
Abstract
The aetiology of cluster headache is still not yet completely understood, but the potential relevance of genetic factors has been recognized during recent years. Nitric oxide (NO) plays a critical role in the regulation of vasodilation, neurotransmission, inflammation and many other events throughout the body. NO also appears to be an important mediator of vascular headache pathophysiology. In this study we have performed an association analysis of five polymorphic micro-satellite markers in the three different NO synthase (NOS) genes; nNOS (NOS1), iNOS (NOS2A) and eNOS (NOS3). Ninety-one cluster headache patients diagnosed according to International Headache Society criteria and 111 matched controls were studied. Phenotype and allele frequencies were similarly distributed in patients and controls except for an iNOS (NOS2A) pentanucleotide repeat allele which was significantly more common in controls. We observed a higher phenotype frequency of this allele in our control group compared with rates in control groups of other studies, whereas the frequency in our patients was similar to that in controls from previous reports. Thus, we conclude that it is unlikely that genetic variations within the NOS genes contribute greatly to cluster headache susceptibility.
Introduction
Cluster headache (CH) is a painful primary headache disorder in which heredity previously has been regarded as being of minor relevance. Several studies have reported a positive family history of CH (1–7). CH has been reported in five pairs of monozygotic twins in which all pairs were concordant (8–11). An increased risk of CH in first and second degree relatives has been shown in several studies (2, 4). This indicates that genetic factors are of importance for CH. Most cases are sporadic cases but about 4–5% of CH patients belong to families which might have a dominant inheritance (5, 12, 13). Most likely, several genes are of importance in sporadic cases, and this genetic predisposition together with other factors may ultimately give rise to the disorder. The molecular genetic background to CH has been studied in only a few investigations so far. Two recent reports ruled out the importance of the CACNA1A gene in a group of sporadic CH patients and in a family with CH (14, 15).
The aetiological and pathophysiological background in CH is not yet completely understood, but it is presumed to involve an activation of hypothalamic and trigeminovascular systems (16). Altered levels of neurohormonal substances suggest a central activation (17–22), and May (19) found support for this hypothesis in PET studies showing abnormal function in the hypothalamic grey matter.
Nitric oxide (NO) is a small messenger molecule, a potent short-lived and highly reactive free radical, with many different physiological effects (23, 24). NO plays a critical role in the regulation of vasodilation, neurotransmission and inflammation. NO is formed by the action of different isoforms of the enzyme nitric oxide synthase (NOS), which converts L-arginine to L-citrulline. Olesen first suggested NO as the common mediator in vascular headache (25–27). Already when Horton studied the CH disorder he observed that histamine induced headache attacks (28, 29). This effect is probably due to release of NO subsequent to activation of endogenous NOS. Ekbom first described how nitroglycerin produces classical CH attacks in active periods (30). This effect is probably mediated through release of NO which causes vasodilation. Increased NO plasma concentrations have been demonstrated in CH patients, both during the active phase and during remission (31). Another study has shown nitrite accumulation in peripheral blood mononuclear cells during bouts, but not during remission (32). NO has been proposed to be the final promoting factor in CH, since it is involved in both the central and the peripheral activation systems; NOS function in the hypothalamus can itself be modulated by NO-ergic routes, and NO is also, as mentioned above, involved in vasodilation. There might thus be a genetically determined predisposition for abnormal function of the NO system in CH patients (33).
There are three different forms of NOS encoded by three different genes: nNOS, iNOS and eNOS. The gene coding for nNOS (neuronal NOS, expressed by neurons and involved in neurotransmission and neuroendocrine functions) is called NOS1 and is located on chromosome 12q24.2. Inducible NOS (iNOS) is up-regulated by several cytokines (including IFN-γ, tumour necrosis factor-alpha and IL-1β) in a complex manner, and NO itself induces the production of several cytokines. IL-1β levels have been shown to be increased in CH patients (34). The gene encoding iNOS, NOS2A, is located on chromosome 17p13.1. The gene coding for eNOS (endothelial NOS, expressed by endothelial cells and implicated in vascular smooth muscle relaxation through a CGMP-mediated signal transduction pathway) is designated NOS3 and is located on chromosome 7q35-q36.
A linkage and association study investigating the possible involvement of a NOS3 polymorphism in migraine did not support a role for the NOS3 gene in migraine sufferers (35). Nor did an association study of a functionally relevant bi-allelic polymorphism in the promotor region of the NOS2A gene in migraineurs and controls support an association between this polymorphism and migraine (36). However, CH and migraine are not believed to have the same pathophysiological background and the three different NOS genes could possibly be of importance in CH aetiology. The aim of this study was to investigate the above mentioned NOS3 and NOS2A polymorphisms, in addition to a second polymorphism in the latter and two polymorphisms in the NOS1 gene in CH patients.
Materials and methods
Blood samples were collected from 91 CH patients (23 females and 68 males). In the patient group there were 92.3% episodic cases and 7.7% chronic cases. Four patients were familial cases. They all clearly fulfilled the International Headache Society (IHS) (37) criteria for CH, and all had attended our department. The control group consisted of 111 healthy individuals (35 females and 76 males). The controls were drawn from a larger group of controls (consisting of blood donors, healthy volunteers and spouses of patients) to match for age and gender. Both patients and controls were of Caucasian origin. For further demographic data see Table 1.
Demographic data of cluster headache (CH) patients and controls

The study was approved by the Local Ethics Committee.
DNA isolation
DNA was extracted from peripheral blood by a simple salting-out procedure (38).
Markers, primers and genotyping
For NOS1 (nNOS) we studied two polymorphic markers; a dinucleotide repeat here referred to as NOS1a and a trinucleotide repeat referred to as NOS1b. The primer sequences and polymerase chain reaction (PCR) conditions for NOS1a and for NOS1b have been described by Xu et al. (39). For NOS2A (iNOS) we studied two polymorphisms located in the promotor region of the gene. One of these markers is a biallelic AAAT/AAAAT repeat sequence extending from 756 to 716 bp 5′ of the main TATA-directed initiation site. For the biallelic repeat we used primer sequence and PCR conditions described by Bellamy et al. (40). The other NOS2A marker, a (CCTTT)n pentanucleotide repeat, has also been mapped to the promotor region. For the pentanucleotide repeat we used primer sequence and PCR conditions described by Xu et al. (41). For NOS3 (eNOS) we genotyped the polymorphic (CA)n repeat in intron 13 of the NOS3 gene using the primers and PCR conditions described by Nadaud et al. (42).
PCR for the different primers was performed in a 7-µl reaction including 50 m
Electrophoresis of amplified DNA fragments was performed on polyacrylamide gels using a ABI 377 DNA sequencer (Applied Biosystems Inc.; Foster City, CA, USA). Fragment sizes were determined by comparison with internal lane standards and the results were finally analysed using the GENESCAN/GENOTYPER software (version 1.1; PE Biosystems; Norwalk, CT, USA).
Statistical analysis
Phenotype frequencies for patients and controls were compared by Fisher's exact test. Probability values for 2 × k contingency tables, in which allele frequencies for patients and controls were compared, were calculated using a χ2 test. The level of significance was chosen as P < 0.05. P-values were not corrected for multiple comparisons. However, the large number of comparisons performed should be kept in mind when evaluating the deviations observed. We estimate that we have at least 80% power to detect a factor with a relative risk of 2.5 with a phenotype frequency of 12% in controls.
Results
For the dinucleotide NOS1a marker we found 13 alleles ranging in size from 186 to 214 base pairs (bp). Phenotype and allele frequencies for the NOS1a marker are shown in Table 2. For the NOS1b marker we found nine alleles ranging in size from 395 bp to 419 bp. Results for the NOS1b marker are shown in Table 3. As for the bi-allelic polymorphism in the NOS2A (iNOS) promotor region, we found two different alleles with sizes 313 bp and 317 bp with homozygosity for 313 as the most common genotype. Genotypes were distributed according to the Hardy–Weinberg equilibrium. Results are shown in Tables 4a and 4b. For the NOS2A pentanucleotide marker nine alleles were detected ranging in size from 179 bp to 219 bp, corresponding to 8–16 repeats. Results for the NOS2A (CCTTT)n marker are shown in Table 5. The carriage of NOS2A 194-bp allele was significantly more common in controls (P = 0.01, odds ratio (OR) = 0.45, 95% confidence interval (CI) = 0.25–0.82). For the dinucleotide marker in NOS3 20 alleles were found ranging in size from 146 bp to 184 p. Phenotype and allele frequencies of the NOS3 marker are shown in Table 6. We found that the distribution of phenotype and allele frequencies was similar in patients and controls except for the carriage of the NOS2A 194-bp allele, which was significantly more common in controls. Genotypes for the 194 marker allele were distributed according to the Hardy–Weinberg equilibrium in both groups.
Phenotype and allele frequencies of NOS1A alleles in cluster headache patients and controls

NS, Non-significant.
∗A probability value for a 2 × 13 contingency table was calculated using a χ ∗ test for comparison of global allele counts in patients and controls. The comparison was not significant.
†Refers to numbers of alleles.
Phenotype and allele frequencies of NOS1b alleles in cluster headache and controls

NS, Non-significant.
†A probability value for a 2 × 9 contingency table was calculated using a χ2 test for comparison of global allele counts in patients and controls. The comparison was not significant.
†Refers to numbers of alleles.
Genotype frequencies and allele frequencies of NOS2A insert alleles in cluster headache patients and controls
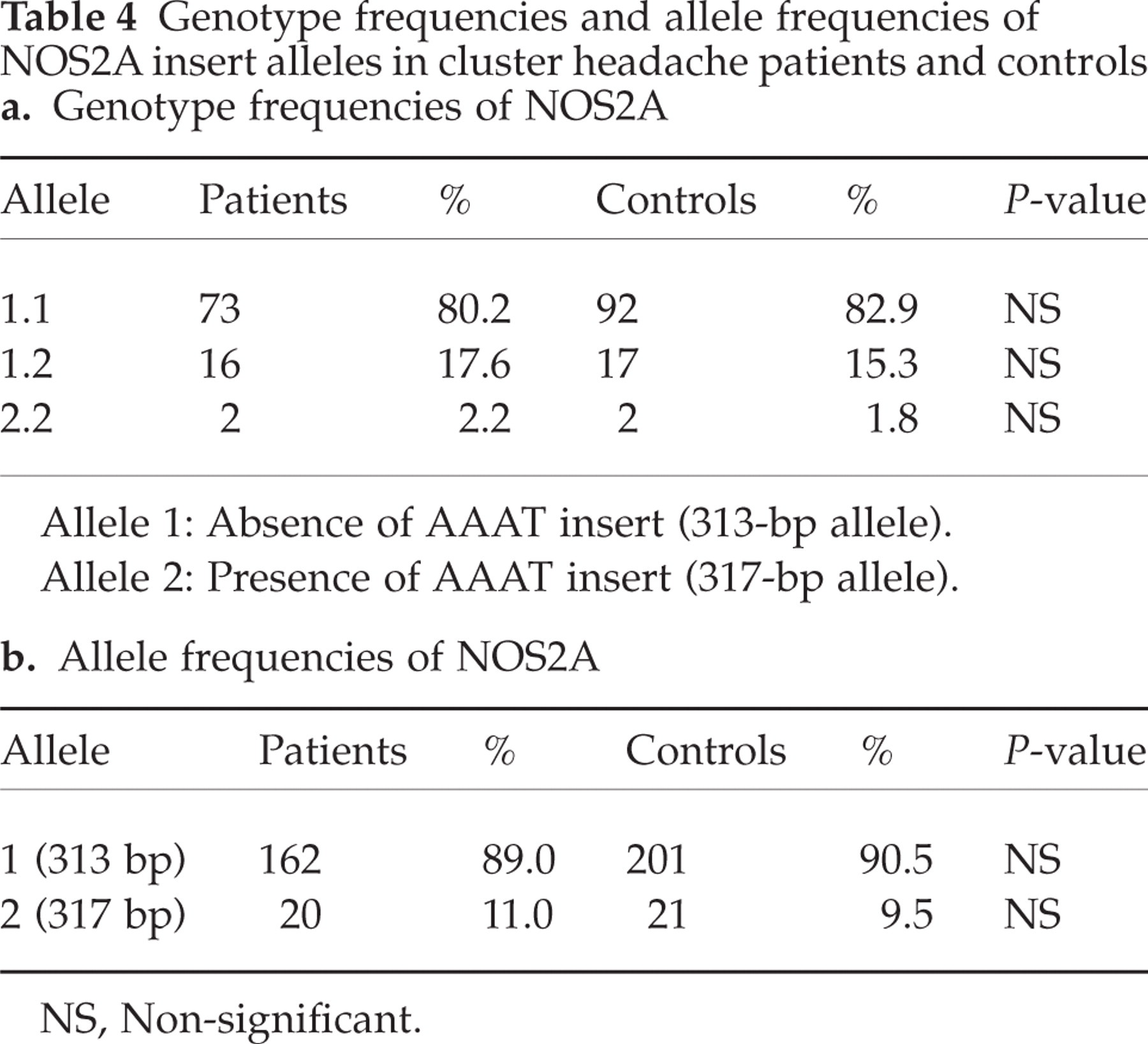
NS, Non-significant.
Phenotype and allele frequencies of NOS2A pentanucleotide repeat in cluster headache patients and controls

NS, Non-significant.
∗A probability value for a 2 × 9 contingency table was calculated using a χ† test for comparison of global allele counts in patients and controls. The comparison was not significant.
†Refers to numbers of alleles.
Phenotype and allele frequencies of the NOS3 (CA)n polymorphism in cluster headache patients and controls

NS, Non-significant.
∗A probability value for a 2 × 15 contingency table was calculated using a χ2 test for comparison of global allele counts in patients and controls. In this table counts for uncommon alleles were combined. The comparison was not significant.
†Refers to numbers of alleles.
The familial CH cases did not show any specific pattern, but the number was small, which makes it difficult to draw firm conclusions.
Discussion
Nitric oxide is believed to play a major role in CH aetiology. We have analysed five polymorphic markers of the three NOS genes assuming that they should be of importance in CH aetiology. However, this study offers no support for an association between these markers and CH.
We found a significant difference between the phenotype frequency of one allele (a 194-bp allele) of a pentanucleotide repeat in the promotor region of the NOS2A gene in patients and controls. Our patient group has about the same frequency of this allele as control groups in previously studied Caucasian populations (43–47), and thus we are hesitant as to the significance of this result, which is likely to be due to random variation. On the other hand, the control group in this present study was matched for age and gender, suggesting that the observed negative association may represent a true protective influence of this allele.
Only a few investigations regarding the molecular genetic background of CH have been performed, and there are as far as we know no other NOS gene studies in CH. Two studies have shown the lack of importance of the genes encoding eNOS and iNOS in migraine. Since migraine and CH are not supposed to share the same pathophysiology, but still have many similarities, we found it reasonable to study the importance of the NOS genes in CH.
We conclude that genetic variations of the three NOS genes do not contribute greatly to CH susceptibility. NO may still be the common mediator in CH, but it might be due to other mechanisms than genetic variations within the NOS genes. However, we cannot completely rule out that these genes have a small influence for the risk of CH.
Footnotes
Acknowledgements
This study was supported by grants from Glaxo Wellcome and the Karolinska Institute.