
Abstract
This prospective multicentre, double-blind, randomized, parallel-group, placebo-controlled trial evaluated the efficacy and safety of a single dose of eletriptan 20 mg, 40 mg and 80 mg in Japanese migraineurs. A total of 402 adult Japanese migraineurs were diagnosed using International Headache Society (IHS) criteria. At 2 h after a single dose, the headache response rates of eletriptan 20 mg, 40 mg, 80 mg and placebo were 64%, 67%, 76% and 51%, respectively, with all doses significantly superior to placebo (P< 0.05). Eletriptan had a statistically significant dose response for headache relief and pain-free response at 2 h post-dose (P = 0.0011 and P = 0.0291, respectively). Most all-causality adverse events were mild and there were no deaths or discontinuations. Saliva samples were used to assess serum eletriptan levels 2 h post-dose. Pharmaco-kinetic evaluations showed no clinically significant differences between Japanese and Western subjects. Eletriptan was shown to be efficacious, safe, and well tolerated in Japanese migraineurs.
Introduction
This study assessed the efficacy, safety and tolerability of a single dose of eletriptan 20 mg, 40 mg and 80 mg in Japanese migraineurs. Eletriptan is a potent, selective 5-HT1B/1D/1F agonist that has been shown to provide rapid and effective relief of migraine symptoms in Western populations (1–4).
Japan and Western countries both use International Headache Society (IHS) criteria to define and diagnose migraine and prescribe therapy (5, 6). The prevalence of migraine is similar in both Japan and the West (5). However, as the pathophysiology of migraine is unknown, we do not know whether therapies shown to be efficacious, safe and well tolerated in Western populations will be as beneficial in treating migraineurs in a population with many ethnic and cultural differences from the West.
In studies to date, eletriptan clinical data have been collected in populations with a composition typical for Europe and the United States. Rather than duplicate entire clinical programmes to determine safety and efficacy for specific ethnic groups, it is now possible to extrapolate or ‘bridge’ clinical data from one population to another. The International Conference on Harmonization (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use facilitates the registration of drugs and biologics among the European Union, Japan and the United States. The ICH has established the bridging concept to evaluate the impact of ethnic factors on the efficacy and safety of a drug or biological product using an established dosage and regimen (7). A bridging programme using the ICH strategy should accelerate the availability of the new and beneficial treatment for migraineurs in areas outside Europe and the United States, e.g. Japan, and should lessen the need to expose patients to unnecessary duplicate placebo-controlled trials.
To gain Japanese acceptance of eletriptan, a bridging study was designed to extrapolate Western efficacy and safety data to the Japanese patient population in accordance with ICH guidelines. This study was the first step in evaluating the clinical response of Japanese migraineurs to eletriptan therapy, and it is intended to be used in future comparisons with similarly conducted Western studies. In addition to clinical efficacy and safety comparisons, pharmacokinetic and pharmacodynamic evaluations must also be conducted to identify any clinically significant differences between Japanese and Western patients.
Cooper et al. have shown that a high pressure liquid chromatography (HPLC) assay of a patient's saliva can be used to quantify the amount of eletriptan and a metabolite, UK-135,800, which correlates with the patient's serum levels (8). This non-invasive procedure was used in this study because it allowed for a pharmacokinetic evaluation of eletriptan in an outpatient setting.
Methods
Ethical conduct
This study was conducted in accordance with the ethical principles originating from the Declaration of Helsinki (1989). The study protocol was reviewed and approved by appropriate independent ethics committees. Written informed consent was obtained from patients before they participated in the study. Investigators would remove a patient from the study if continued participation was believed to be detrimental based on clinically significant adverse events or laboratory abnormalities.
Patients
Randomized patients were males or females who were neither pregnant nor breast-feeding and ranged in age from 18 to 64 years. Based on their previous medical history, patients were entered if they reported at least one acute migraine attack with or without aura every 6 weeks. Patients had to have a headache of severe or moderate intensity prior to dosing and were not permitted to use any analgesics (including dimetotiazine mesylate) or antiemetics within 6 h before or within 2 h post-dosing. Furthermore, they could not have received ergotamine or an ergotamine-like agent within 24 h before or after dosing and/or if dosing occurred less than 6 h after the onset of their headache. Patients were excluded if they had severely limited gastrointestinal absorption (e.g. total gastrectomy). To ensure reliable ‘bridging’, inclusion and exclusion criteria were identical to those used in previous clinical studies.
The intent-to-treat (ITT) population consisted of patients with a baseline efficacy evaluation and at least one efficacy evaluation post-dose.
Design and procedure
This was a phase II, multicentre, double-blind, randomized, parallel-group, placebo-controlled, single-dose, single-attack study. Patients were adult migraineurs diagnosed according to IHS criteria (6). Oral doses of eletriptan 20 mg, 40 mg and 80 mg were compared with placebo. In addition, a 2-h post-dose pharmacokinetic evaluation was performed using saliva assays.
Assignment to treatment regimens was determined by a computer-generated pseudo-random code using a method of random permuted blocks. Pre-packaged drug kits were supplied using these randomization codes. The person who had the responsibility for randomization had sealed drug-blinding codes for use in case of an emergency.
Patients were screened to confirm compliance with inclusion and exclusion criteria and to give written informed consent. Medical history and demographic and diagnosis data were collected. Physical examination, laboratory safety tests, 12-lead electrocardiograms (ECGs), and blood pressure and pulse rate measurements were made, along with a urine pregnancy test for female patients when appropriate. Patients were instructed in how to assess their migraines and then record their assessments in a diary. They were also instructed in how to collect saliva samples for pharmacokinetic assays.
Patients returned home with the randomized drug. When they experienced a migraine headache of moderate to severe intensity, they completed the baseline evaluation section of their diary and subsequently took their study drug, as soon as possible within 6 h of the onset of their migraine. At 0.5, 1, 2, 4 and 24 h after dosing, they recorded their assessments of migraine headache severity and functional impairment. The presence or absence of nausea, vomiting, photophobia and phonophobia were also recorded in their diary.
At 2 h post-dose, patients collected a saliva sample in a specimen container provided by the investigator for a pharmacokinetic evaluation. They were also supplied with chewing gum to facilitate saliva production.
Patients recorded their global impression of efficacy of the study drug over 24 h post-dose and noted any adverse events or concomitant medications during the evaluation period. They also responded ‘yes’ or ‘no’ to the question: Next time you receive a therapeutic medication for migraine, would you choose this drug (the study drug) before another headache therapeutic medication?
If patients did not experience sufficient relief 2 h post-dose, rescue medication (except ergotamine or ergotamine-like drugs) was allowed. Medication used and the time it was taken were recorded in the diary.
Patients were followed for 7–14 days post-dose or after 12 weeks if no attack occurred. Unused medication and diaries were collected and reviewed to determine treatment compliance. The final study visit included a physical examination, blood pressure, pulse rate, ECG and laboratory tests. Final clarification of adverse events and of rescue and concomitant medications was also provided.
Measurement of efficacy
Patients assessed their response to treatment at baseline before dosing and at 0.5, 1, 2, 4 and 24 h post-dose in their diaries. The primary endpoint was headache response at 2 h post-dose for the ITT population, defined as a change from severe or moderate headache at baseline to a mild or absent headache post-dose. Pain-free response at 2 h post-dose was defined as a change from a severe or moderate headache at baseline to an absent headache post-dose and was a secondary endpoint. A sustained pain-free response was defined as achieving a pain-free response at 2-h post-dose, with no headache recurrence, and no use of rescue medication within 24 h after dosing. Other secondary endpoints were functional response 2 h post-dose, headache recurrence, use of rescue medication, global impression and subject acceptability. Headache recurrence was defined as a headache that responded with treatment by 2 h post-dose, but then worsened to become moderate or severe within 24 h post-dose. Associated symptoms (nausea, vomiting, photophobia and phonophobia) were also secondary endpoints, although the incidence of vomiting (<10%) was too low for any meaningful statistical comparisons to be made. Functional response was assessed as an improvement in a patient's ability to perform tasks as he or she normally would without a migraine headache.
Measurement of safety
Safety evaluation included adverse events reported by patients in their diaries by investigators. Investigators recorded any adverse events onto the appropriate case report form (CRF). Additionally, changes in blood pressure, pulse rate, 12-lead ECGs, and changes in laboratory tests performed at screening and at study follow-up visits, were also assessed. Adverse events were detected by indirect subject questioning, physical examination and from laboratory safety data and entries in subject diaries.
Statistical methods
Data for headache response at 2 h for each treatment group were analysed using logistic regression. Analysis of covariance was used to carry out each treatment comparison with placebo. The pairwise comparison of each eletriptan dose and placebo was based on a step-down procedure (9). Similar analyses were used for pain-free response and associated symptoms (i.e. nausea, vomiting, photophobia and phonophobia). Functional response and patients' global impression were analysed using logistic regression techniques for ordinal data. Time to rescue medication and time to headache recurrence were not analysed using formal statistical tests, but rather, were tabulated and plotted by treatment group.
Statistical Analysis System® (SAS) version 6.09 was employed for all statistical analyses.
Sample size calculation
The sample size was based on the requirement to demonstrate a statistically significant difference at the primary endpoint, headache response at 2-h post-dose, between eletriptan 40 mg and placebo after the first dose based on at least 80% power at the 5% two-tailed level of significance. Because the 80 mg dose has consistently shown efficacy that is equivalent or greater than the 40 mg dose, the study also had sufficient power to demonstrate a significant difference for eletriptan 80 mg vs. placebo. The statistical comparison between eletriptan 20 mg and placebo was planned, but the study was not powered for this comparison.
Results
Patient characteristics and disposition
This 12-week study was performed by 55 principal investigators at 54 sites. The mean number of patients per site was 7.4, with a range of 1–32 patients per site. The first patient was enrolled on 20 October 1998, and the last patient completed the study on 10 January 2000.
The mean age of participants ranged from 35 to 36 years, and the majority of patients were women (68–79%) across treatment groups. The majority of patients suffered moderate headache intensity at baseline. Patients' baseline demographic characteristics and baseline headache assessments are summarized in Tables 1 and 2, respectively. There were no major differences between treatment groups.
Baseline demographic characteristics

Baseline headache assessments
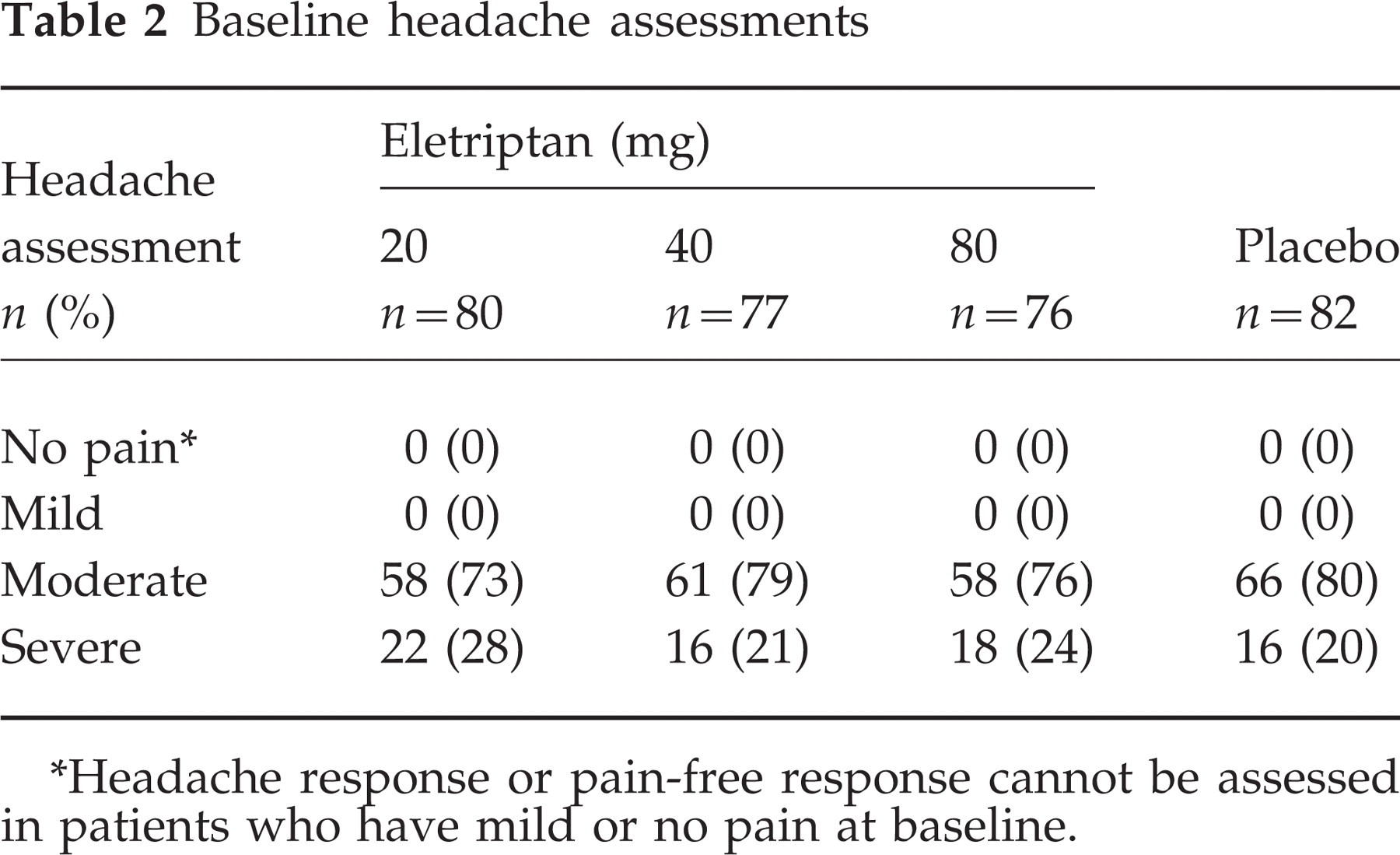
∗Headache response or pain-free response cannot be assessed in patients who have mild or no pain at baseline.
A total of 402 patients were randomized to study treatment. Of these, 76 did not take their assigned study medication because they either did not treat a headache attack or did not experience a migrainous headache of moderate or severe intensity during the study's 12-week duration. Another five patients were excluded from analysis because they did not return for follow-up. Two of these five patients took their study medication and did not experience any adverse events. The other three patients were lost to follow-up and investigators could not determine whether or not they took their study medication. The remaining 321 patients were included in the safety analysis; 318 in the laboratory data analysis, 307 in the efficacy evaluable population and 309 in the analysis of the study's primary efficacy endpoint of headache response at 2 h post-dose (Fig. 1).

Allocation of patients to treatment groups.
Efficacy
Relief of headache
At baseline, all treatment groups reported comparable incidences of moderate or severe headaches. The percentages of patients reporting a severe headache at baseline for the eletriptan 20 mg, 40 mg and 80 mg and placebo groups were 28%, 21%, 24% and 20%, respectively, and the percentages reporting a moderate headache at baseline were 73%, 79%, 76% and 80%, respectively.
Headache response rates at 2 h post-dose for the eletriptan 20 mg, 40 mg and 80 mg and placebo groups were 64%, 67%, 76% and 51%, respectively. There was a statistically significant dose response at 2 h post-dose (P=0.0011). All eletriptan doses were shown to be statistically significantly superior (P<0.05) to placebo (Fig. 2 and Table 3).
Headache and pain-free response at 2 h post-dose for the ITT population

∗Compared with placebo: P<0.05.
†Logistic regression.

Headache response at 2 h post-dose for the ITT population (P=0.0011 for dose response). ∗P<0.05 vs. placebo.
In addition, the pain-free response rates at 2 h post-dose for the eletriptan 20 mg, 40 mg and 80 mg and placebo groups were 24%, 22%, 28% and 13%, respectively. There was a significant dose response for pain-free response rate at 2 h post-dose (P=0.0291) (Fig. 3 and Table 3). Furthermore, pain-free response increased with higher doses of eletriptan, and all doses of eletriptan resulted in a greater increase in pain-free response than placebo at 2 h post-dose. Eletriptan 80 mg was shown to be statistically significantly superior to placebo (P=0.0129) (Fig. 3 and Table 3). The sustained pain-free response was 21%, 18%, 26% and 9% for the 20 mg, 40 mg, 80 mg and placebo groups.

Pain-free response from baseline to 24 h post-dose for the ITT population. Eletriptan 20 mg □, 40 mg ○, 80 mg •, placebo ▪. (P=0.0291 for dose response).
Improvement in functional impairment
Patients treated with eletriptan showed a greater dose-related increase in functional response (P=0.0160) at 2 h post-dose. At baseline, all treatment groups reported comparable percentages of patients with moderate and severe functional impairment. The percentages of patients in the eletriptan 20 mg, 40 mg and 80 mg and placebo groups reporting severe functional impairment at baseline were 25%, 27%, 24% and 28%, respectively, and the percentages reporting moderate functional impairment at baseline were 65%, 58%, 61% and 56%, respectively. The functional response rates at 2 h post-dose for the eletriptan 20 mg, 40 mg, 80 mg and placebo groups were 65%, 65%, 75% and 54%, respectively. Furthermore, patient function increased with higher doses of eletriptan, and all doses of eletriptan resulted in a greater increase in patient function than placebo at 2 h post-dose. Eletriptan 80 mg was shown to be statistically significantly superior to placebo (P=0.0125).
Headache recurrence
The headache recurrence rate for all eletriptan-treated patients was lower than the rate for patients treated with placebo (Table 4). However, a statistical analysis did not show a statistically significant dose response with eletriptan. Additionally, there were no statistically significant differences in headache recurrence between eletriptan and placebo. Statistical significance for a dose response with headache recurrence was not expected as the study was not designed to detect a difference in headache recurrence, but in headache response, the primary efficacy parameter.
Headache recurrence and use of rescue medication results by treatment group for the ITT population

∗Compared with placebo: P<0.01.
† n is the number of patients who had a response within two hours post-dose.
‡Logistic regression.
Use of rescue medication
Rescue medication was permitted in the event of insufficient response 2 h after the study drug was taken. The percentage of patients taking rescue medication was similar for all eletriptan-treated patients, and all of the eletriptan groups required less rescue medication than placebo-treated patients (Table 4). The percentages of patients in the eletriptan 20 mg, 40 mg, 80 mg and placebo groups who took rescue medication at least 2 h post-dose were 21%, 23%, 23% and 43%, respectively. All doses of eletriptan were statistically significantly superior to placebo (P<0.05). There also was a significant dose relationship (P=0.0242). Correspondingly, the median time to rescue medication was shortest for placebo-treated patients and longest for those who took eletriptan 20 mg (i.e. 4.5 h vs. 8.0 h, respectively). The median times to rescue medication for eletriptan 40 mg and 80 mg were 7.4 h and 6.0 h, respectively.
Relief of associated symptoms
Eletriptan was more effective than placebo in relieving associated migraine symptoms (Table 5). At baseline, all treatment groups reported comparable incidences of nausea, vomiting, photophobia and phonophobia. At 2 h post-dose, the eletriptan groups reported a lower percentage of associated symptoms compared with placebo.
Incidence of associated symptoms (nausea, vomiting, photophobia and phonophobia) reported at baseline and 2 h post-dose for the ITT population

Global impression
Patients reported that they preferred eletriptan to placebo for the relief of their headache. Patients' global impressions of eletriptan providing headache improvement 24 h after dosing were 82% for both eletriptan 40 mg and 80 mg, 73% for eletriptan 20 mg and 67% for placebo.
Tolerability and safety
Eletriptan was shown to be safe and well tolerated in Japanese patients. The analysis of safety and tolerability included all 321 patients treated. There were no deaths or discontinuations related to adverse events or clinical laboratory test abnormalities.
Reported adverse events were predominately mild in severity and transient in nature and did not require treatment. Adverse events reported by 3% or more of patients are summarized in Table 6 and include somnolence, nausea, asthenia, vomiting, malaise, thirst, back pain, paresthesia, dyspnea and vasodilatation.
Incidence of treatment-related adverse events experienced by ≥3% patients in each treatment group

Two serious adverse events (i.e. resulting in hospitalization) were reported and both patients recovered without sequelae. One patient reported an exacerbated vascular headache within 24 h after administration of eletriptan 80 mg that was considered to be study drug-related. The other serious adverse event (also a headache that required hospitalization) occurred 14 days after dosing (in the 80 mg group) and was considered by the investigator not to be study drug-related.
The incidence of abnormalities in clinical laboratory test results (e.g. creatine kinase, glucose, bilirubin) ranged between 28% and 36% of patients treated and was similar for both the eletriptan and placebo groups. However, none of these results was considered to be treatment-related.
There were few changes in ECG, blood pressure and pulse rate evaluations reported from baseline as compared with the end-of-study visit. The incidences of these changes were similar in all eletriptan and placebo treatment groups. ECG changes were reported in one patient treated with eletriptan 20 mg and four patients treated with placebo. An independent cardiologist did not consider any of these reports of ECG changes to be clinically significant. There were no reports of ECG changes in patients treated with either eletriptan 40 mg or 80 mg.
Pharmacokinetics
Based on established clinical methods, saliva eletriptan concentrations are believed to correspond with plasma eletriptan levels in Japanese patients (8). The saliva eletriptan levels increased with dose and were 5.62 ng/ml, 11.99 ng/ml and 28.6 ng/ml in patients taking 20 mg, 40 mg and 80 mg eletriptan, respectively. When comparing dose-normalized eletriptan saliva concentrations from two previous western studies with those obtained in this study, dose-normalized saliva eletriptan levels were found to be 13% lower in Japanese patients. The mean dose-normalized eletriptan saliva concentrations in the two Western studies were 14.46 ng/ml (mean ± 2 SE: 12.98, 15.94) (Pfizer Inc., data on file) and 14.24 ng/ml (mean ± 2 SE: 12.09, 16.39) (Pfizer Inc., data on file), respectively, compared with 12.53 ng/ml (mean ± 2 SE: 11.24, 13.81) in this study. However, this clinical finding was not considered to be clinically relevant.
Discussion
This study shows that eletriptan is an effective and safe treatment for Japanese patients suffering migraine attacks. All three doses of eletriptan were effective in relieving headache and associated symptoms by 2 h post-dose and helped to improve patients' functional abilities.
Headache response, pain-free response and functional response at 2 h post-dose all showed a statistically significant dose response (P<0.05). For headache response at 2-h post-dose, eletriptan 20 mg and 40 mg achieved 13% and 16% greater rates of pain relief than placebo, respectively. However, eletriptan 80 mg was shown to be the most effective dose by achieving a 9% greater response than eletriptan 40 mg and a 25% greater response than placebo for this same primary endpoint.
Although the placebo response was 51% in this study, this is within the guidelines established by the IHS for the development of migraine therapies (10). While the placebo response rate observed in this study is high, and the corresponding therapeutic gain low, it should be noted that eletriptan still achieved a statistically significant dose response for both 2 h headache response rate and 2 h pain-free rate. Furthermore, each dose offered significant improvement over placebo for 2 h headache response (Table 3), despite the high placebo rate. All of these findings are consistent with published findings in western patients (1), indicating that eletriptan is as effective in Japanese patients as in those in the West.
The lack of an observed dose response for headache recurrence as well as rescue medication usage can be attributed to the fact that this study was not powered for these analyses. Rather, the study was designed to determine the statistical difference for 2 h headache response. These findings are of interest and may warrant further investigation.
Eletriptan was shown to be safe and well tolerated in Japanese patients. The overall incidence and pattern of adverse events was similar to other triptans administered to Western patients in other clinical studies (11–14). Adverse events reported by 3% or more of Japanese patients included somnolence, nausea, asthenia, vomiting, malaise, thirst, back pain, paresthesia, dyspnea and vasodilatation. There were also no clinically relevant changes in laboratory assays, vital signs or ECG evaluations.
Eletriptan saliva levels in Japanese migraine patients were found to be slightly lower than saliva levels of similarly dosed western patients. However, this pharmacokinetic difference appears to have no clinically relevant impact on the efficacy, safety or tolerability of eletriptan in Japanese patient populations.
Eletriptan was shown to be effective, safe and well tolerated in Japanese migraineurs at doses of 20 mg, 40 mg and 80 mg. In addition, the pharmacokinetic evaluation in this study showed no clinically significant differences between Japanese and Western patients. This would confirm that data from Europe and the United States of America regarding treatment with eletriptan can be applied to medical practice in Japan.
Footnotes
Acknowledgement
This work was supported by a grant from Pfizer Ltd.
Eletriptan Steering Committee members
Keio University: Yasuo Fukuuchi. Kitasato University: Fumihiko Sakai. Tokyo Women's Medical University: Makoto Iwata. Hokkaido University: Kunio Tashiro. Tohoku University: Yasuto Itoyama. Juntendo University: Toyo Miyazaki. Showa University: Kiyoshi Matsumoto. Nagoya University: Gen Sobue. Yamaguchi University: Mitsunori Morimatsu. Kyushu University: Jun-ichi Kira.