dimerization, and tyrosine phosphorylation of EGFR/ErbB, followed by the binding and activation/phosphorylation of an adaptor/target protein such as Src, has been developed by Kholodenko et al.[42, 43]. Two kinetic modules may be considered here, one concerning the dimerization of EGFR/ErbB on activation with its subsequent phosphorylation, and the other module the binding and activation/phosphorylation of the target protein, in this case Src. Considering first the second module concerning the target protein: this is bound and phosphorylated by EGFR/ErbB through activated EGFR/ErbB kinase and a coupled cycle of phosphorylated and dephosphorylation of the target protein by specific tyrosine phosphatase. Other considerations of these kinetic schemes are given in [42], 44–46.
Kinetics of Src family kinases (Src, Fyn) interaction with NR2B subunit of the NMDA receptor
There have been no quantitative models of Src and Fyn interaction and subsequent tyrosine phosphorylation of the NMDA receptors, although such sites are identified. Kinetic parameter constants have been determined for calcium/calmodulin-dependent protein kinase (CaMKII) binding to specific residues on NR2B subunit [47]. An appropriate kinetic model, however, has probably been provided by [48]. This gives the following kinetic modules: dimerization transphosphorylation of ErbB to give the activated receptor (Figure 5); subsequent phosphorylation (dephosphorylation) of the target/adaptor protein given by Src (or Fyn or other Src family kinase (SFK)); then binding of Shc (or Fyn or other SFK) to the NR2B subunit of the NMDA receptor (Figures 2, 5).
Quantitative measures of ErbB4 control of NMDA receptor activity
Early studies examined the acute effects of NRG-1 application on NMDA receptors and reached the conclusions that NRG-1 inhibits synaptic plasticity 49–51 and reduces NMDA receptor activity and synaptic transmission. These studies used tonic application of a functional domain, such as the epidermal growth factor (EGF) domain, of particular isoforms of recombinant NRG-1 [49], 51–53, but the function of endogenous NRG-1 at synapses may be different. The location or duration or physiological concentration of these functional domains is not known [54], nor their action on different ErbB receptors. Indeed, NRG-1-deficient mice show reduction in NMDA receptor activity, compared with wild type, rather than the opposite effects that would be expected from the acute studies [55]. Recently, Li et al. have shown that preventing NRG-1/ErbB4 signalling by using either ErbB4 knockout mice or ErbB4 RNAi leads to loss of NMDA receptor currents in hippocampal neurons and this is accompanied by a decrease in spine size and number [56].
If Fyn is the SFK under consideration, then the Fyn kinase activity as a consequence of the NRG-1 action on ErbB4 increases to a peak in approximately 7 min, well after the ErbB4 kinase activity reaches a maximum at 1 min. The dose–response curve for NRG-1 increasing kinase activity of ErbB and of Fyn indicates that they have similar ED50s but quite different activation levels [57].
Changes in the ErbB4-NMDA receptor pathway in schizophrenia
ErbB4 hyperphosphorylation in schizophrenia
In schizophrenia there is a large increase in activation of ErbB4 in the prefrontal cortex by NRG-1, with such hyperactivation traced to a substantial increase in ErbB4–PSD-95 interaction [58]. Although those authors reported that, in post-mortem brain slices, NRG-1 stimulation suppressed NMDA receptor activation in human prefrontal cortex, such experimental observations are open to criticisms concerning interpretation of the effects of exogenous application of NRG, as recently presented by Li et al.[56].
PSD-95 is a multiprotein complex containing various protein kinases with the PD21, PD22, PD23, SH3 and GuK domains. The PSD-95 binds SFKs such as Fyn, as well as ERbB4/ ErbB2 and the NR2B subunit of the NMDA receptor, so enhancing their interaction. It is claimed that in subjects with schizophrenia ErbB in this complex is hyperphosphorylated and this leads to hypophosphorylation of the NMDA receptor by the SFKs (Figure 6) [58, 59]. The question arises as to how increases in ErbB hyperphosphorylation leads to decreases in phosphorylated SFKs, which in turn lead to decreases in phosphorylation of the NR2B subunit of the NMDA receptor (Figure 6). It will be noted in this scheme that it is not NRG-1 that is abnormal, but the state of phosphorylation of the ErbB4 receptor because, it is suggested, of its enhanced interaction with the PSD-95 complex, although it might be claimed that enhanced or reduced delivery of normal NRG-1 leads to a long-standing change in ErbB4 –PSD-95 interaction.
The ErbB hyperphosphorylation hypothesis for schizophrenia. This suggests that ErbB phosphotyrosines are hyperphosphorylated following the action of neuregulin leading to failure of phosphorylation of sites on the NR2 subunits of the N-methyl-d-aspartate (NMDA) receptor. The sites shown as no longer phosphorylated here (×), are purely speculative. Compare this Figure with Figure 4.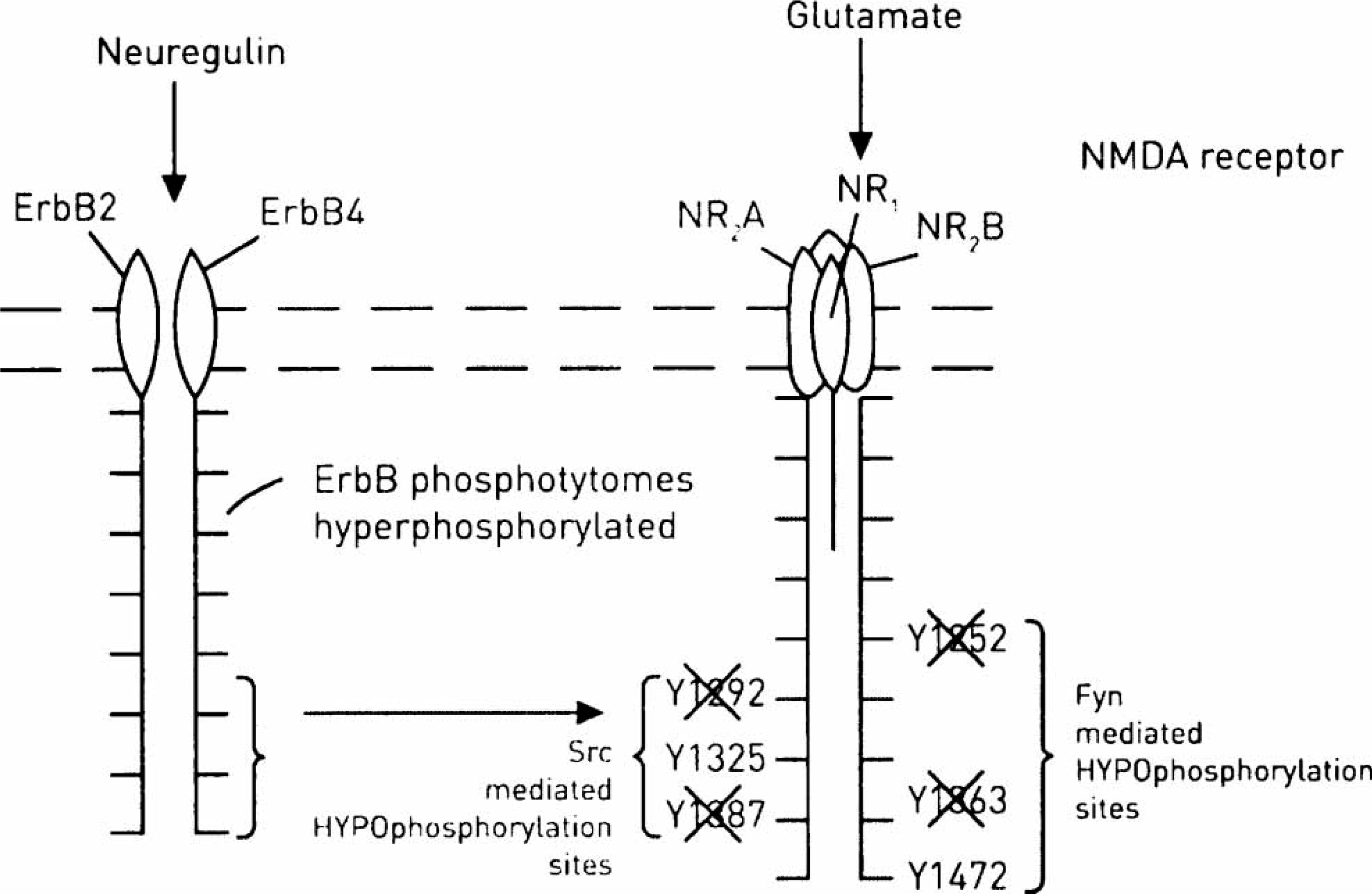
Changes in splice variants of ErbB4 in schizophrenia
Stimulation of ErbB4 by NRG-1 promotes receptor ectodomain cleavage by the metalloprotease tumor necrosis factor –α-converting enzyme (TACE) [60]. The intracellular fragment containing the transmembrane and cytoplasmic domains may be subsequently cleaved by presenilin/γ-secretase, so releasing its intracellular domain (E41CD) into the cytosol from where it can be translocated into the nucleus [61].
Alternative splicing of the ErbB4 gene generates two isoforms that differ in their extracellular juxtamembrane (JM) regions as well as their sensitivity to proteolytic cleavage, thus having different capacities to signal through regulated intramembrane proteolysis. ErbB4 JMa is cleaved by TACE and presenilin, whereas ErbB4 JMb isoform is not [62, 63]. Interestingly, mRNA for ErbB4 JMa/CYTI is elevated in patients with schizophrenia [64], suggesting that dysregulation of splice-variant specific expression of ErbB4 is the basis of the association of this gene with schizophrenia. Indeed, consideration of the cleavage products of the ErbB4 protein (at full length, 180 kDa), namely at 21, 55 and 60 kDa in the prefrontal cortex of the brains of patients with schizophrenia, shows that the ratios 21 kDa/180 kDa and 55 kDa/180 kDa are significantly reduced, with the full-length protein at 180 kDa increased by 30% [65]. Whether these ratios are reduced simply because of the elevation of the full-length 180 kDa protein is not clear.
Polymorphisms in Fyn and Src in schizophrenia.
In Fyn knockout mice, there are very few spines on the dendrites of cortical neurons [66]. There is a relationship between particular polymorphisms of the Fyn gene and performance on neuropsychological tests of prefrontal cortex activity, namely the Wisconsin Card Sorting Test, in patients with schizophrenia [67] (although in another study it was claimed that there is no evidence for involvement of genomic Fyn gene mutations in schizophrenia [68]). It is interesting in this context that Src inhibitions protect the injury to retrospinal cortical neurons by NMDA antagonists, such as ketamine and MK-801, and this effect of Src inhibitors is mimicked by atypical antipsychotics such as clozapine [69].
Clozapine modulation of ErbB4 and NMDA receptors
The atypical neuroleptic clozapine increases NMDA receptor-mediated excitatory currents in most prefrontal cortical neurons, facilitating long-term potentiation, whereas the typical neuroleptic, haloperidol, does not [70]. This potentiation is selective for the NR2B subtype-containing NMDA receptors in the nucleus accumbens and is blocked by selective SFK inhibitors [71]. Interestingly, clozapine increases the protein levels of ErbB-4 receptors in the hippocampus [72].
Conclusion
The NMDA hypofunction hypothesis for schizophrenia suggests that agents should be sought that enhance the activity of this receptor. NMDA agonists are not likely to be useful because of their leading, among other things, to excitotoxic cell death of the neurons consequent on excess calcium entry. Approaches using the allosteric site on the NMDA receptor complex that bind glycine, a necessary condition for operation of the receptor, are promising. Indeed, chronic treatment of rodents with glycine does not produce excitotoxicity [73]. Multiple small trials with this approach have in general produced positive cognitive enhancement [8, 74]. Another possible approach is to prevent reuptake of glycine, so enhancing its extracellular concentration and NMDA receptor activity [75]. Of particular interest is the NRG-ErbB4 pathway in NMDA regulation. Rather than directly attempting to upregulate the NMDA receptor, it is suggested that compounds should be sought that prevent hyperphosphorylation of ErbB4 or the subsequent hypophosphorylation of NMDA receptors through changes in the recruitment of the SFKs. This paper and previous ones emphasize that hallucinations, delusions and cognitive decline can all be related to loss of synaptic spines and hence functioning synapses in particular parts of the brain [3, 4, 76]. NMDA hypofunction, with subsequent loss of spines, emphasizes the need for fast-throughput assays to identify compounds that will restore the functioning of NMDA receptors and hence synapses with the resultant re-establishment of neural circuitry, holding out the possibility of some rehabilitation of those suffering from schizophrenia.