
Abstract
To determine the tolerability and efficacy of eletriptan in patients who had discontinued oral sumatriptan due to lack of efficacy or intolerable adverse events (AEs) during previous clinical treatment (not a controlled trial). Eletriptan is a potent, selective 5-HT1B/1D receptor agonist with beneficial pharmacokinetic properties compared with sumatriptan. In a double-blind, parallel group, placebo-controlled multicentre study, patients with and without aura (n = 446) were randomized to 40 mg eletriptan (E40, n = 188), 80 mg eletriptan (E80, n = 171) or placebo (n = 87) for treatment of up to three migraine attacks. Two-hour headache response, based on first-dose, first-attack data, was 59% for eletriptan 40 mg, 70% for eletriptan 80 mg, and 30% for placebo (P < 0.0001 for both doses of eletriptan vs. PBO; P < 0.05 for E80 vs. E40). Onset of action was rapid, with 1-h headache response rates significantly superior for E40 and E80 vs. placebo (40%, 48%, 15%; P < 0.0005). Both E40 and E80 were significantly superior to placebo, based on first-dose, first-attack data, for 2-h pain-free response (35%, 42%, and 7%; P < 0.0001). Both E40 and E80 demonstrated significant consistency of response, with headache relief rates at 2 h on at least two of three attacks in 66% and 72% vs. 15% on placebo (P < 0.001). AEs were mild to moderate in severity and dose related. The most commonly reported AEs included nausea, vomiting, asthenia, and chest symptoms. E40 and E80 produce an effective response in patients who had previously discontinued treatment with sumatriptan due to lack of efficacy or side-effects.
Introduction
In the early 1990s, sumatriptan was a major breakthrough in acute migraine treatment (1). Despite high efficacy in randomized controlled clinical trials (on average, 59% of attacks with sumatriptan 100 mg) (2), there is still a segment of migraine patients who are not satisfied with sumatriptan because of side-effects or less than optimal efficacy (3, 4). Also, headache recurrence has been related to the short plasma half-lives of some triptans, and is reported in 30–40% of patients in clinical trials of sumatriptan, rizatriptan and zolmitriptan (1, 3, 5).
There is still a need for improvement in acute migraine therapy. Despite large sales of triptans in some countries, only a minority of migraine patients receive the beneficial therapy of a triptan. While many patients require a triptan for optimal treatment, some of these migraine patients will not have an optimal response to initial treatment. It is general clinical practice that if a patient does not respond to one triptan, then it is worthwhile to try treatment with another related medication. However, this important aspect of drug treatment has not been tested thoroughly for triptans in the past. There is only one double-blind treatment trial evaluating naratriptan in the treatment of patients who had responded poorly to oral sumatriptan (4). The study showed that poor responders to sumatriptan can benefit from migraine therapy with an alternate triptan.
The triptans differ from each other in pharmacokinetic and pharmacodynamic properties (2, 6). Eletriptan has better oral bioavailability (50% vs. 14%) and longer plasma half-life (4–5 h vs. 2 h) than sumatriptan (7–9). Therefore, the aim of the present study was to provide clinical evidence on whether a new triptan, eletriptan, previously shown to have superior efficacy to sumatriptan (10), might be effective in patients who had discontinued sumatriptan either due to lack of efficacy or due to side-effects.
Patients and methods
Ethical conduct
The clinical trial was conducted according to the 1996 revision of the Declaration of Helsinki (revised South Africa, 1996) and local laws and regulations relevant to the use of new therapeutic agents in the country of conduct. The study protocol was approved by the Ethics Review Committee in each participating country. Patients signed consent forms signifying a complete understanding of the study's purpose, the procedures involved, and the potential benefits and risks of participating in the study.
Patient enrolment
Male and female subjects age ≥ 18 years were invited to participate if they met the International Headache Society (IHS) diagnostic criteria for migraine, with or without aura, and could reasonably expect to suffer at least one acute attack of migraine every 6 weeks. Patients were required to have discontinued therapy with oral sumatriptan at least 2 weeks, but not longer than 2 years, prior to the screening visit. In general, subjects had been in the practices of the investigators for a significant period of time and their lack of sufficient response to sumatriptan was documented in the patient notes, along with the use of other therapies, including other triptans, during the last 6 months before screening. Patients were asked to give one of the following reasons for stopping the treatment with sumatriptan: slow onset of action, inconsistent response, poor overall efficacy, recurrence or tolerability. Female subjects were required to be adequately protected against pregnancy.
Exclusion criteria included pregnancy or breast-feeding, known coronary artery disease, significant arrhythmias, heart failure, significant ECG abnormalities, and uncontrolled hypertension. Any significant systemic, organ, neurological, endocrine, metabolic, and psychological disorders reported by the patient or discovered during the physical examination also resulted in exclusion. Patients considered to have atypical migraine such as frequent attacks, prolonged aura or any migraine that was considered atypical were not included in the study. Patients who, during the course of the trial, required treatment with sumatriptan or any other 5-HT1B/1D agonist in addition to study medication were also excluded.
Design and procedure
The study was a double-blind, parallel-group, placebo-controlled, multicentre study comparing the efficacy, safety, and tolerability of oral eletriptan 40 mg (E40), oral eletriptan 80 mg (E80) and placebo (PBO). A total of 446 patients were randomized in a 2 : 2 : 1 ratio to E40 (n = 188), E80 (n = 171), or placebo (n = 87). The patients were recruited over a 14-month period from approximately 50 headache treatment centres in Norway, Sweden, Denmark, Finland, and the Netherlands. Patients were enrolled in the study until they had treated three migraine attacks or until they reached the maximum duration of 18 weeks. Study medication consisted of one or two 40-mg tablets of eletriptan or matching placebo. No other 5HT1B/1D agonists could be taken during the study. Upon experiencing a ‘typical’ migraine headache, subjects were instructed to take their medication as soon as possible and within 6 h. The migraine headache had to be of at least moderate severity and not decreasing in intensity. Subjects should not have taken analgesics or anti-emetics within the preceding 6 h, or ergotamine or ergotamine-like agents in the previous 48 h. Patients were to record information in a diary regarding migraine symptoms for up to three attacks. If, after 4 h and within 24 h, the migraine recurred, the patient could take a second dose of the study medication. If patients experienced an inadequate response after 4 h they were allowed to take rescue medication (which could not include an ergotamine-like compound or another 5HT1B/1D medication). Subjects were asked to record their migraine symptoms in the diary at baseline (immediately predose), 30 min, 1, 2, 4, and 24 h after dosing with the study medication.
A complete medical history and physical examination were performed at the initial visit. At the initial clinical visit, a record of any concomitant medication was established. Pregnancy screening tests were performed and home pregnancy kits were provided for use prior to dosing. Study medication could not be taken if a positive pregnancy test result was obtained.
Randomization of subjects into study groups was done by a computer-generated method. The blinded randomization list was provided by the sponsor to the investigators, along with numbered blister packs containing eletriptan or matching placebo tablets. Investigators could unmask individual subjects only in the case of an emergency by contacting the sponsor. Once the study was completed, the randomization code was released by the sponsor.
Measurement of efficacy
Subjects recorded the intensity of each migraine headache in the diary based on a 4-point scale: 0, pain absent; 1, mild pain; 2, moderate pain; 3, severe pain. A response was considered a change from severe or moderate pain to mild or absent at various time points after dosing with study medication. The primary efficacy endpoint was 2-h headache response after taking the first dose of study medication for the first attack. Recurrence was defined as a return of headache to moderate or severe intensity within 2–24 h if the subject had an initial response within 2 h of taking the study medication. A post hoc sustained response analysis was also conducted. Sustained response was defined as achieving a headache response within 2 h post dose, with no recurrence or rescue medication needed. Nausea and vomiting were recorded as being either present or absent at baseline and at 1, 2, 4 and 24 h. After 24 h, subjects were asked to consider if, upon developing a migraine, they would choose this or another medication for treatment. At the final clinical visit, subjects who had treated at least one attack were asked ‘Overall, how would you rate the study medication?’ in order to determine the global assessments of the study medication, a measure of overall patient satisfaction. Responses were made based upon a 5-point scale: 0, poor; 1, fair; 2, good; 3, very good; 4, excellent.
Assessment of tolerability
At screening, 12-lead ECG, pulse, and blood pressure assessments were performed. Further ECGs were done only if deemed clinically necessary. All adverse events (AEs), whether related to the medication or not, were reported to the study investigators. At the final visit, performed within 14 days after treating the third or final attack, subjects were physically examined and pulse and blood pressure measurements were taken. Patients were questioned about AEs and concomitant medications and diaries were returned and reviewed with the patient.
Power calculation and statistical analysis
The primary efficacy endpoint was the headache response rate at 2 h after the first dose of study treatment for the first attack. Assuming headache response rates of 30% for placebo, 50% for eletriptan 40 mg, and 64% for eletriptan 80 mg, with at least 80% power to detect at least a 20% difference between the eletriptan and placebo groups on a two-sided test at the 0.05 level of significance (α ≤ 0.05), at least 465 patients were needed. All analyses were performed on the intent-to-treat (ITT) population, defined as all randomized patients who took study medication with a valid baseline and at least one post-baseline evaluation for each attack.
Headache response rates were analysed using a categorical linear model (SAS procedure PROC CATMOD). CATMOD procedure was also used to analyse the pain-free response rates and the incidence of nausea, vomiting, and headache recurrence. Analysis of variance (SAS Institute Inc., Cary, NC, USA GLM procedure) was used to analyse the global evaluation of medication and global acceptability. Tolerability data were summarized descriptively for all patients who received at least one dose of the study medication.
Results
Patients
There were 188 patients randomized to E40, 171 to E80, and 87 to PBO. The treatment groups were similar in terms of gender, age, race, migraine diagnosis and baseline symptoms at the day of the first attack (Table 1a,b). Patients were recruited between January 1999 and March 2000. Over 80% of the study subjects in each of the groups were female, consistent with previously reported data (11, 12). For all three groups, the mean age of study subjects was approximately 41 years and the predominant diagnosis was migraine without aura. During the trial period, 427 patients (96%) experienced and treated at least one migraine attack with study medication (E40, n = 179; E80, n = 167; PBO, n = 81). For the first attack, 218 (51%) reported nausea at baseline, while 14 (3%) reported vomiting. At baseline, 293 patients (69%) described their headache as moderate, with the other 134 (31%) rating their headache pain as severe (Table 1b). Three hundred and fifty-six patients (80%) treated two out of three attacks and 305 patients (68%) treated all three migraine attacks (Fig. 1). The proportion of patients treating all three attacks was much higher for eletriptan 40 mg (72% (135/188)) and 80 mg (75% (128/171)) than for placebo (48% (42/87)). Reasons for not treating all three attacks for the combined eletriptan groups vs. placebo were as follows: AEs, 5.8% (21/359) vs. 3.4% (3/87); insufficient response, 4.7% (17/359) vs. 33.3% (29/87); and miscellaneous other reasons, 12.8% (46/359) vs. 13.8% (12/87).

Patient disposition.

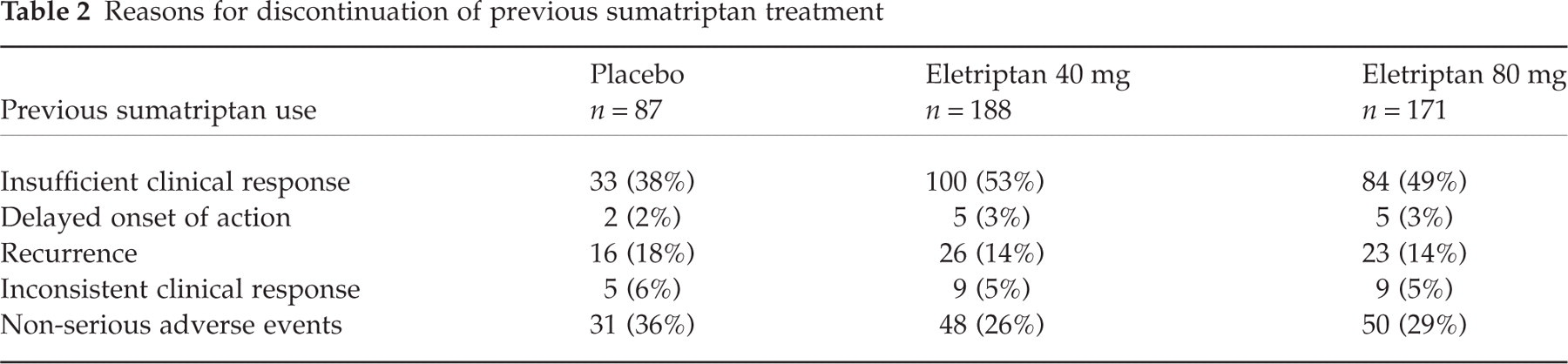
The majority of randomized patients, 317 (71%), had discontinued sumatriptan due to lack of clinical efficacy, with most reporting insufficient overall clinical response (53% (100/188), 49% (84/171), and 38% (33/87) for E40, E80, and PBO, respectively) (Table 2). Other reasons given were recurrence of headache followed by inconsistent clinical response and delayed onset of action. The proportion of the patients who had discontinued use of oral sumatriptan due to the occurrence of non-serious AEs was similar across the three study treatment groups: 26% (48/188), 29% (50/171), and 36% (31/87). There was a trend difference in reasons for sumatriptan discontinuation (Table 2), with more patients in the eletriptan treatment groups discontinuing prior sumatriptan for efficacy reasons, and more patients on placebo discontinuing due to AEs. This difference would appear to disadvantage the eletriptan treatment groups, suggesting that a somewhat more treatment-resistant group was randomized to eletriptan. However, we have explored the possible effect of the ‘sumatriptan discontinuation rate’ on the 2-h post dose headache response by including this effect as a covariable in the statistical model. The covariable effect for discontinuation from sumatriptan was not statistically significant and the overall 2-h treatment response with and without the covariable was consistent.
Reasons for discontinuation of previous sumatriptan treatment
Efficacy
First-dose, first attack results
The 2-h headache response rates (first-dose, first attack) for E40 and E80 were both found to be significantly superior to placebo (59% (91/154), 70% (106/152) and 30% (22/74), respectively; P ≤ 0.0001) (Fig. 2). In addition, more subjects in the E80 group had a 2-h headache response than those assigned to E40 (P < 0.05). Both doses of eletriptan had significantly superior 1-h headache response rates compared with placebo (40% (63/158), 48% (77/159) and 14% (11/76), respectively; P < 0.0005). E80 was also found to provide significantly superior benefit vs. placebo within 0.5-h post dose (19% (30/160) vs. 8% (6/75); P < 0.05).

Headache response in patients who had previously discontinued sumatriptan: first-dose, first attack data. □, Placebo (n = 81); ▨, Ele 40 mg (n = 179); , Ele 80 mg (n = 167).
The 2-h pain-free response (first-dose, first attack) was significantly greater after E40 and E80 than after placebo (35% (54/154), 42% (64/152), and 7% (5/74); P ≤ 0.0001) (Fig. 3). E80 provided significantly greater pain-free response to placebo at 1 h as well (15% (24/159) vs. 3% (2/76); P < 0.05).
Pain-free response in patients who had previously discontinued sumatriptan: first-dose, first attack data. □, Placebo (n = 81); ▨, Ele 40 mg (n = 179); , Ele 80 mg (n = 167).
Headache recurrence (return of headache from pain free or mild intensity between 2 h and 24 h after administration) was 26% (20/77), 32% (26/82), and 50% (11/22) for eletriptan 40 mg, 80 mg and placebo, respectively, and fewer patients who received eletriptan required rescue medication (24% (42/179), 14% (23/167), 63% (51/81)). To measure further the efficacy of treatment, a post hoc analysis of sustained headache response was conducted. Both doses of eletriptan achieved a significantly higher sustained response vs. placebo (39% (55/140), 45% (56/124) and 14% (10/74); P < 0.0005). For each of the three attacks, both headache response and pain-free response at 2 h were significantly greater with E40 and E80 (Table 3).

∗P ≤ 0.0001 eletriptan vs. placebo;
†P < 0.0005 eletriptan vs. placebo;
‡P < 0.05 eletriptan 40 mg vs. eletriptan 80 mg. Note that the sample size for each attack is smaller than the sample size reported in Fig. 1 due to missing data at the 2-h time point.

∗P < 0.005 eletriptan vs. placebo
†P < 0.0005 eletriptan vs. placebo
‡P ≤ 0.0001 eletriptan vs. placebo
§P < 0.01 eletriptan 40 mg vs. eletriptan 80 mg. Note that the sample size for each attack is smaller than the sample size reported in Fig. 1 due to missing data at the 2 h time-point.
There was a significantly lower incidence of nausea after eletriptan treatment than after treatment with placebo at 1 h (41% (65/159), 44% (69/158), 62% (47/76); P < 0.05) and 2 h (30% (47/155), 33% (50/151), 52% (39/75); P < 0.01).
Both 40 mg and 80 mg eletriptan doses were rated acceptable vs. any other previous medication used to treat migraine in 62% (197/317) of patients (P < 0.0001) in comparison with only 20% (15/76) for placebo. Eletriptan patients had significantly higher mean global evaluation scores than patients treated with placebo (mean score 1.8, 1.9, 0.4; P = 0.0001). Fifty-eight percent of eletriptan 40 mg patients (106/184) and 65% of eletriptan 80 mg patients (110/169) rated their treatment as good or better in comparison with 13% in the placebo group (11/83).
Consistency of response results
In terms of intrapatient response, a high level of consistency was achieved with eletriptan across three treated attacks (Fig. 4). The percentage of subjects who reported a response in two out of three headaches was significantly higher on E40 (66% (74/112)) and E80 (72% (72/100)) compared with placebo (15% (5/34); P ≤ 0.001). Using stringent criteria requiring response in all three attacks resulted in significantly higher rates for E40 (38% (43/112)) and E80 (41% (41/100)) compared with placebo (6% (2/34); P ≤ 0.001).
Consistency of response across three attacks: percent of patients responding to ≥ 1, ≥ 2, or all three attacks. □, Placebo (n = 34); ▨, Ele 40 mg (n = 100); , Ele 80 mg (n = 184).
Tolerability
Both doses of eletriptan were associated with more side-effects than placebo. There were no deaths or serious treatment-related AEs. The majority of AEs were mild to moderate in severity and transient in nature. The most commonly reported AEs were asthenia, nausea, chest symptoms, and vomiting (Table 4). A pairwise comparison demonstrated that there was no statistically significant difference for either of the two doses of eletriptan (E40, E80) vs. placebo (P-value of 0.417 and 0.105, respectively). It should be noted, though, that the study was not powered to detect differences in the incidence of adverse events.
Incidence of treatment-related adverse events experienced by ≥3% of patients in any one treatment arm
Discussion
While there is extensive literature on the different options for the acute treatment of migraine, there is only one reference to a double-blind triptan trial investigating a second treatment option after initial treatment failure (4). In the present study, eletriptan 40 mg and 80 mg were effective and well tolerated in the treatment of migraine headache in patients who were previously dissatisfied with sumatriptan treatment. Headache responses for both doses of eletriptan E40 (59%) and E80 (70%) were superior to placebo (30%) and there was a significant dose-related increase in headache response at 2 h. The 2-h pain-free response was also dose-dependent and better than placebo response. E80 pain-free response was significantly better after 1 h compared with placebo. The 70% headache response rate achieved with the E80 dose is particularly striking in contrast to the original lack of efficacy experienced by the majority of subjects when previously treated with oral sumatriptan. Patients gave insufficient clinical response (71%) and non-serious AE (29%) as the major reasons for having discontinued sumatriptan.
Eletriptan was safe and well tolerated. Among the more common AEs, the incidence of nausea was somewhat elevated but this was true for all treatment arms. Of subjects receiving placebo, 8% reported an incident of nausea. Previous double-blind placebo-controlled trials of eletriptan have reported a frequency of treatment-related nausea of 1–4% with placebo (10, 13, 14). The higher incidence reported here may be the result of variability between studies or of technicalities in the methodology such as differentiating between diagnosis as an AE or an actual symptom of the migraine itself. The frequency of nausea observed with eletriptan shows a 6% increase between the 40-mg and 80-mg doses. However, this increase in frequency does not appear to result from a dose response as the difference in frequency of nausea observed between the 40-mg and 80-mg doses in other trials has ranged from 1% to 5% (10, 13, 14).
The 80-mg dose was used because we thought that it might be important to provide aggressive therapy for this treatment-resistant group of patients, though in some regions, such as Europe, use of a second 80-mg dose of eletriptan within 24 h is higher than the regulatory authorities recommend.
In the past 4 years, a number of second-generation triptans have been developed, including naratriptan, rizatriptan, zolmitriptan, almotriptan, frovatriptan and eletriptan (2, 6). Studies have demonstrated a range of pharmacological and pharmacokinetic differences among these compounds, with a potential for improved clinical effects (2, 15–17). Compared with sumatriptan, eletriptan is a more potent agonist of 5-HT1B/1D receptors; has higher lipophilicity; and has faster association rates and slower dissociation rates than sumatriptan at the human recombinant 5HT1D receptor (16, 18). Notable pharmacokinetic differences between eletriptan and sumatriptan include higher oral bioavailability (50% vs. 14%) and a longer elimination half-life (4–5 h vs. 2 h) (7–9).
For patients in whom sumatriptan does not provide effective relief, one clinical trial has suggested a rational approach for switching to naratriptan (4). In 347 patients who did not respond to sumatriptan, it was found that 25% of patients improved significantly at 2 h post dose when administered naratriptan for subsequent attacks (4). Pain relief with naratriptan at 4 h post dose was also effective (41%, P < 0.01) (4). However, as the patient population in the naratriptan trial was tested for a lack of response to sumatriptan, a direct comparison with this study cannot be made.
In the present study, patients who had previously failed and discontinued adequate treatment with oral sumatriptan were recruited. A potentially important study limitation is that the historical report of sumatriptan treatment failure was not confirmed by prospective treatment. Another important study limitation is the lack of a sumatriptan control group. The study results suggesting the efficacy benefit of eletriptan in a subgroup of patients who had failed sumatriptan treatment would have been strengthened if the study design had used a parallel sumatriptan treatment group, and if this sumatriptan group could have been shown to have a significantly lower response.
With these limitations in mind, the headache response achieved with eletriptan is impressive, particularly in light of the relatively high intrapatient consistency of response.
These results show that eletriptan provides rapid and effective relief from migraine, with a well-tolerated side-effect profile, in patients with previous poor response or intolerance to sumatriptan. The response rates for eletriptan seen in this study are consistent with the response rates in a number of randomized placebo-controlled trials vs. other triptans (10, 19). The results of the present study confirm that eletriptan may have an important role in the clinical armamentarium for the acute treatment of migraine, including the treatment of patients who have stopped treatment with sumatriptan due to poor response or tolerability.
Footnotes
Acknowledgements
This study was supported by Pfizer Ltd.
Appendix
The study was conducted by the following sites and their respective personnel. Denmark: F. W. Bach MD, Aarhus Kommunehospital, Aarhus; A. Korsgaard MD, Private Practice, Odense; M. Langmark MD, Hilleroed Sygehus, Hilleroed; O. Neubauer MD, Private Practice, Nykoebing; J. Olesen MD, KAS, Glostrup; K. Pedersen MD, Private Practice, Hjorring; M.-J. Rasmussen MD, Central Sygehuset i Esbjerg, Esbjerg; D. Rasmussen MD, Bispebjerg Hospital, K⊘benhavn. Finland: M. Färkkilä MD, PhD, Helsinki Headache Centre, Helsinki; H. Havanka MD, Haukiputaan Laakarikeskus, Haukipudas; M. Ilmavirta MD, Torikeskuksen Laakariasema, Jyvaskyla; E. Kinnunen MD, Hyvinkaa Aluesairaala, Hyvinkaa; J. Liukkonen MD, Mikkelin Paansarkypoliklinikka, Mikkeli; E. Sako MD, Turku Headache Centre, Turku; T. Jolma MD, Porin Laakarikeskus, Pori. The Netherlands: J. P. ter Bruggen MD, PhD, Jeroen Bosch Hospital, Hertogenbosch; W. Linssen MD, Sint Lucas Andreas Ziekenhuis, Loc. Sint Lucas, Amsterdam; P. Schiphof MD, Bernhoven Ziekenhuis, Oss; W. Verhagen MD, Nijmeegs Interkonfessioneel Ziekenhuis, Canisius-Wilhelmina, Nijmegen. Norway: K.-F. Amthor MD, Volvat Medisinske Senter, Oslo; T. Hauge MD, Fylkessjukehuset I Molde, Molde; I. Monstad MD, Hedmark Sentralsjukehus, Elverum; K. Nestvold MD, Sentralsykehuset I Akershus, Nordbyhagen; O. Rosjo MD, Nevrologgruppen, Oslo; L. J. Stovner MD, Regionsykehuset I Trondheim, Trondheim; S. B. Strandquist MD, Private Practice, Toensberg. Sweden: J. Albo MD, Lakarhuset Vallingby, Vallingby; S. Boes Hansen MD, Kronobergskliniken, Vaxjo; B. Borre MD, Neuro Kliniken, Helsingborg; L. Brattstrom MD, Lanssjukhuset, Neurologmottagningen, Kalmar; R.-M. Brinkeborn MD, Luthagsgarden, Uppsala; C. Dahlöf MD, Sociala Huset, Göteborg; L. Edvinsson MD, Medicinkliniken Universitetssjukhuset, Lund; I. Eilenberg MD, Limhamnslakargrupp, Tarnan, Malmo; Y. Hallstrom MD, St Gorans Sjukhus, Neurocentrum, Stockholm; A. Henriksson MD, Lakarhuset Hermelinen, Lulea; B. Jacobsson MD, Vardcentralen, Molkom; B. Janzon MD, Medicinskt Centrum Foretagsservice, Norrkoping; G. Johansson MD, Primarvarden, Finspang; R. Johansson MD, Sjukhuset I Kristinehamn, Kristinehamn; G. Malmqvist MD, Neurologsektionen, Helsingborg; E. Riman MD, Lundsbysjukhus Neurologiska Kliniken, Göteborg; O. Sydow MD, Neurologmottagningen Danderydssjukhuset, Danderyd.