
Abstract
Tumour necrosis factor-alpha (TNF-alpha) and interleukin-6 (IL-6) have recently been found to have a pain-mediating function in addition to their immunological, proinflammatory function. According to the hypothesis of neurovascular inflammation in migraine, these two cytokines could contribute to migraine pain generation. We analysed IL-6 and its soluble receptors sIL-6R and sgp130 as well as TNF-α and its soluble receptor sTNF-RI in 27 migraine patients and eight headache-free controls. Migraine patients tended to have less sTNF-RI (794 ± 158 pg/ml) than controls (945 ± 137 pg/ml). No differences in cytokine concentrations were observed. If TNF-α plays a role in migraine physiopathology, migraine patients may lack sufficient antagonistic sTNF-RI to neutralize hyperalgesic TNF-α during a migraine attack.
Keywords
Introduction
Some cytokines have recently been found to have a pain-mediating function in addition to their known immunological function. Tumour necrosis factor-alpha (TNF-α), for example, causes hyperalgesia, when administered centrally or peripherally (1, 2), and elicits ectopic activity in nociceptive primary afferent neurones (3). Similarly, interleukin-6 (IL-6) induces hyperalgesia (2, 4). Moreover, IL-6 knock-out mice exhibit reduced mechanoallodynia and hyperalgesia (5, 6).
Even though some previous studies have investigated the possible role of TNF-α or other cytokines in migraine physiopathology, no definite role was found (7–9).
The mechanisms leading to the typical headache in migraine are still not understood. In general, headache is mediated through the trigeminal nociceptive fibres of dural and meningeal vessels and meninges. However, the activation of brainstem structures lasts longer than the pain experience of migraine (10) and thus cannot explain the headache period. A centrally triggered neurovascular inflammation with isolated release of nonalgesic calcitonin gene-related peptide (CGRP), which is documented for humans during a migraine attack (11), would also not account for the painful sensation. Thus, the hypothesis of cytokines as possible pain mediators in neurovascular inflammation offers a potential mechanism for the generation of migraine pain.
Methods
To determine whether TNF-α and/or IL-6 are involved in the generation of migraine pain, we analysed TNF-α and its antagonistic soluble receptor sTNF-RI as well as IL-6 and its soluble receptors sIL-6R and sgp130 in serum samples of 27 migraine patients (25 women, two men; mean age 42.4 ± 11 (23–77) years) and eight healthy headache-free controls (six women, two men; mean age 36.4 ± 18 (20–74) years) using commercially available ELISA-Kits (R&D Systems). Patients were recruited in the headache clinic of the Department of Neurology; controls were recruited from medical and non-medical staff of the hospital. Nineteen women (mean age 44.0 ± 11 years) had a migraine without aura (defined by International Headache Society criteria), eight (six women, two men; mean age 36.5 ± 10 years) a migraine with aura. Patients with an analgesic-induced headache were not included. Blood samples were taken from six women with migraine without aura (age 36.8 ± 11 years) during a migraine attack. The mean duration of the attack was 2.1 days (range 5 h and 7 days). Four women with migraine without aura (mean age 36.5 ± 9.7 years) had taken non-steroidal analgesic drugs (NSAID) the week before blood sampling. Two migraine patients were taking β-blockers prophylactically. The last migraine attack had occurred a mean of 8.6 days prior to blood sampling. Controls were considered headache-free if they had no primary headache and no or less than four tension-type headaches per year.
Blood was drawn from the antecubital vein in the morning, centrifuged, and stored at −20°C until the assay. Exclusion criteria were an ongoing infection (documented with a blood cell count and C-reactive protein determination), pain of other origin, and autoimmune disorders in the patient history.
Statistical analysis was performed using
Results
The
Cytokine and soluble receptor levels in patients and controls

MANVOA: P = 0.52 compared with controls.
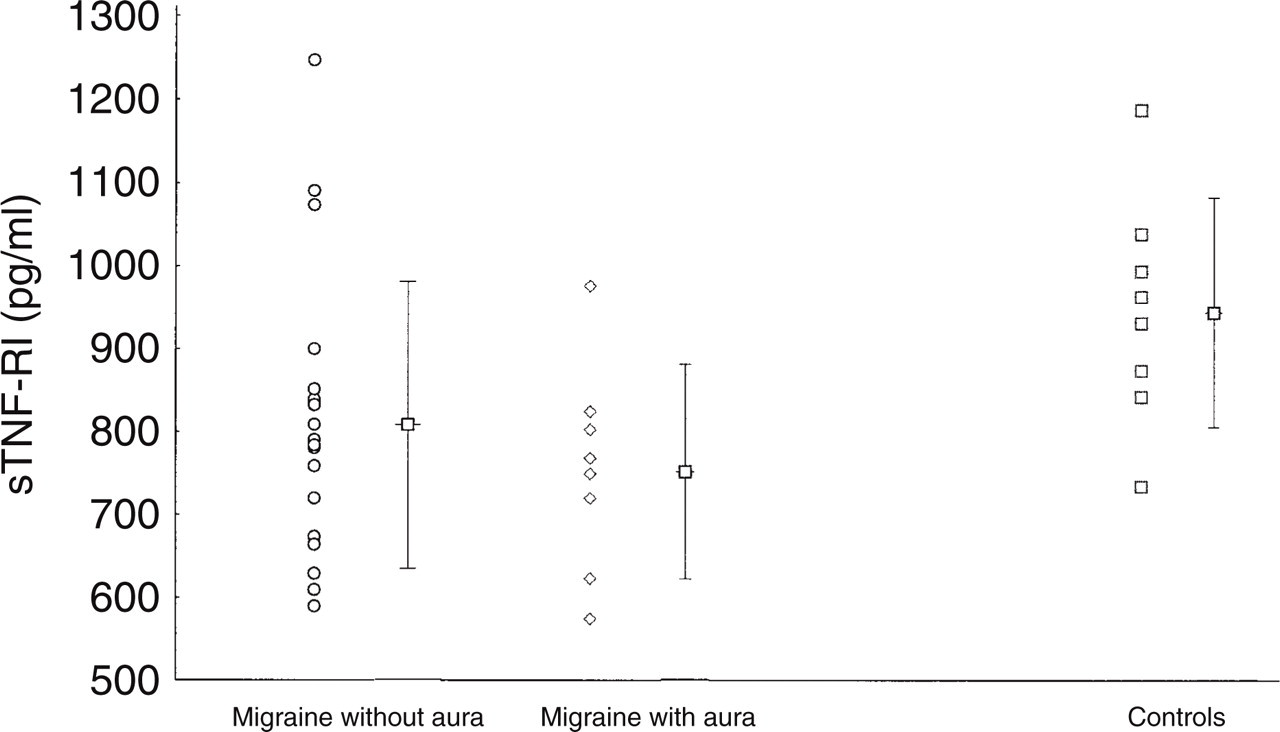
The scatterplot shows the values of sTNF-RI in migraine patients without aura, with aura, and in controls. The bar in the middle of the whisker represents the mean; the whisker, the upper and lower standard deviation.
In order to evaluate the possible effects of being in an actual migraine attack or taking NSAID on the cytokine levels, we also calculated the means for patients during an actual attack as well as for those who had taken NSAID the week before blood sampling. No differences could be found between the values of all cytokines and soluble receptors in patients’ samples taken during an attack compared with the values between attacks (see Table 1). In particular, there was no trend indicating an increase, except for a marginal increase in IL-6 concentration. The levels of the four patients who had taken analgesics the week before blood sampling did not differ from those of the other 23 patients, with the exception that their sTNF-RI values were lower (741.1 ± 101 pg/ml vs. 803.5 ± 166 pg/ml).
There was a significant correlation of sTNF-RI with age in migraine patients (r = 0.41; P < 0.05), which was not detected in controls (r =−0.34; NS). Correlations of other cytokines or soluble receptors with age were not observed in the patient or the control group.
Discussion
In this study we could not demonstrate that migraine patients differ in serum concentrations of TNF-α, IL-6, and their soluble receptors from controls. However, contrary to our expectations, migraine patients tended to have decreased sTNF-RI. Following the hypothesis of neurovascular inflammation, we would have expected increased sTNF-RI. Surprisingly, our finding is in line with another, also unexpected observation that a subgroup of menstrual migraineurs showed decreased TNF-α production (12).
If one assumes a role for TNF-α or a TNF-α deficiency in migraine physiopathology, one could hypothesize that less total sTNF-RI would result in less buffer capacity for locally released hyperalgesic TNF-α during a migraine attack, as sTNF-RI are antagonistic to TNF-α (13). Since sTNF-RI increase the half-life of TNF-α by a delayed excretion of bound TNF-α, a decrease of sTNF-RI would result in a reduced half-life, but greater effect of TNF-α and quicker and perhaps higher excretion of TNF-α (13). Accordingly, a limited local release of unbuffered TNF-α, for example, produced by endothelial cells that express CGRP receptors (14–16), would lead to hyperalgesia of meningeal vessels and result in a pulsatile headache that is typical for migraine.
Our failure to detect an elevation of TNF-α or IL-6 during a migraine could be due to the fact that the concentration of cytokines in serum samples does not reflect local processes that are characterized by limited, not systemic release of a short-lived cytokine. Such a limited release would be hidden by a systemic dilution effect, as is also known for CGRP. An elevation of CGRP during a migraine attack, for example, could be shown only if the samples were collected from the jugular vein (11), not the antecubital vein.
We can only speculate on the reasons for the decrease of sTNF-RI in migraine patients. TNF-α polymorphism could be one reason, since a common single base pair polymorphism in the promoter region of the TNF-α gene has been attributed to decreased TNF-α levels (17). However, a common TNF-α gene polymorphism was not related to migraine (18). It is not known if a TNF-α gene polymorphism also influences sTNF-RI levels. sTNF-RI levels themselves remain intra-individually stable over a lifetime despite considerable interindividual variation (13), a phenomenon that could be due to gene polymorphism. As the group sizes were small, an imbalanced distribution of a gene polymorphism unrelated to migraine could bias our results.
Another possible cause of a decrease of sTNF-RI could be the more or less regular intake of analgesics such as NSAID in migraine patients. Under such circumstances a decrease of sTNF-RI would be an epiphenomenon of migraine rather than a physiopathological condition. Nevertheless, it is difficult to evaluate the influence of NSAID on TNF and sTNF-RI production. Since the same signals are able to stimulate TNF-α and sTNF-RI release (19), a short-term decrease of sTNF-RI could be ascribed to NSAID intake, as was shown for TNF-α (20, 21). In contrast, daily treatment with NSAID (acetylsalicylic acid, diclofenac) resulted in an increase of TNF production during long-term observation (22, 23). Whether regular intake of NSAID can be regarded as equivalent to daily intake remains to be determined. Thus, although a decrease of sTNF-RI could be explained by recent NSAID medication, regular treatment with NSAID would explain the positive correlation of sTNF-RI with age only in migraine patients, not in controls.
In conclusion, it remains to be determined whether possible changes of TNF-α or its soluble receptor are related to migraine pathophysiology or simply an epiphenomenon. Our preliminary results do not favour a contributory role of IL-6 and its receptors to migraine pain generation. Future studies should focus on the analysis of TNF-α from samples taken during an attack and directly from the jugular vein in order to elucidate whether TNF-α or other cytokines are the missing link between neurovascular inflammation and migraine pain.
Footnotes
Acknowledgements
We wish to thank Mrs J. Benson for carefully reading the manuscript.