
Abstract
During adolescence there is a loss of approximately 30% of the synapses formed in the cortex during childhood. Comprehensive studies of the visual cortex show that this loss of synapses does not occur as a consequence of less appropriate projections being eliminated in favour of more appropriate ones. Rather it seems that synapses with low efficacy for transmission are eliminated in favour of those with higher efficacy. The loss of low-efficacy synapses is known, on theoretical grounds, to enhance the function of neural networks, but large synapse losses lead to failure of network function. In the dorsolateral prefrontal cortex (DLPC) of those suffering from schizophrenia the number of synapses is relatively very low, approximately 60% lower than that observed in normal childhood. It is not known if this is due to an additional loss over that during normal adolescence or whether it results from a failure to form a normal complement of synapses during childhood. The first study of synapse loss in the mammalian nervous system was made on the neuromuscular junction at Sydney University in 1974. Since then this junction has provided principal insights into the molecular basis of synapse formation and regression, so providing a paradigm for investigations of these phenomena in the DLPC. For example the molecules muscle-specific receptor tyrosine kinase (MuSK), agrin and neuregulin have been identified and their critical roles in the formation and maintenance of synapses elucidated. Loss of function of MuSK or agrin leads to failure of neuromuscular synapse formation as well as a loss of approximately 30% of excitatory synapses in the cortex. Similar synapse loss occurs on failure of neuregulin in vitro and of neuroligin in vivo. It is suggested that three important questions need to be answered: first, over what development period are the synapse numbers in DLPC of subjects with schizophrenia lower than normal; second, what are the relative importance of MuSK/agrin, neuregulin/ErB and neurexin/neuroligin in synapse formation and regression in the DLPC; and third, to what extent have these molecules gone awry in schizophrenia.
The first description of single axon terminal growth cones forming synapses in a mammal during development was given in 1974 [1]. The preparation chosen was the innervation of the rat diaphragm muscle at 14 days in utero because this is the most accessible of structures for investigation of axon growth cone movement in situ. Growth cones were observed to form synapses at a single site on the growing myotubes, and this site was subsequently innervated by other growth cones, until by birth each muscle cell received a multiple innervation [2] of up to four axon terminals [1] (for a review see [3]). All of these terminals but one then reverted to a growth-cone type bulb and retracted from the synaptic site in the first 2 weeks after birth [1]. These observations, and many others on the formation and regression of synapses in both developing and reinnervated muscles after lesion of their motor-axons, were explained in quantitative terms by a relatively simple theory called the dual constraint hypothesis. This integrated into a single conceptual framework the function of molecules subsequently discovered as important in neuromuscular synapse formation and regression [4], [5].
In 1979 Huttenlocher reported changes in density of synapses in supragranular levels (level 3) of human dorsolateral prefrontal cortex (DLPC) during development, using the phosphotungstic method of labelling [6]. He showed that synapse density reaches a peak before adolescence, and then decreases by at least 30% during the adolescent years, remaining constant thereafter until later life (>70 years). This has been recently confirmed using the synaptic vesicle-associated protein synaptophysin as a marker of synapses [7]. Most interestingly, Glantz and Lewis have shown a greatly increased loss of synapses in the DLPC of subjects with schizophrenia over the approximately 30% loss that normally occurs during adolescence, with no such enhanced loss occurring in the visual cortex of such subjects [8]. The purpose of this paper is to show how our substantial knowledge of the phenomenon of synapse formation and regression of neuromuscular synapses, together with the dual constraint theory that integrates this knowledge, can be used to illuminate the mechanisms of synapse formation and regression in the cortex, identify the putative molecules involved, and point out what might have gone awry with these mechanisms and molecules in schizophrenia.
What does the regression of neuromuscular synapses achieve?
The regression of synapses that accompanies the loss of synapses at the neuromuscular junction is accompanied by refinement of the topographical map between the spatial position of motoneurons in the muscles motor pool in the spinal cord and the spatial position of muscle cells within the muscle (e.g. figure 12 in [9] and figure 6 in [10]). But this is essentially a matter of refinement of an existing topographical projection pattern, and so cannot claim to be establishing the projection pattern itself.
Muscles consist of varieties of different proportions of muscle cell types (types I–III), characterized by their different myosin isoenzyme content or myosin adenosine triphosphate, each innervated by different classes of motoneurons in mature muscles. These different muscle cell types are established very early during development [11], independently of the nervous system [12]. It seems likely that motoneurons of a particular type consolidate their terminals on muscle cells of the appropriate type very early during development, before excess synapses are eliminated from most muscle cells [13]. Regression of synapses on muscle is not then associated with establishing appropriate matching of motoneuron and muscle cell types.
The regression of synapses on muscle cells is dependent on impulse traffic. Blocking such traffic with tetrodotoxin arrests synapse regression [14], as does blocking synaptic transmission [15]. In contrast, increasing the rate of impulse traffic increases the rate of regression of synapses [16]. Of particular interest is that stimulation of one of two nerves that innervate a muscle during the period of synapse regression greatly increases the size of the motor units of the stimulated nerve, with a concomitant decrease in the size of the motor units of the other nerve [17].
The principal function of excess synapse formation during early development is most likely that it operates as a safety factor to ensure that the bulk of the muscle cells in a muscle receive an innervation from a terminal and that after the regression of synapses is complete this terminal possesses a high efficacy for transmission. The safety factor for synaptic transmission at the mature neuromuscular junction, namely the ratio of the threshold for initiating an action potential in the muscle to the size of the synaptic potential, varies between approximately 2 and 5, depending on the muscle cell type [18]. The safety factor for muscle innervation during development is approximately 3–4, that is, an excess exists of three or four terminals at a single junction [11]. According to dual constraint theory, even with this innervation safety factor, a few muscle cells (approx. 6%) will not be innervated and therefore degenerate if all terminals are initially randomly distributed among all the developing muscle cells (figure 7 in [5]). According to this theory the early formation of excess synapses achieves two objectives. First it ensures that the target cell receives synapses from neurons in the network in which it participates. Second it guarantees that these synapses have the highest impulse firing rate and are so destined to possess the highest efficacy for transmission; such synapses are favoured for stabilization over the cohort of other appropriately connected synapses that are destined to be eliminated. A fully wired-up network with the most efficient synapses is guaranteed by this process.
Formation and regression of synapses during development of connections in the primary visual cortex
The question then naturally arises as to whether the regression of synapses in the cortex is associated with the refinement of the positions of projecting axons or is rather, as suggested for neuromuscular synapses, a process for ensuring a safety factor for the innervation of neurons by high-efficacy terminals. The most carefully and fully investigated area of cortex examined in this context is the primary visual cortex, which receives projections from the lateral geniculate nucleus. Area 17 of visual cortex in adults receives a definite pattern of ocular dominance projections so that, as viewed from above, this cortex possesses narrow bands of transition between right and left eye-dominated cortex [19], [20]. These patterns of ocular dominance were not thought to be present during early development, the two ocular projections at first overlapping, with distinct columns emerging only after birth and following visual experience [19]. It was then natural to attribute the process of establishing ocular dominance to the paring away of excess synapses in inappropriate areas of the visual cortex. It is now known, however, that ocular dominance columns are established well before the regression of synapses occurs (in the mouse at postnatal days (P)12–P20 [21] with regression beginning at approximately P28 [22]; in the ferret at P12–P18 [23], [24] with regression beginning at approximately P49 [25]; in the cat at P19–P39 [26] with regression beginning at approximately P70 [27]; and in the monkey 3 weeks before birth [28] with regression beginning at approximately P1500 [29]).
The critical period for ocular dominance plasticity, whereby closing one eye leads to the other eye spreading its synaptic influence into the territory of area 17, normally the prerogative of the closed eye, begins later than the period of establishment of the ocular dominance columns and ends before synapse regression begins (namely approx. P20–P32 in the mouse [21]; approx. P35–P49 in the ferret [23], [25]; approx. P28–P42 in the cat [27] and approx. P14–P42 in the monkey [30]). That is, the critical period is over before the period of synapse regression. It follows that neither the formation of ocular dominance columns nor the critical period are served in an important way by the process of synapse regression. Orientation columns also emerge before the period of synapse regression (at P30 in the cat and at P35 in the ferret [31]), so these are not related to the phenomenon of synapse regression either.
Formation and regression of synapses during development of connections in the DLPC
Glutamatergic synapses, the dominant excitatory synapses in the brain, are made on dendritic spines, which in general receive a single nerve terminal bouton. These spines are found on the dendrites of excitatory neurons throughout all layers of the cerebral cortex, from the relatively superficial supragranular layers that receive most of the cortico-cortical synapses (layers 2,3), the granular layer (layer 4), which receives most of the thalamic input, as well as the subgranular layers (layers 5,6) that project to subcortical nuclei. In mice there is approximately a 20% loss of synaptic spines that initially form on dendrites in the DLPC. This occurs from an initial peak at P28 to decline over a 4 week period between 1 and 2 months after birth, that is, in the adolescent period for these animals [22]. It is observed in the supragranular layers and is not confined to the DLPC but occurs throughout the cortex [22]. The total number of spines (all with synaptic terminals) is stabilized after 4 months, although there is a continual turnover of spines of between 3% and 5% (for a review see [32]).
Nearly 30 years ago Huttenlocher reported changes in density of synapses in supragranular levels (L3) of human DLPC during development and showed that the number of synapses reaches a peak at between 1 and 5 years of age, and then decreases during adolescence until approximately 15 years, remaining constant thereafter until later life (>70 years; Figure 1) [6]. This account of synapse loss in DLPC has been reinvestigated using the synaptic vesicle-associated protein synaptophysin as a marker of synapses [7]. The greatest decline in synapse numbers occurred between 6–10 years and 11–15 years, remaining stable thereafter, so that the peak numbers occur later in this study, but although substantial loss is still noted during the adolescent years.
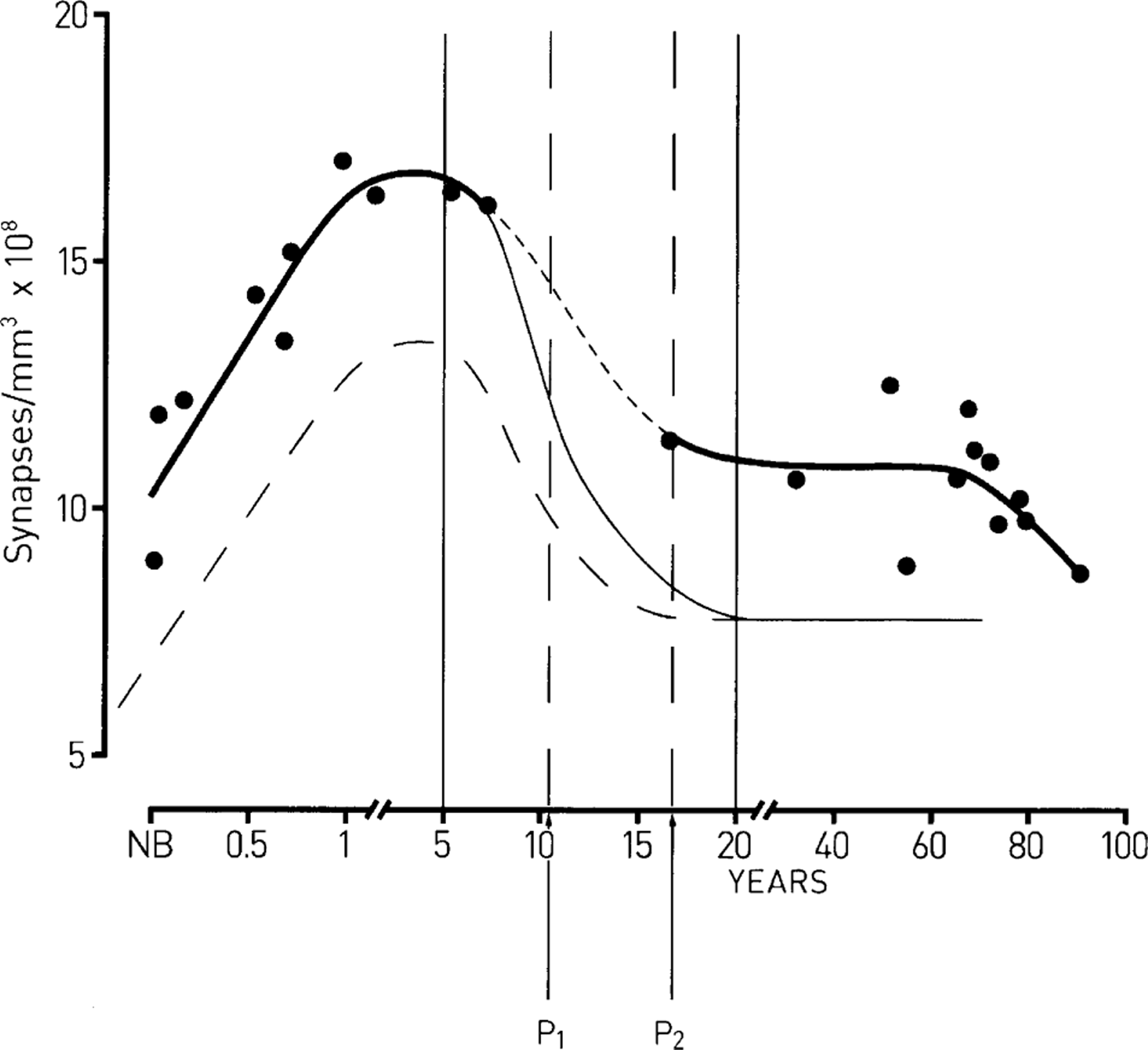
The formation and regression of synapses in the human dorsolateral prefrontal cortex during development. The ordinate gives the synapse density and the abscissa the time in years after birth. (––) Best fit to the observations (•) of Huttenlocher [6], according to which synapse formation occurs during childhood, between birth and approximately 5 years, with a subsequent regression of approximately 30% of these synapses, mostly in the adolescent period. (––) Supposition that an excess regression of synapses occurs in schizophrenia, mostly in the adolescent period, such that a net 60% of synapses are lost; this leaves 30% less synapses than normal in maturity. (—) Supposition that only 60% of the normal complement of synapses form during childhood in schizophrenia, with this followed by a normal rate of synapse regression such that the number of synapses at the end of the adolescent period is 30% less than normal. The first continuous vertical line indicates the time of maximum synapses in the cortex and the second continuous vertical line the time when synapses have stabilized in maturity. The vertical broken lines (P1 and P2) indicate the period over which there is maximum synapse regression in the adolescent years.
What does the regression of synapses in the cortex achieve?
All of the observations on primary visual cortex suggest that excess numbers of synapses in the cortex is, like that at the neuromuscular junction, concerned with ensuring elimination of an excess of appropriately connecting synapses to target cells in such a way as to optimize the efficacy of the stabilized synapses remaining when regression is complete.
The well-defined critical period for ocular dominance columns, during which time synaptic spines display a dynamic in which synapse formation occurs involving active projections from the non-deprived eye and is accompanied by synapse regression from the inactive projections of the deprived eye, is by definition an activity-driven process (for a review see [33]). This plasticity of the spines displayed in synapse formation and regression is clearly restricted in time for terminals that subserve ocular dominance, with such plasticity much less evident for the spines that subserve orientation selectivity [34]. The existence of critical periods for such spine plasticity does not impact on the suggestion that the major regression of synapses that occurs in the cortex during adolescence is due to the elimination of an excess of appropriately connecting synapses under the influence of impulse activity. This is not a process of synapse formation and regression, as occurs in the critical period indicative of plasticity, but only one of synapse regression and therefore of net spine loss.
Formation and regression of synapses in the DLPC of subjects with schizophrenia
There are fewer synaptic spines in the DLPC of mature subjects with schizophrenia compared with normal subjects [8], [35]. No such changes in spine numbers are detected in the visual cortex of patients versus controls [8]. It is not known if there is an additional loss of spines during the adolescent period (between 6–10 years and 16–20 years; Figure 1) of schizophrenia subjects over the normal 30% spine loss or whether there is a loss of spines in the DLPC of such subjects after a stable spine number is reached at 16–20 years in normal subjects. There is then no developmental series at present available describing changes in spine numbers of schizophrenia subjects. Evidence that this excess loss of spines in the DLPC in subjects with schizophrenia does occur during adolescence comes from recent magnetic resonance imaging (MRI) studies. These show that changes in grey matter of DLPC of normal adolescents follow the same time course as the loss of synapses in the neuropile [36]. Because there is no substantial loss of neurons during adolescence the grey matter loss is likely due to synapse loss, although this is by no means certain because of the possibility of astrocyte loss. Recent observations with MRI showing a progressive excess of grey matter loss in DLPC of adolescent subjects who are in transition to schizophrenia may be interpreted as showing an excess loss of synapses in these subjects [37]. It is, however, still possible that the decreased number of spines in schizophrenia subjects arises because of a failure of synapse formation during childhood (Figure 1).
Layer 3 pyramidal neurons in DLPC receive cortico-cortical synapses, such as those from parietal cortex as well as hippocampus, thalamus and synapses from lintrinsic collaterals [38]. There is evidence that these thalamic projections are affected in schizophrenia [39], as are those from the hippocampus [40], [41]. Taken together, these observations suggest the hypothesis that there is an excessive regression of synapses in the DLPC of adolescent subjects suffering from schizophrenia, especially on layer 3 pyramidal neurons receiving cortico-cortical and thalamic–cortical projections (Figure 1). The mechanisms underpinning this excess loss of synapses is of great interest because decreasing connectivity below a certain value in the neural networks of the brain is expected, on theoretical grounds, to lead to rapid loss of function.
Dual constraints on the formation and regression of synapses during development of neuromuscular connections
A theory that accounts in quantitative terms for synapse formation and regression is as follows [4]. Suppose that the collaterals of an axon, constituting for example part of the motor-axon projection to a muscle, have an equal chance to come into contact with a muscle cell. Each of these muscle cells (m) has synapse-formation molecules (Rm) that it can make available and each motoneuron, n, has synapse formation molecules (Tn) that are transported to its axon collateral terminals where they are incorporated into the terminal membrane (Figure 2a). The reaction between the Tn of a terminal on a muscle ‘m’, namely (Tnm) and Rm, gives rise to the formation of new synaptic terminal, which is made in amounts directly proportional to the product (Snm) of the reaction (Figure 2a). Several collaterals, each from a different motoneuron, may initially form terminals at the same site on a muscle cell (Figure 2b,c).
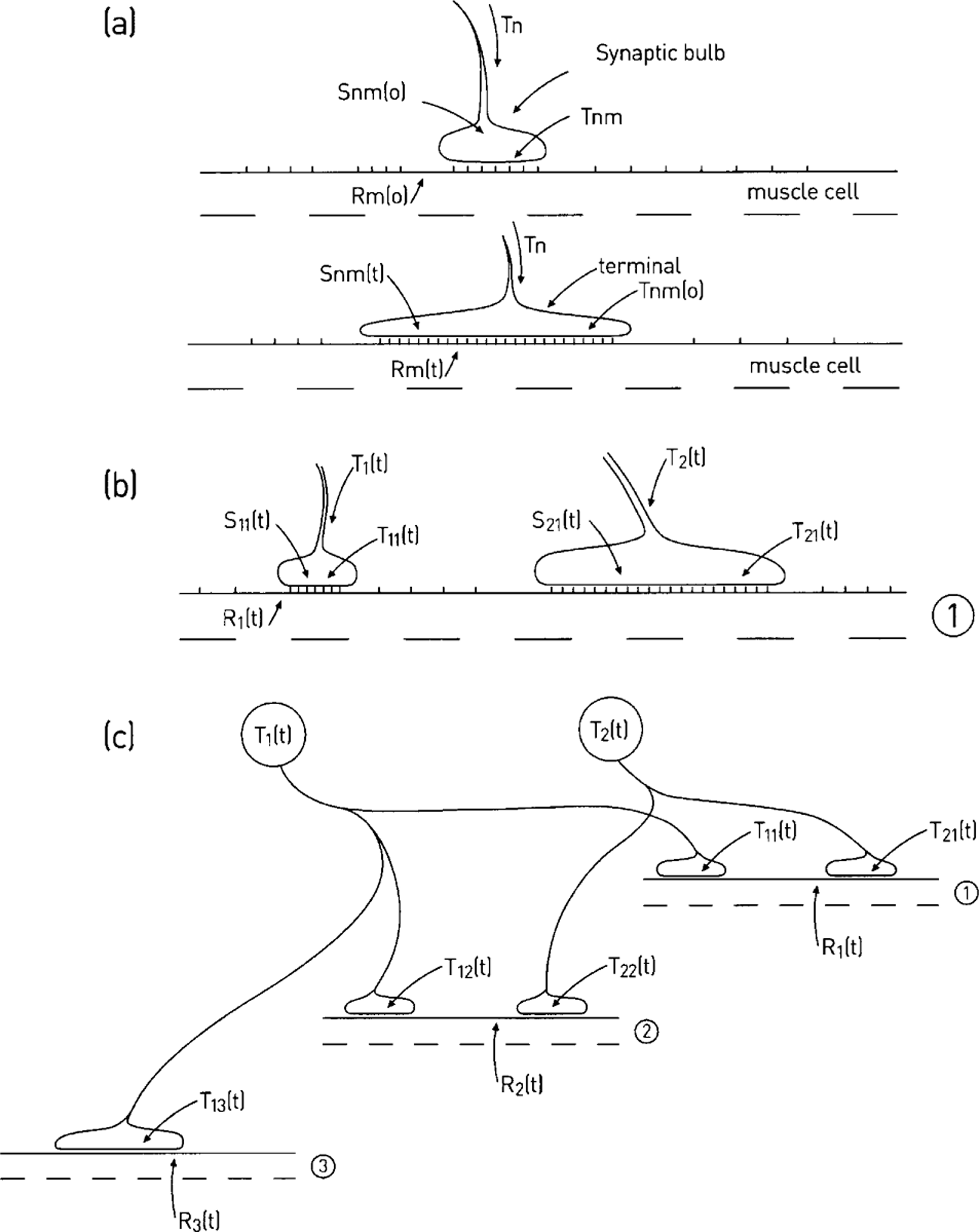
Dual constraint theory of synapse formation and regression at the neuromuscular junction. (a) Motor growth cone (size proportional to Snm(0)), containing synapse-formation molecules, Tnm(0), makes initial synaptic contact (t = 0) with a pool of synapse-formation molecules in the muscle (Rm(0)); at some time later (t) the growth cone has developed to form a terminal as a consequence of the reaction between the synapse formation molecules Rm(t) and Tnm(t). (b) The terminals of two neurons (1 and 2) have established themselves at a synaptic site on muscle cell 1 by time t; each terminal receives synapse-formation molecules T1(t) and T2(t), which are incorporated into the presynaptic membrane (T11(t) and T21(t)) where they react with the postsynaptic synapse-formation molecules (R1(t)); the size of the terminals (S11(t) and S21(t)) is then proportional to the products of this reaction. (c) The connections between the two motoneurons with synapse-formation molecules T1(t) and T2(t) and three muscle cells with synapse-formation molecules R1(t), R2(t) and R3(t) at time t [4].
At time t the reaction between R and T for a particular collateral is
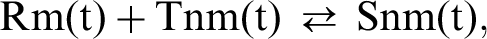
in which the area of terminal is directly proportional to Snm(t); knm and K are the forward and reverse rate constants of this process. At time zero, when the synaptic terminal first forms on the muscle cell, the Tnm molecules are free to move in the presynaptic membrane and Rm molecules at the synaptic site of the post-synaptic membrane. The probability of collision between a Tnm molecule and an Rm molecule is taken as proportional to the products of the amounts of these, so the back reaction is proportional to KSnm(t). If there is an autocatalytic effect, whereby the amount of Snm(t) produced accelerates the availability of Rm(t), then αn gives an index of the autocatalytic effect so that:
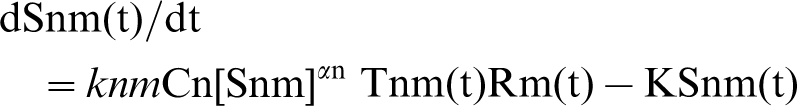
The factor Cn allows for changes in the affinity for axon n and is generally put equal to 1. The equations derived for this model and their numerical solutions for growth or regression of terminals over time are described in [4]. Ramussen and Willshaw have made a complete analysis of this model [42]. They termed it the ‘dual constraint’ theory because there are two kinds of competition, namely competition for a presynaptic resource (T molecules in the terminal) and for a postsynaptic resource (R molecules in the muscle). Postsynaptic competition is required to remove synapses and presynaptic competition explains experimental observations on the formation and elimination of nerve terminals, as revealed by the very extensive set of investigations carried out on this phenomenon at the neuromuscular junction [5], [43]. Rasmussen and Willshaw introduced the important notion that the amount of Tnm in a terminal is dependent on the relative extent of anterograde and retrograde transport of Tn between the neuron cell body (in this case the motoneuron) and the synaptic terminals of the neuron in question [42].
Identification of dual constraint molecules at the neuromuscular synapse
Have the molecular identities of T and R been identified? Four molecules are of particular interest in this regard, namely muscle-specific receptor tyrosine kinase (MuSK), agrin, neuregulin and ErbB, a receptor tyrosine kinase of the epidermal growth factor (EGF) family. MuSK, together with the dystroglycan/utrophin/dystrophin complex (of which dysbindin is a component) in muscle, is the principal junction formation molecular complex (R). Dysbindin-1 is a component of the dystrophin-associated protein complex (comprising dystroglycans, dystrobrevins, sarcoglycans and syntrophins). It binds to β-dystrobrevin and acts as an adaptor protein linking the complex to intracellular signaling cascades [44]. MuSK is activated by the multi-domain proteoglycan agrin released from motor-nerve terminals to generate junction-specific transcription (for a review, see [45]). Agrin is a principal terminal (T) junction formation molecule. The neuromuscular junction has an extracellular matrix-bound form of agrin (LN form) that possesses an amino-terminus required for secretion and binding to laminin in the extracellular matrix. In agrin-deficient and MuSK-deficient mutant mice post-junctional differentiation is drastically impaired, with neither clustered acetylcholine receptors nor other synaptic specializations detectable (for a review, see [46]). Conditional inactivation of MuSK in adult mice leads to loss of acetylcholine receptors, disassembly of the postsynaptic apparatus and retraction of the synaptic terminal [47].
Neuregulin (NRG) plays an additional role as a T factor for controlling the number of acetylcholine receptors, with ErbB being its R factor [5]. The neuregulin–ErbB and agrin–MuSK pathways are spatially separated at the neuromuscular junction, with MuSK concentrated in the postsynaptic primary cleft, together with acetylcholine receptors, and ErbB2/4 found only in the depths of the subsynaptic folds [48]. There are four NRG genes in the mammalian genome: nrg-1, nrg-2, nrg-3 and nrg-4[49–51]. Alternative splicing and differential promoter usage generates multiple NRG isoforms (Types 1–6). The NRGs activate the receptor tyrosine kinases ErbB2–4. Whereas ErbB4 binds NRGs and has catalytic activity, NRGs binding to ErbB3 lack tyrosine kinase activity and NRGs are unable to bind to ErbB2 at all. ErbB4 is the only receptor that can bind NRGs as a homodimer, leaving ErbB2 and ErbB3 requiring other ErbBs to form heterodimers before binding. ErbB4 is the preferred receptor binding NRG-2, NRG-3 and NRG-4 (for a review, see [52]). Prejunctional NRG-1 interacts with its postjunctional ErbB2/4 receptors to activate both intramuscular ras MAPK and PI3 kinase, so altering the expression of acetylcholine receptors at the junction (for a review see [53]). But this NRG-1/ErbB interaction is not required for the clustering of acetylcholine receptors because clusters still form in the absence of ErbB2 and ErbB4 in the muscle, although this interaction is required for the differentiation of the postjunctional apparatus and for maintaining the synaptic terminal at the junction [54–56].
NRG-3 is found at terminal Schwann cells of the neuromuscular junction [57], where it interacts with postjunctional ErbB4 receptors to cluster acetylcholine receptor (AchR) receptors [58], [59]. Terminal Schwann cells express only ErbB2 and ErbB3, with activation of these by NRG-1 released from the nerve terminal maintaining the viability of these cells [60–62]. Thus ErbB knockouts do not maintain terminal Schwann cells [61] that under these conditions migrate from the terminals towards any extra-junctional sources of NRG-1 [63].
Disrupted in schizophrenia 1 (DISC1) is found in peripheral neurons, both sensory and sympathetic-like, that form autonomic neuromuscular junctions [64]. DISC1 interacts with fasciculation and elongation protein zeta-1 (FEZI) and nuclear disruption element-like-1 (NDELI/NUDEL) and with cyclic adenosine monophosphate degrading phosphodiesterase (PDE4B) [65], [66]. The kinesin-1 motor complex has been implicated in the DISC1 and NDEL1/NUDEL interaction as well as in interactions of DISC1 and lissencephaly-1 (LIS1; necessary for axon extension of some neurons) [67]. DISC1 then regulates the localization of a NDEL1/NUDEL/LIS1 complex into the axons and growth cones as a cargo receptor for axon elongation. Specific interactions between DISC1 and NDEL1/NUDEL also occur in a dynein motor complex necessary for the formation of neurites in sympathetic-like neurons [68], such that genetic variants of DISC1, proximal to its NDEL1 binding site, affect the interaction leading to modulation of neurite formation [68], [69] (for a review see [70]). Cytoplasmic dynein is the motor protein that drives the transport of microtubules from the centrosome in the soma into the axon and to the growth cone [71]. The dynein motor complex is essential for normal microtubular dynamics [72] and has a key role in the transport of microtubules from the centrosome to axons and neurites [71], [73]. DISC1 is, among other things, likely to be of considerable importance in providing the microtubule system to axons for the transport of T factors (e.g. neuregulin) into the terminals. It follows that any mutations in the genes for these molecules will disrupt synapse formation and/or regression according to the dual constraint hypothesis.
Neurexin is a presynaptic receptor at the neuromuscular junction that binds α-latrotoxin, a major component in black widow spider venom, to produce a massive release of transmitter [74]. Neurexin belongs to a family of highly variable transmembrane proteins [75] that in mammals are encoded by three genes, each of which includes independent promoters for the long α-neurexins and the shorter β-neurexins [74]. α-Neurexin does not possess a post-junctional partner at the mammalian neuromuscular junction but is required for efficient transmitter release, probably through regulation of calcium influx into the motor-nerve terminal [76]. It appears then that neurexin does not play a role as a T factor in synapse formation and regression at this junction where it was originally discovered
Identification of dual constraint molecules at central synapses
It is evident that the dual constraint theory can be applied to the formation and regression of synapses in the cortex (Figure 3). Application of the theory to cortical synaptic spines is similar to that for the neuromuscular synapse. Are the molecules identified with the T and R substances at the neuromuscular synapse the same as those to be identified with the T and R substances at synaptic spines? All the T and R molecules at the neuromuscular junction discussed here have been localized to cortical neurons and their processes.
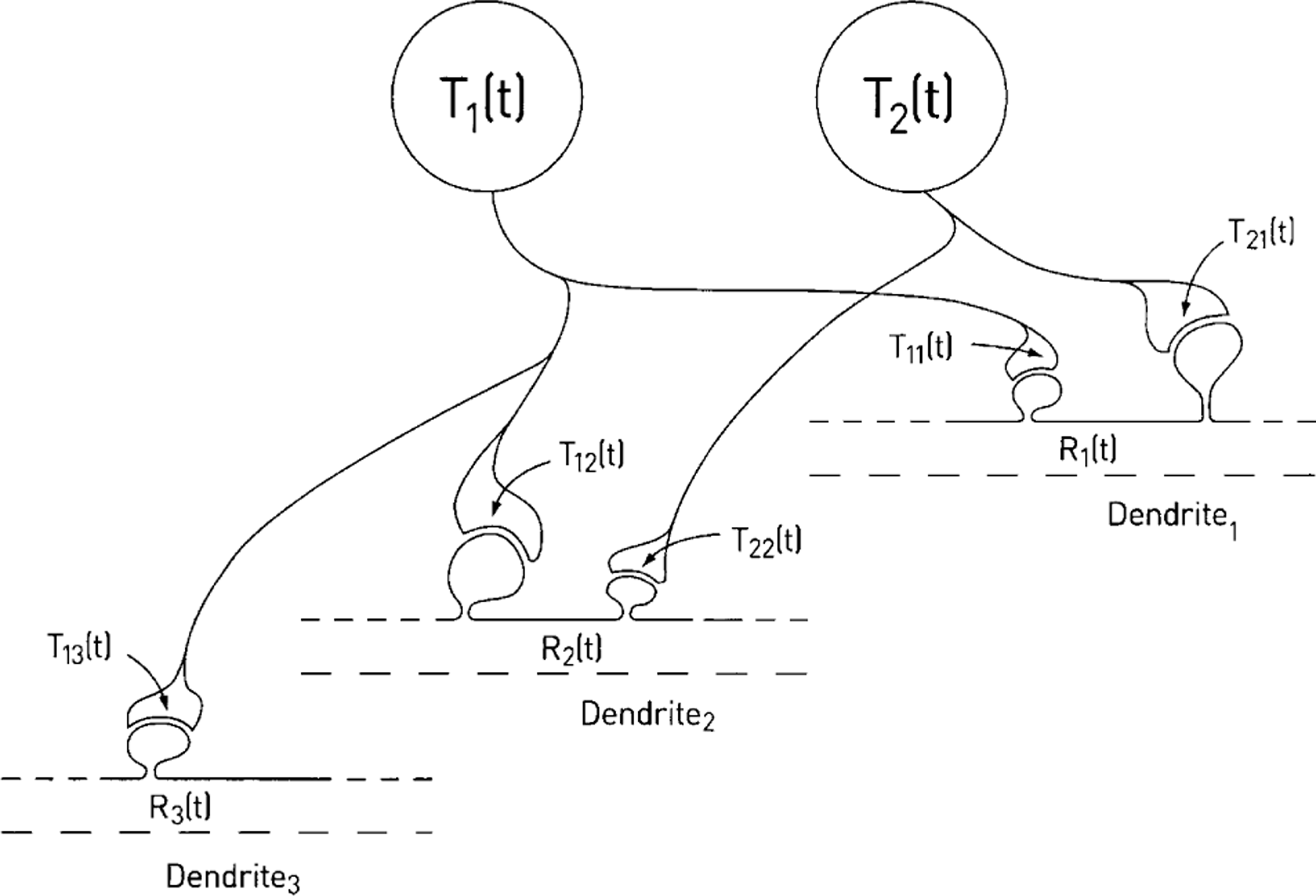
Dual constraint theory of synapse formation and regression at synaptic spines on neurons in dorsolateral prefrontal cortex. Compare this with Figure 2(c) and see the Figure 2 legend for definitions of symbols on this figure.
NRG-1 is localized in cortical neurons and their synaptic terminals on spines, as it is in motoneurons and their terminals at synapses on muscle cells, where it is a T factor [77] (Figure 4). The NRG-1 receptor ErbB4 interacts with domains of the postsynaptic density (PSD-95), as does the N-methyl-D-aspartate (NMDA) receptor, so that they become co-localized and apposed to synaptophysin-positive presynaptic terminals [78]. The release of NRG-1 from nerve terminals modulates transmission at many central excitatory synapses by modifying postsynaptic receptors [79–81]. The ErbB4 receptor is recruited to synaptic spines by action potentials that open NMDA receptors, a process that is blocked by Mg+ +[82]. Over-expressing ErbB4 selectively enhances alpha-amino-2-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) synaptic currents and increases synaptic spine size. Preventing NRG1/ErbB4 signalling, using ErbB4 ribonucleic acid interference (RNAi), destabilizes AMPA receptors and leads to loss of synaptic NMDA currents and a decrease in spine size [82]. (Interestingly, NRG-1 is postsynaptic and ErbB is presynaptic at inhibitory synapses (for a summary see [83].)
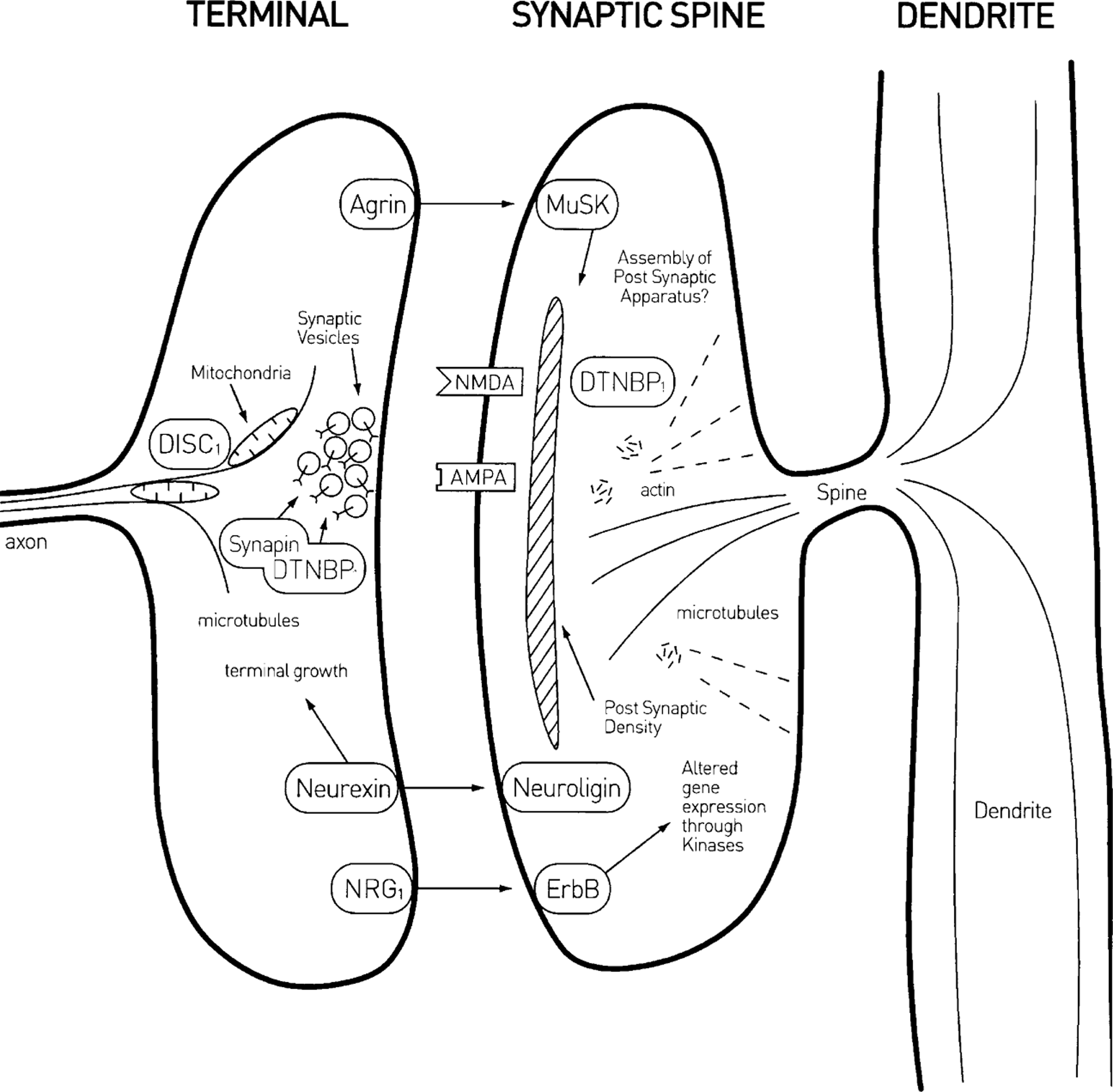
Spatial distribution of molecules that have been implicated in both synapse formation and regression. It is proposed that the principal among these are (inappropriately called) ‘muscle specific receptor tyrosine kinase’ and its nerve terminal released ligand ‘agrin’ (MuSK/agrin), first discovered at the neuromuscular junction, as were nerve terminal released ‘neuregulin’ and its receptor ‘ErbB’ (NRG/ErbB) and ‘dysbindin’ (dystrobrevin binding protein 1; DTNBP1) but not the molecule ‘disrupted in schizophrenia’ (DISC1) that is involved in microtubule formation, among other things.
NRG-3 is expressed by astrocytes, and is involved in glial–glial and glial–neuronal interactions that are mediated by ErbB1–4 receptors on the astrocytes [84].
Dysbindin (dystrobrevin binding protein 1; DTNBP1) is found in the cortex associated with microtubules and postsynaptic densities of spines at certain synapses [85], as well as in glutamatergic nerve terminals in association with the protein synapin, which primes synaptic vesicles for exocytosis [86] (Figure 1). So a principal molecule engaged in the assembly of the postsynaptic apparatus and its mechanical stability at the neuromuscular synapse (DTNBP1), and therefore part of an R factor, is found at synaptic spines. Knockouts of synapin result in decreased exocytosis of transmitter at nerve terminals [86]. Over-expression of DTNBP1 leads to enhanced glutamate release at synapses, whereas under-expression does the opposite [87], reflecting perhaps the influence of DTNBP1 on synapin control of vesicle priming.
The principal R factor molecule involved in neuromuscular synaptic development, MuSK, has recently been found in neurons throughout the cortex [88]. Its T factor ligand, agrin, is secreted by nerve terminals at the neuromuscular synapse where it binds to the extracellular matrix; but this is not possible at central synapses because they do not possess an extracellular matrix. This difficulty is overcome through cortical neurons secreting an isoform of agrin (termed SN) that possesses a variant amino terminus that converts agrin from a secreted, matrix-associated protein to a type II transmembrane protein [89]. Agrin and the specific tyrosine kinase MuSK are found only at excitatory synapses in the brain, and in agrin-deficient mice there is a >25% loss of such synapses accompanied by dysregulation of components of the MuSK pathway [90]. It therefore appears very likely that the agrin–MuSK pathway plays an important role in synapse formation at central synapses [91].
DISC1 immunoreactivity is found at synapses of dendritic spines in human prefrontal and parietal cortex [92] (Figure 4). The most common labelling at synapses consists of an unlabelled axon terminal forming an asymmetric, presumably excitatory, synapse with a spine that possesses immunoreactivity [92]. The presence of DISC1 in dendritic spines, and indeed in dendritic shafts as well, is consistent with its role in neuronal process formation. Iterative yeast-two hybrid screens have identified many of the proteins that interact with DISC1, with these predominantly found at synapses, consistent with the immunohistochemical localization of DISC1 there [93]. DISC1 is localized to mitochondria in cortical neurons [94] and its complex with FEZI is necessary for transport of these mitochondria from soma to axon and dendrites; without these mitochondria cortical neurons do not form neurites [95], [96]. In developing mice DISC1 is expressed in neurons within the cortex and hippocampus, as well as other brain areas, as early as embryonic day 10 (E10), with peaks at E13.5 (active neurogenesis period) and P35 (corresponding to the period of puberty in humans [97]). Speculatively, these periods of enhanced DISC1 expression may indicate the importance of this for neuronal migration (at E13.5) and changes in dendritic spines during adolescence (after P35). It seems very likely then that DISC1 plays an important role in ensuring the delivery of T substances to the growing nerve terminals.
Neurexins do not appear to play a role in synapse formation and regression at the mammalian neuromuscular junction, as noted here, but they may well do so at central synapses. At these synapses neurexins constitute a family of presynaptic cell-surface receptors that possess extracellular domains which resemble those of agrin and laminin; the endogenous postsynaptic receptors for these neurexins are neuroligins (for a review, see [98]; Figure 4). Different splice forms of postsynaptic neuregulin bind to presynaptic β-neurexin to generate synapse formation, with binding to α-neurexin ensuring terminal growth [99], [100]. Presynaptic neurexin binds to PDZ domains in the terminal to organize Ca2 + channels of the N and P/Q type, necessary for transmitter release [101], [102]. There is a 20% loss of neuropile in α-neurexin knockout mice; this appears to be primarily due to loss of inhibitory type II synapses [103]. Interestingly, although there is no postsynaptic receptor for neurexins at the mammalian neuromuscular junction there is at the Drosophila neuromuscular junction, where postsynaptic neuregulin interacts with presynaptic β-neurexin to localize the presynaptic scaffold to which the exocytosis apparatus is anchored, that is, the active zone [104], [105]. Thus although there appears to be no possibility of identifying neurexin as a dual constraint molecule at the neuromuscular junction it is quite possible that it is a T molecule with neurexin as its R receptor at central inhibitory synapses.
Loss of function of dual constraint molecules in the DLPC of subjects with schizophrenia
NRG-1a is significantly decreased in DLPC grey matter of subjects with schizophrenia, although NRG-1b may be increased [106–108]. DTNBP1 mRNA is significantly decreased in DLPC supragranular layers 2,3 and infragranular layers 5,6 but not granular layer 4 of subjects with schizophrenia, whereas no differences in mRNA for a variety of other proteins (spinophilin, synaptophysin, cyclophilin) have been detected in any layer of DLPC [106]. What effects would be expected as a consequence of loss of these dual constraint molecules at synapses in DLPC? As noted here, Malinow et al. have shown that erbB4 is critical for maintaining spine size and density through its mediation of the function of NRG-1 released from terminals on the synaptic spines [82]. It follows that the loss of NRG-1a in DLPC of subjects with schizophrenia would be expected to contribute to an excess loss of synaptic spines.
Single nucleotide polymorphisms within the 140 kb gene for DTNBP1 are strongly associated with schizophrenia [109–117]. DTNBP1 mRNA in the DLPC is significantly reduced in subjects with schizophrenia [106]. Two polymorphisms in DTNBP1 have been linked to schizophrenia and shown to be associated with changes in event-related potentials elicited during a continuous performance test, a measure of prefrontal function in the healthy human population [118]. There is also a reduction of DTNBP1 in the hippocampus of subjects with schizophrenia that is accompanied by loss of glutamatergic synapses and reduction in its size [119]. As Numakawa et al.[87] have shown, the loss of DTNBP1 results in considerable decrease in glutamate release from synapses so that the reduction in DTNBP1 in subjects with schizophrenia probably leads to a loss in the efficacy of excitatory synaptic transmission.
The neurexin-neuroligin molecules are not implicated in schizophrenia [121]. The Scottish translocation that led to the discovery of DISC1 removes interaction between NDELI/NUDEL with DISC1 [122], [123]. This would be expected to lead to a failure of nerve terminal growth at synapses. Indeed, single nucleotide polymorphisms in the DISC1 gene associated with schizophrenia give rise to reduced hippocampal grey matter volume [124] as well as reduced prefrontal cortex grey matter volume [125], [126], consistent with the loss of synaptic dendritic spines and indeed of entire basilar and apical dendrites as previously mentioned [124]. But, because mutations of DISC1 can lead to failure of migration of neurons from the ventricular plate along radial glial cells during development of the cerebral cortex, in addition to failure of the development of dendrites and therefore of synaptic spines by those neurons that do migrate [69; see also [127], the relative contributions of neuron loss versus synapse loss to this decrease in grey matter is not clear. A specific mutation in the mouse DISC1 gene (at L100P) gives rise to schizophrenia-like behaviour with profound deficits in, for example, prepulse inhibition, that can be reversed with antipsychotic treatment [128]. Very interestingly, atypical antipsychotics such as danzapine and risperidone, given in clinically relevant doses to mice, increase DISC1-mRNA expression in frontal cortex, whereas typical antipsychotics such as haloperidol do not, indicating the possibility of enhanced synaptic spine formation induced by the atypical antipsychotics [129]. DISC1's role as a cargo receptor for neurite growth places it at the centre of the delivery system for T molecules required for synaptic terminal growth.
A recent discovery of great interest is that consolidation of central synapses, both nerve terminals and dendritic spines on which they abut, is related to the development of an encasing extracellular matrix of chondroitin sulphate proteoglycans together with the establishment of an actin cytoskeleton in the spine. This is triggered by intracellular calcium influx into the spine, probably through NMDA receptors [130]. Thus mononuclear deprivation during the critical period, involving the elimination of spines in supragranular layers of area 17 that subserve the inactive deprived eye pathway, requires that these spines lose their mature proteoglycan encasement, so that they can become relatively mobile and are able to regress [120]. This loss of proteoglycan is dependent on the plasmin system tissue-type plasminogen activator, (tPA)-plasmin, which breaks down the proteoglycan [131]. Of great interest is that release of tPA is under the control of inhibitory basket cells involved in discriminating between competing sensory inputs from each eye in supragranular layers during the critical period. tPA release is then governed by the activity of inhibitory GABAergic neurons, which ultimately determine the extent of consolidation or mobility of synaptic spines [131]. We have then part of the process by which synaptic spines are consolidated in supragranular layers in the critical period (at least for the binocular area in area 17). Blocking GABAergic transmission prevents the release of tPA and so prevents plasticity, whereas accelerating GABAergic synapse maturation (mimicked using diazepam) increases the release of tPA, the enzymatic breakdown of chondroitin sulphate proteoglycan, and so allows spine mobility with the consequences that the critical period is moved earlier [131]. It is very interesting in this regard that one susceptibility gene for schizophrenia is proline dehydrogenase (PRODHZ) that controls the extent of GABAergic neurons in different areas of cortex [132]. Individuals with schizophrenia show a loss of GAD67 mRNA expression and GABA synthesis in subpopulations of GABAergic neurons in the DLPC [133], [134]. Whether these changes in GABAergic neurons in the DLPC of subjects with schizophrenia have implications for the regression of synapses there is not known.
Dual constraints on synapses in schizophrenia
This paper focuses on how our knowledge of synapse formation and regression at the neuromuscular junction might lead to insights into the failure of synapses to form, or excess loss of synapses in the DLPC of subjects suffering from schizophrenia. Why concentrate on DLPC? Andreasen et al. have argued that the DLPC is a key node in a core feedback circuit that includes the cerebellum and the thalamus, which is disrupted in schizophrenia, leading to difficulties in prioritizing, processing, coordinating and responding to information [135]. This is called the ‘cognitive dysmetria’ hypothesis. A recent meta-analysis of neuro-functional correlates of vulnerability to psychoses, using functional MRI (fMRI), shows unequivocally that there is decreased activation in DLPC of those at risk of schizophrenia as well as of first-episode patients during tasks that normally activate DLPC, such as the Wisconsin Card-Sorting Test [136–138]. Indeed most positron emission tomography (PET) studies show that the Wisconsin Card-Sorting Test fails to produce much activation of the DLPC in patients with schizophrenia, who perform at best poorly on the test [136]. The decrease in DLPC activity in patients with schizophrenia during appropriately designed cognitive tasks reflects a decrease in grey matter there as determined on MRI [139], [140]. When a primate is required to remember a visual cue location through a delay period of seconds between the stimulus and a memory-guided behavioural response, neurons in the DLPC show location-tuned increased firing throughout the delay period [141]. Furthermore, the firing of DLPC neurons is enhanced if the primate focuses attention inside the receptive field of the neuron (for a theoretical treatment, see [142]). Such spatial working memory procedures, dependent on normal activity in DLPC, are severely compromised in schizophrenia (for a theoretical treatment, see [143]). Thus MRI, fMRI, PET and behavioural studies confirm the cognitive dysmetria hypothesis of Andreasen et al.[144], [145]. The DLPC then figures as the principal cortical region for consideration in this paper.
Dual constraints on synapse formation and regression in depression
Is there any evidence that the hypothesis for schizophrenia described in this paper could also be applicable to patients with major mood disorders (MMD)? The most consistent areas of the brain to show loss of grey matter and function in those suffering from MDD, using MRI and PET, are the subgenual and perigenual cingulate cortex as well as the amygdala (for a review see [146]). There is loss of grey matter volume in the perigenual cingulate of patients with elevated risk of MDD [147–149] (for a review see [150]). PET studies show abnormalities in resting regional cerebral blood glucose metabolism in the rostral anterior cingulate of depressed individuals ([151], [152]; for reviews see [153–155]).
Neural networks have been proposed, involving the subgenual/perigenual anterior cingulate and the amygdala, which might go awry in MDD [156], [157]. In particular stress may involve a network that incorporates interactions between the amygdala, subgenual anterior cingulate and the raphe nucleus [149], [156], [157]. The emphasis on such circuits arises from consideration of subjects that are asked to restrain their emotions in a particular challenging social situation. In this case there is activation of prefrontal anterior cingulate cortex that is thought to inhibit activity in the amygdala, which normally supports emotional responses [158]. In a recent summary of brain imaging, depression was shown to be accompanied by decreased activity in dorsal anterior cingulate cortex, with a concomitant increase in activity in the amygdala ([159]; but see [160]).
There is a decrease in the extent of the synaptic vesicle-associated protein synaptophysin in the anterior cingulate cortex of subjects with mood disorders, probably reflecting a loss of synapses [161]. This is accompanied by a large loss of neurons in layers III and V of the ventromedial prefrontal cortex, which includes the orbitofrontal cortex projecting to other cortical regions and to the amygdala [162], [163]. MDD is also accompanied by changes in the hippocampus, such as a decrease in spine density on apical dendrites of CA3 pyramidal cells as indicated by a loss of the spine marker spinophilin [164], as well as decrease in spine density in the subiculum [165]. Very interestingly, a loss of neurons in the hippocampus of MDD patients can be offset by an increase of neurogenesis there due to an action of antidepressants such as desipramine, fluoxetine and imipramine [166], or of electroconvulsive shock therapy, which triggers the release of brain-derived neurotrophic factor [167].
There is evidence that at least two dual constraint molecules may function abnormally in MDD, namely neuregulin-1a, which is reduced in neurons of prefrontal cortex in depressed subjects [168], and DISC1, which is distributed abnormally, changing from a location predominantly in the postsynaptic density and mitochondria of spines to a nuclear location [169]. In summary it appears likely that there are significantly fewer synapses in the anterior cingulate of subjects with MDD and that there are abnormalities in at least two dual constraint molecules. But far more research needs to be carried out in order to ascertain that the hypothesis entertained here for schizophrenia is applicable to subjects with MDD.
Conclusion
It has been argued that regression of synapses in the DLPC is, like that at the neuromuscular junction, concerned with ensuring elimination of an excess of appropriately connecting synapses to target cells in such a way as to optimize the efficacy of the stabilized synapses remaining when regression is complete [170], [171]. In the absence of regression a large number of synapses, all of very low efficacy and therefore of low safety factor for transmission, would be stabilized. According to the present hypothesis this process of synapse formation and regression at the neuromuscular junction is dependent on a phalanx of molecules such as MuSK, agrin, neuregulin, ErbB and so on. It is highly likely that these molecules are also important in synapse formation in DLPC during childhood and perhaps during synapse regression in adolescence. But we have seen that if these molecules are not expressed then approximately 30% of synapses fail to form in the cortex at all. This being the case, it is expected that a failure of many synapses to form during childhood rather than an excess loss of synapses during adolescence might characterize schizophrenia.
Answers to the following questions are urgently required: (i) does the loss of grey matter in the DLPC during normal adolescence occur as a consequence of synapse regression and/or glial cell loss; (ii) does the excess grey matter loss in the DLPC of schizophrenia subjects occur as a consequence of synapse regression and/or glial cell loss; (iii) does the excess loss of synapses in DLPC of schizophrenia subjects occur before, during or after the adolescent period; (iv) to what extent does the excess loss of synapses that occurs in the DLPC of schizophrenia subjects also occur in other parts of the brain; and (v) to what extent do key dual constraint molecules such as MuSK, agrin, and others, play a role in determining synapse regression in the brain and the stabilization of high efficacy synapses during adolescent development.
Different clinical stages have been identified during the prodromal period for schizophrenia [172]. Are these related to synapse loss in the DLPC? It is difficult to see how this could be the case if the relatively smaller number of synapses in the DLPC arises from a failure of synapse formation during childhood. In contrast, such a relationship might arise as a consequence of the vulnerability of an already small number of synapses to further regression when such prodromal individuals are subjected to either psychological stress or that due to infection. This could lead to a loss of synapses below the level at which DLPC networks can function.
The present paper highlights an inferred role of some dual constraint molecules in the process of synapse formation during childhood and synapse loss during adolescence, together with the consequence of genetic defects in these molecules. It has not considered the impact of environmental factors, be they identified as due to psychological stress or infection. This will be forthcoming.