
Abstract
The mechanism for headache in patients with acute ischaemic stroke are not completely understood. We analysed the relationship between headache and the early worsening of neurological symptoms in patients with acute ischaemic stroke, and we studied the possible biochemical mechanisms implicated. Headache at the onset of ischaemic stroke predicted progression with a sensitivity, specificity, and positive predictive value of 56%, 99%, and 98%, respectively. CSF concentrations of glutamate, Interleukin-6, and NO-m were significantly greater in patients with progressing stroke than in patients with nonprogressing stroke, and these biochemical markers were also significantly higher in patients with headache than in those without headache. Results of this study suggest that headache at the onset of ischaemic stroke is an independent predictor of neurological worsening and we hypothesize that headache might be a surrogate marker of the molecular mechanisms involved in neurological worsening after acute stroke.
Introduction
Headache is present in 10% to 39% of patients during the acute phase of cerebral infarction (1–5). The mechanisms for headache in patients with ischaemic stroke are not completely understood (5). Prevailing views include dilation of collaterals, focal distension of the artery, local ischaemia of the arterial muscle, and irritation of the pain-sensitive arterial wall by atheroma (6). Serotonin and other vasoactive peptides released from the junctional elements of the trigeminal vascular system may also play a role (7). Recently, excitatory amino acids (EAAs) have been related to the pathophysiology of headache that occurs at the onset of stroke (8).
In acute stroke, excitotoxicity (9, 10), inflammatory reactions (11), and nitric oxide generation (NO) (12) are the biochemical mechanisms underlying early clinical worsening that occurs in 26–34% of patients with acute ischaemic stroke (10, 13–19). The rise of neuroexcitatory amino acids in plasma and CSF in patients with headache during the acute phase of cerebrovascular diseases (8), supports the hypothesis that headache at the onset of ischaemic stroke could be a good predictor of neurological deterioration.
In this study we analysed the relationship between headache and the early worsening of neurological symptoms in patients with acute cerebral ischaemia, and we studied the possible biochemical mechanisms implicated.
Patients and methods
We studied 241 patients with a first-ever acute ischaemic stroke. The patients were selected from a larger cohort of 271 consecutive patients with hemispheric infarction admitted within the first 24 h duration from symptoms onset. The characteristics of the group and inclusion criteria for the whole series of patients have already been published (9, 10). In summary all patients had a persistent focal neurological deficit and absence of mass effect or cerebral haemorrhage on the cranial CT carried out before inclusion. The presence of headache at stroke onset was particularly evaluated in these patients. For the purposes of this study we excluded 30 patients in whom headache was not recorded due to communication difficulties. The total number of patients were included in a prospective clinical investigation conducted between October 1992 and December 1996 to analyse clinical, biochemical, and radiological factors related to progressing stroke. The protocol was approved by the Ethics Committee of Hospital Clínico Universitario de Santiago de Compostela, and informed consent was given by the patients themselves, or by their relatives.
On admission, blood pressure and body temperature were recorded and blood chemistry studies, chest X-ray, ECG and nonenhanced cranial CT were performed. Stroke subtype was classified according to the TOAST definitions (20). We did not include patients with transient ischaemic attack. Stroke severity was assessed by a neurologist immediately after these tests with the Canadian Stroke Scale (CSS). The CSS measures level of consciousness (alert, 3; drowsy, 1.5), speech (normal, 1; expressive deficit, 0.5; receptive deficit, 0), orientation (orientated, 1; disoriented or non applicable, 0), facial paresis (none, 0.5; present, 0), weakness in arm, hand, and leg (none, 1.5; mild, 1; significant, 0.5; total, 0; scored individually for each item), on a total score from 1.5 (maximal deficit) to 10 (absence of deficit) (21). Following already published criteria (22), progressing stroke was diagnosed when the CSS score decreased 1 or more points between admission and 48 h. This difference represents the change with the highest sensitivity, retaining good specificity (23).
All the CT examinations were carried out with a CT System 3000 plus (GEC) scanner with a 512×512 display. Early signs of infarction were carefully evaluated during the first examination; these signs included hypodensity consistent with the clinical picture, and the indirect signs of cerebral infarction like obscuration of the lenticular nucleus, obscuration of the cortex, and mass effect on the cortical sulci and structures of the median line. On days 4–7 after clinical onset, a second nonenhanced brain CT was performed to asses the volume (in cubic centimetres) of the infarction, according to the formula 0.5×a×b×c, where a and b represented the largest perpendicular diameters measured on CT scan and c the slice thickness. Based on the extension and topography of the infarct on CT (24), the patients were grouped as follows: total anterior circulation infarcts (TACI), partial anterior circulation infarctions (PACI), lacunar infarcts (LACI) and posterior circulation infarcts (POCI).
Blood and CSF samples were drawn during the painful period in those patients with headache, and in all patiens without headache; the interval between inclusion and taking the CSF was 2.6±3.7 h. Blood was obtained by venipuncture and collected in crystal tubes containing EDTA-K3. Plasma was prepared by centrifugation (3000×g for 15 min) and stored at −70°C. A lumbar puncture was performed if there were no signs of mass effect on CT. CSF samples were prepared by centrifugation (2000×g for 10 min) and immediately stored at −80°C.
Quantification of glutamate was performed by HPLC following the method described elsewhere (8–10). NO generation was calculated by quantifying the final products of its reactions, nitrates and nitrites, by a colourimetric assay (Cayman Chemical Co, Ann Arbor, MI), following the method described in a previous work (12). IL-6 concentrations in CSF samples were measured with commercially available quantitative sandwich enzyme-linked immunoadsorbent assay (Quantikine) kits obtained from R & D System (Minneapolis, MN, USA), as previously described (11). These measurements were made by technicians from an independent laboratory who were unaware of the clinical outcome and neuroimaging findings. As our original purpose was to determine EAAs, further biochemical markers were only investigated in patients from whom we had obtained and stored (frozen) CSF samples for possible future laboratory determinations. Availability of samples relied exclusively on the amount of CSF obtained at the time of inclusion and not on other clinical and biochemical factors.
Statistical analysis
Continuous variables were expressed as mean±one standard deviation, or as median [quartiles], when they were not normally distributed. Categorical data were analysed with the χ2 test with the Fisher correction when necessary. Comparison between two groups of continuous variables was performed by using the Mann–Whitney test.
The importance of headache for subsequent early neurological deterioration (0=no, 1=yes), was assessed by logistic regression analysis based on the maximum likelihood ratio. A model was fitted to assess the adjusted OR of progressing stroke for those baseline clinical and radiological variables which reached a P-value <0.15 in the bivariate analysis. The resulting model was further adjusted by the ultimate infarct volume. In order to test the interaction between headache and the CSF biochemical markers on neurological deterioration, we calculated the odds ratios for headache in three separate logistic models with a further adjustment for glutamate, IL-6, and NO-m, respectively.
Results
Of the 241 patients studied, 73 (30.6%) complained of headache at onset of stroke. The main characteristics of the studied population are shown in Table 1. Patients who experienced headache had longer delay to hospital admission, higher stroke severity, systolic blood pressure, frequency of early CT signs of infarction on admission, and higher infarct volume than those who did not have headache. The frequency of posterior territory infarctions was similar between the two groups. Concentrations of glutamate, IL-6, and NO metabolites (NO-m) in CSF were significantly higher in patients with headache than in those without headache.
Clinical characteristics and biochemical markers in cerebrospinal fluid of patients with and without headache at stroke onset

CSS, Canadian Stroke Scale; CSF, cerebrospinal fluid.
∗Glutamate was determined in 67 patients with headache and in 145 patients without headache; IL-6 was determined in 30 patients with headache and in 50 patients without headache; NO-m was determined in 37 patients with headache and in 62 patients without headache.
Progressing stroke occurred in 84% and 20% of patients with and without headache, respectively. Patients who showed stroke progression had longer delay from stroke onset to hospital admission, higher stroke severity, systolic and diastolic blood pressure, stronger acute-phase response (increased serum glucose and fibrinogen concentrations, and elevated body temperature), and ultimate infarct volume in comparison with those who had not stroke progression (Table 2). The frequency of headache on admission was significantly higher in the group with subsequent neurological worsening. Headache at the onset of ischaemic stroke predicted progression with a sensitivity, specificity, and positive predictive value of 56%, 99% and 98%, respectively.
Clinical characteristics and biochemical markers in cerebrospinal fluid of patients with and without progressing stroke
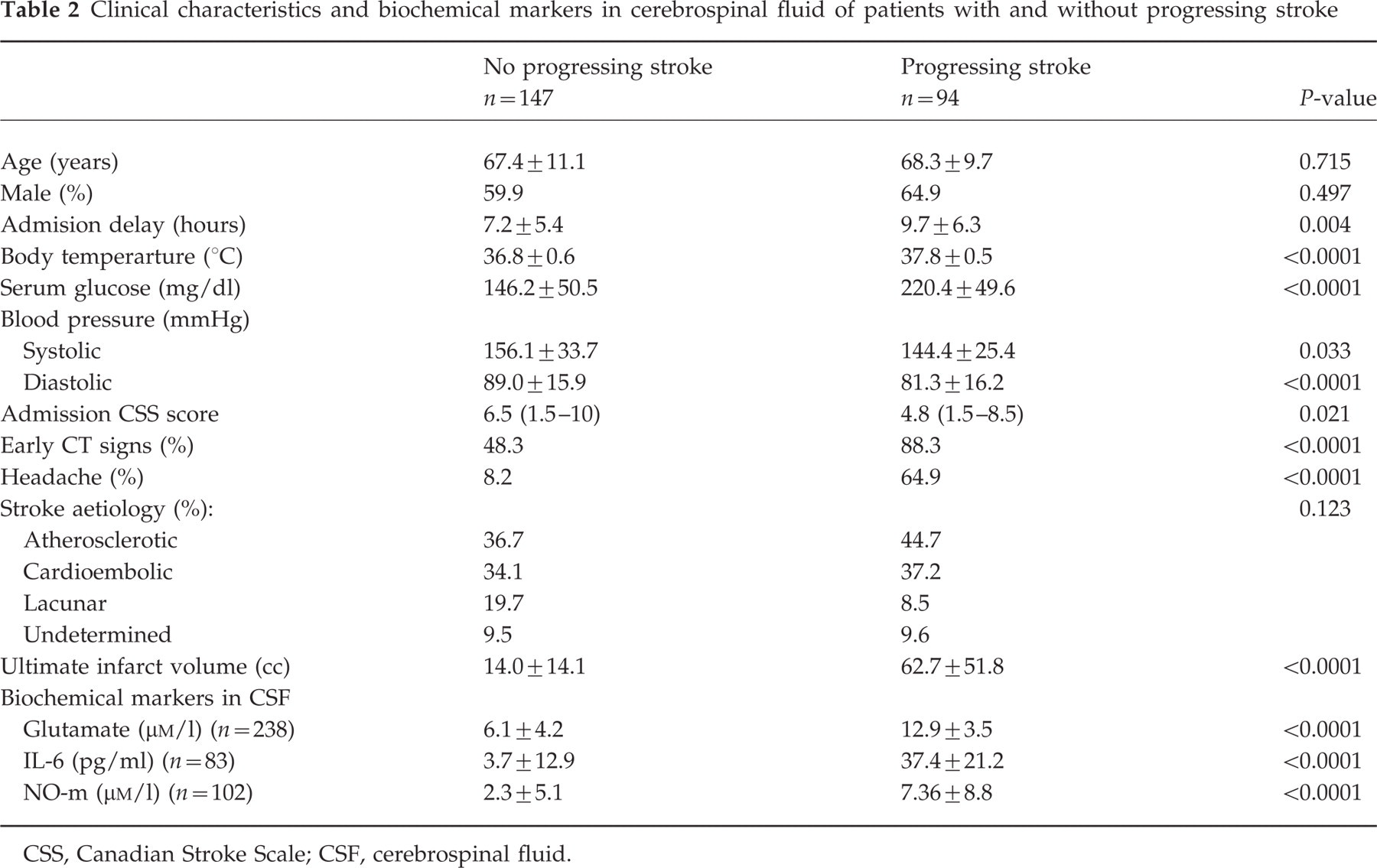
CSS, Canadian Stroke Scale; CSF, cerebrospinal fluid.
CSF concentrations of glutamate, IL-6, and NO-m were significantly greater in patients with progressing stroke than in patients with non-progressing stroke (Table 2).
In the logistic regression analysis, headache was an independent factor (OR 16.01, 95% CI 5.40–47.48, P<0.0001) associated with early neurological deterioration, after adjustment for the baseline related factors in the bivariate analyses (Table 3). When ultimate infarct volume was included as a covariate in a further analysis, it was not selected by the model as an independent predictor of progressing stroke (data not shown). The effect of headache remained significant after further adjustment for glutamate and NO-m, but was markedly reduced by the IL-6 effect (Table 4).
Adjusted odds ratio of early neurological deterioration for baseline clinical and CT variables

Adjusted odds ratio of early neurological deterioration for headache without and with a further adjustment for biochemical markers

(∗)Covariates include body temperature, serum glucose, systolic blood pressure, and early CT signs of infarction, plus glutamate, IL-6 or NO-m in models 1, 2 and 3, respectively.
Discussion
In the last few years, several risk factors of neurological deterioration in acute cerebral infarcts have been described, including clinical, neuroimaging, ultrasonographic and biochemical parameters; some are associated with an increased risk of early neurological deterioration (progressing infarct), while others are mainly related to the development of delayed worsening (25). Clinical and neuroradiological factors predict worsening in less than 65% of cases (19). In contrast, glutamate concentration in plasma higher than 200 µmol/l is the more powerful marker of progressing stroke, able to correctly predict 97% of cases (10), but is time consuming to determine. Given the known association between headache and elevated glutamate levels in CSF (8) we studied the prognostic value of this symptom in stroke progression. The present findings demonstrate that headache at the onset of ischaemic stroke is an independent predictor of neurological worsening, with high specificity and positive predictive value.
The cause of headache in ischaemic cerebrovascular disease is poorly understood. A simple explanation could be a direct effect of the embolus or thrombus on the vascular wall, with or without reflex dilation. However, this theory does not explain differences in the incidence of headache between stroke subtypes or stroke location (26). In some persons the dura in occipital part is supplied by the posterior cerebral artery and not by the external carotid artery. This could explain, why patients with posterior cerebral artery strokes suffered more often from headache. However, in the current study, the frequency of posterior territory infarctions was similar between the two groups of patients with and without headache.
Headache in cerebrovascular disorders has been mostly related to electrochemical stimulation of the trigeminovascular afferent system, since the pial nerve fibers are of trigeminal origin, and the perivascular fibers contain vasoactive neuropeptides (e.g. calcitonin gene-related peptide, substance P) which, upon release into the vessel wall, increase blood vascular permeability (27, 28). What triggers the trigeminovascular system in ischaemic stroke remains speculative, but a disturbance in blood flow or dilation of the pain-sensitive collateral vasculature has been proposed (29). Depolarization of the trigeminal system results in an afferent discharge perceived as pain, and in a release of vasoactive neuropeptides producing subsequent vasodilation (30). Platelet aggregation at the ischaemic site, and possibly at systemic circulation, provides another potential explanation for triggering the trigeminovascular pathway (31). The products of this release reaction include serotonin and prostaglandins.
Biochemical changes have also been implicated in the pathogenesis of headache in ischaemic stroke. Glutamate concentrations in the CSF of patients with headache at onset of cerebral infarction are significantly higher than those of patients without headache, suggesting glutamate participation in the genesis of pain (8). The release of EAAs might not be the cause but rather an epiphenomenom of pain. Although symptomatic spreading depression, and consequently glutamate release may be related to the cortical lesion (32), glutamate increase is more likely associated with the hypoperfusion that develops in the core of infarction and in the surrounding ischaemic penumbral area (33). The present study replicates preliminary results in a larger series of patients (8) and, interestingly, describes new molecular markers, such as IL-6 and NO metabolites, associated with headache in acute ischaemic stroke.
Increased levels of all these biochemical compounds have been associated with progressing stroke in clinical investigations (10–12), therefore we hypothesize that headache might be a surrogate marker of the molecular mechanisms involved in neurological worsening after acute stroke. This fact could be particularly evident with respect to the inflammatory response that accounts for neurological deterioration (11), because the effect of headache on progressing stroke was markedly reduced after adjustment for IL-6 concentrations.
Although serum cytokines studies in headache patients have yielded conflicting results, increased plasma levels of IL-5 and IL-4 have been described in 84.3% and 37.5% of patients with migraine without aura (34). These results support the growing arguments of an immunoallergic mechanism in the pathophysiology of migraine. Other studies have found a significant decrease in the expression of ICAM-1 during migraine crisis experimentally induced by the administration of isosorbide dinitrate (a nitric oxide donor) and in spontaneous migraine attacks (34). It has been hypothesized that ICAM-1 down-regulation may be a ‘defence’ mechanism to inhibit during a migraine attack the critical step of transendotelial migration into the cerebral tissues of activated leucocytes, as proposed in the ‘sterile inflammation’ hypothesis (35).
Although our results suggest that the association of headache with subsequent progressing stroke may also be related to high glutamate and NO generation, other potential mechanisms may be involved since the odds of neurological deterioration for headache were not substantially modified after adjustment for glutamate and NO-m concentrations (Table 4). Headache could be an epiphenomenon of brain oedema and intracranial hypertension leading to neurological deterioration, however, its prognostic effect was independent of the early CT signs of infarction and ultimate infarct volume.
In conclusion, headache is a strong predictor of early neurological deterioration in acute ischaemic stroke. Excitotoxic and inflammatory mechanisms may play a important role in the pathophysiology of headache that accompany cerebral infarction.