
Abstract
Profound nitric oxide release associated with cortical spreading depression (SD), has been implicated in stroke, traumatic brain injury and migraine pathophysiology. SB-220453 represents a mechanistically novel, well-tolerated class of compounds which may have therapeutic potential in the treatment of conditions associated with neuronal hyperexcitability and inflammation. The aim of the present study was to investigate the effects of SB-220453 on the nitric oxide (NO) release associated with SD in the anaesthetized cat. In vehicle treated animals, KCl application for 6 min to the cortical suface produced repeated changes in extracellular direct current field potential with associated NO release. This activity was sustained for a median duration of 55 min (25–75% range, 32–59 min) and 59 min (25–75% range, 34–59 min), respectively. SB-220453 (1, 3 and 10 mg/kg i.p.) produced a dose-related inhibition of this activity and at the highest dose tested, the median duration of changes in extracellular field potential and NO release were reduced to 4 min (25–75% range, 4–5min) and 5 min (25–75% range, 5–5min), respectively. No effect was observed on basal systemic haemodynamic parameters or resting cerebral laser Doppler blood flux at any of the doses of SB-220453 tested. SB-220453 therefore represents a novel compound to assess the potential benefit of inhibiting SD associated nitric oxide release in neurological disease.
Introduction
Current hypotheses of migraine pathophysiology suggest that a hyperexcitable brain is the basis for elicitation of migraine attacks [1, 2, 3]. In migraineurs the interictal excitability of the brain is thought to be defined in part by genetic factors. These may include channelopathies such as missense mutations in P/Q type calcium channel CACNL1A4, observed in unrelated familial hemiplegic migraine families [4], defective mitochondrial meta-bolism and abnormal magnesium metabolism [5]. Superimposed upon this genetically defined baseline are environmental factors, e.g. stress, diet, etc., which serve to modulate this set point.
Despite the multifactorial aetiology of migraine, it is proposed that the amplification of an initial triggering event is produced by a common mechanism. This is believed to be similar to cortical spreading depression (SD) [6–10] or spreading depression of Leao observed in experimental animal models [11, 12]. As such, it provides a mechanism by which a migraine trigger which is metabolic in origin can evoke secondary repetitive activation of the trigeminovascular system (reviewed by Read and Parsons) [13] (Fig. 1).
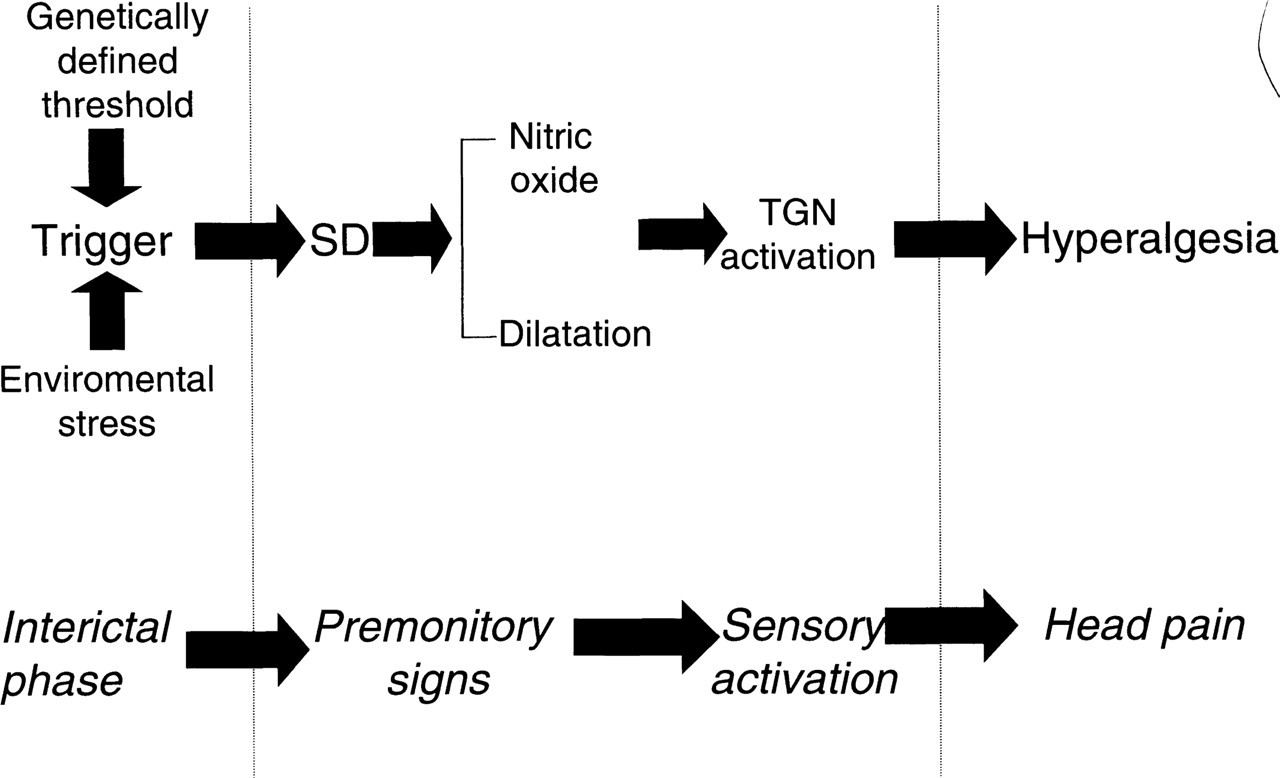
During the profound depolarization induced by SD, local nitric oxide concentrations fluctuate in anaesthetized cats and mediate dynamic coupling of the cerebrovasculature to the increased metabolic demand of SD [14, 15]. However, whilst the role of cortical NO release in cats may in part be vasomotor, local NO concentrations are often not directly correlated to changes in regional cerebral blood flux or pial artery diameter [16]. Additionally, in rats, whilst SD also induces marked increases in local concentrations of NO, there is little if no role in coupling the metabolic demand of SD to changes in local flux [17, 18, 19]. Therefore, it is likely that other ‘non-vasomotor’ actions of released NO may be expressed, which may include modulation of ion channel activity [20, 21] and direct stimulation of nociceptive afferents [22, 23].
The role of NO in mediation of migraine headache has been extensively studied by Olesen et al. [22] and Iversen et al. [24]. Intravenous infusion of the NO donor, glyceryl trinitrate (GTN) into volunteers with no history of migraine and into migraineurs produced a dose dependent and immediate headache. The mean summed immediate headache score in migraine patients was significantly greater than in normal patients, and was followed by a delayed migraine attack suggesting an exaggerated GTN-induced response in migraineurs and implicating a role for NO release [22, 24]. Indeed, in preliminary clinical studies of the non-specific NOS inhibitor L-NG methyl-arginine hydrochloride, a marked amelioration of headache and associated migraine symptoms has been observed [25]. Collectively these results may indicate that an ongoing release of NO occurs during the headache phase of a migraine attack.
SB-220543 is a mechanistically novel, benzoylamino benzopyran, which binds selectively and with high affinity to a unique, stereoselective CNS binding site [26]. SB-220453 is highly effective in inhibiting abnormal levels of neuronal excitability with pronounced activity in rodent seizure and trigeminal nerve stimulated neurogenic plasma protein extravasation models [26]. These observations provided precedent for assessment of SB-220453 in animal models of propagating neuronal hyperexcitability, i.e. SD [27]. Hence, the proposed role of SD in coupling a multifactorial migraine trigger to sensitization of nociceptive afferents and the pivotal role of NO mediation in this process. This study investigates the effect of SB-220453 on SD induced nitric oxide release and associated pial artery hyperaemia in the anaesthetized cat.
Methods
Surgical preparation
All experimental procedures were approved by a local ethics committee and conform to the United Kingdom (UK) Animals (Scientific Procedures) Act 1986. Anaesthesia was induced in overnight fasted male cats (2.6–4.7 kg), using 5% halothane and maintained with α-chloralose(100 mg/kg i.v.). Following a right femoral vein and artery cannulation, for supplemental anaesthetic administration and diastolic, systolic blood pressure-derived heart rate recordings, respectively, a tracheotomy was performed and the animal mechanically ventilated to maintain blood/gas parameters at physiological levels (Ciba Corning 288 blood gas analyser, Dagenham, UK).
Animals were positioned in a stereotaxic frame and a cranial well formed above the superior aspect of the skull. The left suprasylvian gyrus of the parietal lobe was exposed by craniotomy and durectomy and the well area filled with mineral oil(37°C) (Sigma Chemical Co., St Louis, USA). Body temperature was monitored and maintained at 37°C with a homeothermic blanket control unit (Harvard Instruments Ltd, Kent, UK).
Extracellular field potential recordings
Extracellular direct current (d.c.) field potential recordings were made using Teflon insulated, 0.125 mm diameter Ag/AgCl microelectrodes (Advent Research Materials, UK. The recording electrode was placed on the cortex parenchyma, proximal to the pial artery of interest and referenced to the left limb semitendinous muscle. Electrodes were connected to a field effect input stage and amplified using a Neurolog system NL834 4-channel d.c. preamplifier. Signals were low pass filtered and passed to a data acquisition system for off-line analysis of changes in d.c. amplitude (mV) and total duration of activity (min) (Biopac MP100, Linton Instruments, UK).
Electrochemical NO detection
As in previously published studies [15–17, 28], electrochemical NO measurements were made using a Pt/Ir alloy working electrode coated with an NO selective membrane and referenced to a carbon fibre counterelectrode (Model NO-501, NO monitoring device (Inter Medical, UK Ltd)). The recording electrode was located on the cortical surface of the suprasylvian gyrus, whilst the counterelectrode was situated on the parietal bone. The electrodes were calibrated at 37°C, in vitro using aqueous solutions of S-Nitroso-N-Acetyl-d-Penicillamine (SNAP) in 0.1
Regional cerebral blood flux recordings
Estimations of regional cerebral blood flux were determined utilizing laser Doppler flowmetry (rCBFLDF) (Oxford Optronics, UK). Following correction for extraneous light effects, the probe was positioned proximal to the nitric oxide electrode and directly above the pial artery of interest. rCBFLDF is expressed as percentage change from mean pre-treatment value minus the residual laser Doppler signal. The residual laser Doppler signal was taken as the mean value, determined over 5 min, following euthanasia of the animal.
Experimental protocol
For illustration of recording set up see Read et al. [28]. Four separate treatment groups were utilized; vehicle (labrasol, saturated polyglycolysed C8-C10 glycerides), 1 ml/kg (Alfa Chemicals Ltd, UK) (n = 7), SB-220453, cis-(–) -6-Acetyl-4S-(3-chloro-4-fluorobenzoylamino)-3,4-di- hydro-2,2-dimethyl-2H-1-benzopyran-3S-ol (Medicinal Chemistry, Smithkline Beecham Pharmaceuticals, Harlow, UK), 1 mg/kg (n = 4), 3 mg/kg (n = 5) and 10 mg/kg (n = 6). All treatments were administered 90 min prior to SD induction via the intraperitoneal (i.p.) route. SD activity was induced as in previous publications [14, 26], using KCl solid (30 mg) placed directly onto the cortical parenchyma in a region 1 cm distant from the recording site. After a period of 6 min the KCl was removed from the brain surface and recordings were maintained for a further 54 min. Arterial blood samples were removed for determination of blood acid/base status immediately prior to completion of the experiment. Animals were then euthanased by pentobarbitone overdose (300 mg/kg i.v.).
Data analysis
Effects on baseline variables
The effects of SB-220453 or vehicle effects on diastolic and systolic blood pressure, heart rate and rCBFLDF are presented as predose, sampled 1 min before vehicle or drug administration and post-dose, sampled immediately prior to induction of SD.
SD-induced changes
During SD, effects of SB-220453 or vehicle on total duration of activity and amplitude of changes in cortical d.c. potential, nitric oxide release, and rCBFLDF were assessed (Fig. 2). Additionally the number of d.c. depolarizations, NO peaks and rCBFLDF events occurring during the period of SD activity was also ascertained. All data were analysed for normality using the Shapiro-Wilk test, with normally distributed data expressed as mean±standard error of the mean (

Raw data plot of cortical extracellular d.c. field potential (mV), nitric oxide electrode current (pA), regional pial artery laser Doppler blood flu (ldfu) following induction of KCl cortical SD in a vehicle-treated animal (labrasol 1 ml/kg i.p. 90 min prior to SD induction). The figure demonstrates the measurement of each variable amplitude and duration of d.c. and nitric oxide activity. Delay between variables reflects relative distance of probes from site of KCl application, rather than metabolic response.
Results
Haemodynamic parameters pre- and post-dosing of SB-220453 or vehicle
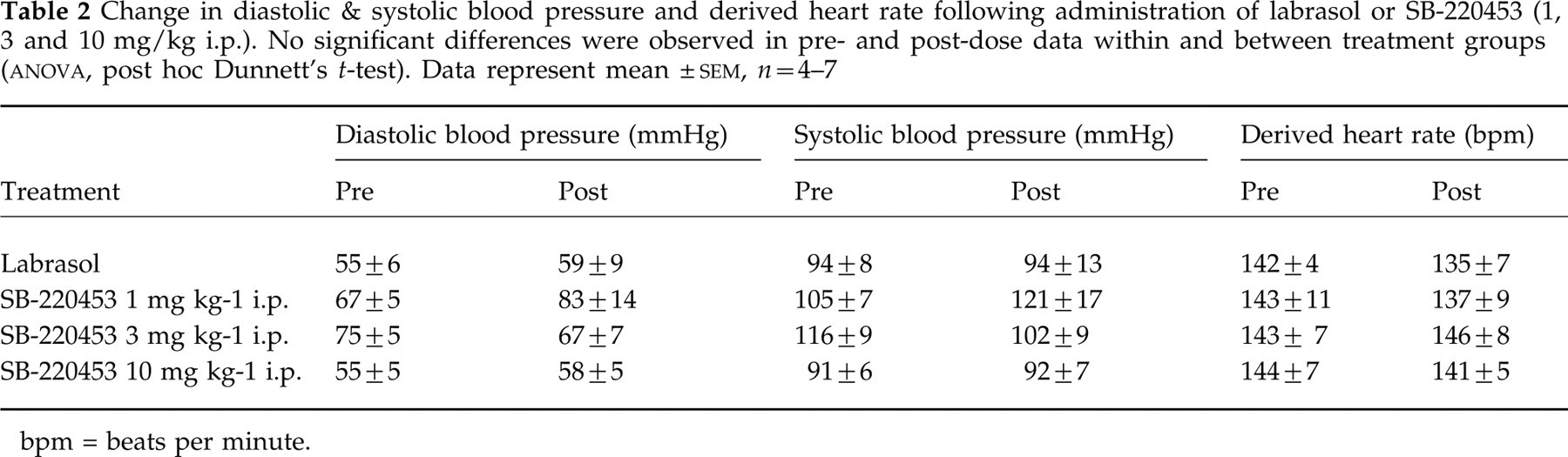
Blood/acid base status, diastolic and systolic blood pressure, and derived heart rate all remained within the normal physiological range throughout the experiment with no significant difference observed within or between treatment groups (

Pre- and post-dose effects of vehicle (Labrasol) 1 ml/kg i.p., and SB-220453 1, 3 and 10 mg/kg i.p. on pial artery cerebral blood flux (rCBFLDF). Data represented as mean ±
Physiological parameters determining blood/acid base status. All values calculated at 37°C. Data are represented as means ±

Change in diastolic & systolic blood pressure and derived heart rate following administration of labrasol or SB-220453 (1, 3 and 10 mg/kg i.p.). No significant differences were observed in pre- and post-dose data within and between treatment groups (
bpm = beats per minute.
Effect of SB-220453 on SD-induced changes in cortical extracellular d.c. field potential, rCBFLDF, and NO release
Application of KCl for 6 min to the cortical parenchyma induced repetitive negative shifts in cortical extracellular d.c. field potential and a multiphasic increase in NO current and hyperaemia as previously described in Read et al. [15] (Fig. 2). In vehicle treated animals a median of 5 d.c. deflections was observed (25%–75% range; 4–6). SB-220453 dose dependently reduced the number of extracellular d.c. depolarizations occurring within each recording period to a median of 2 (25%–75% range; 2–3) (P < 0.05) and a median of 1 (25%–75% range; 1–1) (Mann–Whitney U-test vs. vehicle) (P < 0.01), at doses of 3 mg/kg and 10 mg/kg i.p., respectively. This reduction in d.c. event number corresponded to a progressive decrease in duration of cortical d.c. activity from 55 min (25–75% range, 32–59 min) in vehicle treated animals to 4 min (25–75% range, 4–5 min) in SB-220453 10 mg/kg i.p pre-treated animals.
Similar to the observed decrease in d.c. deflection number, SB-220453 dose-dependently reduced the number of NO peaks and hyperaemic events during the recording period (Fig. 4). For example, the median number of nitric oxide peaks in vehicle treated animals was 4.5 (25–75% range of 3–5). At 10 mg/kg i.p. SB-220453, a median number of 1 nitric oxide peak was observed (25–75% range, 1–1), which represented a significant reduction when compared to control (Mann–Whitney U-test, P < 0.01). In all animals the total duration of cortical nitric oxide release activity, was approximately similar to that of d.c. activity, rapidly ceasing on termination of cortical negative d.c. shifts. In vehicle treated animals, total duration of nitric oxide release activity was 59 min (25–75% range, 34–59min), which was dose-dependently inhibited by SB-220453 treatment. This inhibition reached significance (Mann–Whitney U-test) at 3 and 10 mg/kg, with the duration of nitric oxide release reduced to 23 min (25–75% range, 21–25 min, P < 0.05) and 5 min (25–75% range, 5–5 min, P < 0.01) (Fig. 5).

Box plot of number of SD associated nitric oxide peaks observed after KCl application to the cortex of anaesthetized cats. Animals were pre-treated 90 min prior to KCl application with either vehicle (labrasol) 1 ml/kg i.p. or SB-220453 1, 3 and 10 mg/kg i.p. Data represented as medians with box limits at 25 and 75% of range. Significance differences in distribution vs. vehicle were assessed using Mann–Whitney U-test (∗∗P < 0.01). n = 4–6.
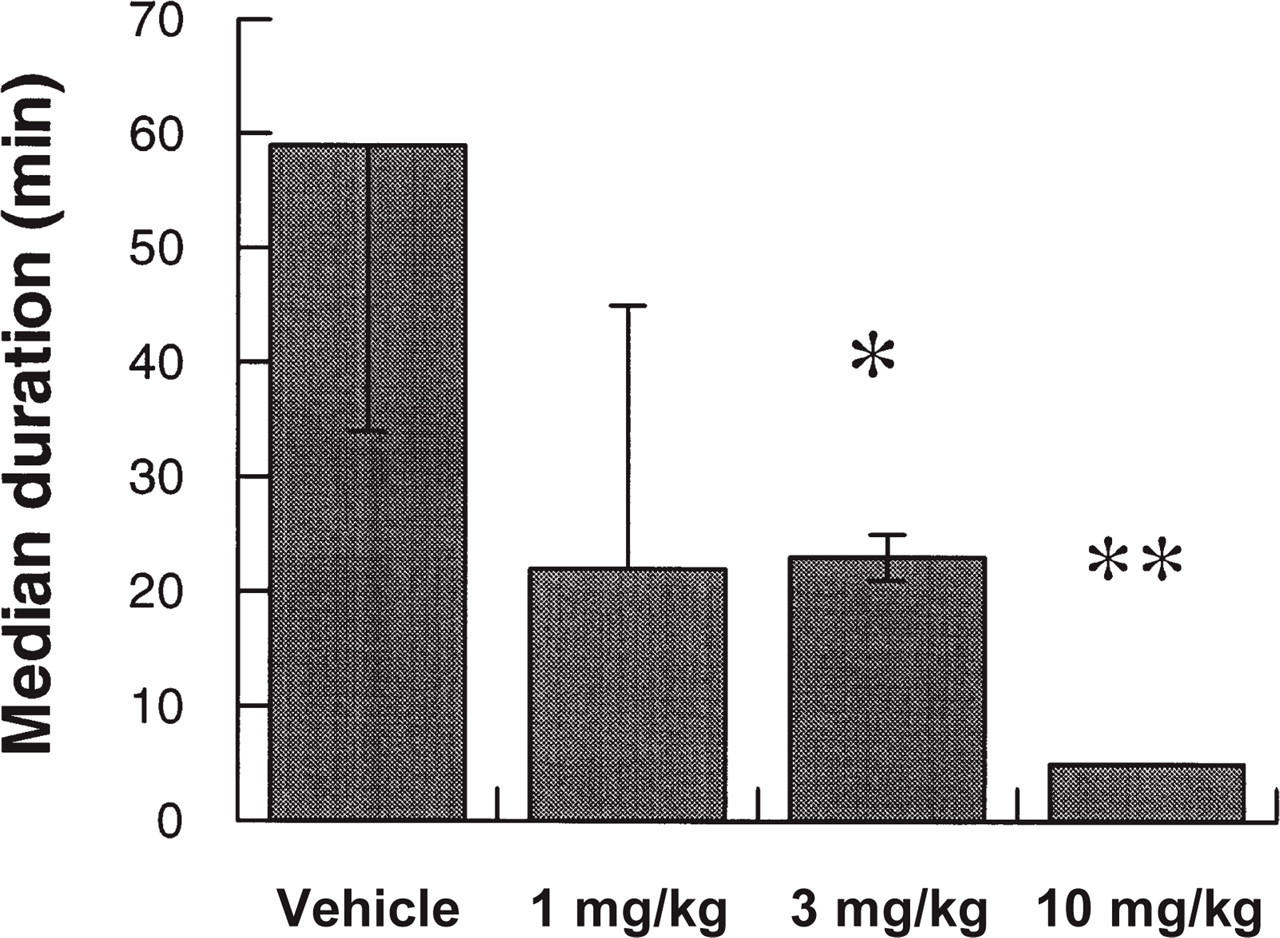
Median total duration of SD induced nitric oxide release activity following application of KCl solid to the parietal cortical parenchyma in the anaesthetized cat. Animals were pre-treated 90 min before induction of SD activity with either vehicle (Labrasol) 1 ml/kg i.p., SB-220453 1 mg/kg i.p., 3 mg/kg i.p. or 10 mg/kg i.p. Significant differences between SB-220453 treated animals and vehicle treated animals were assessed by Mann–Whitney U-test, where ∗∗P < 0.01, ∗P < 0.05, n = 4–7.
Interestingly, the primary d.c. depolarization remained intact in all treatment groups and the mean amplitude of any residual d.c. depolarizations remained unaffected by SB-220453 treatment at all doses when compared to vehicle (
Discussion
SB-220453 represents a mechanistically novel class of compounds, with a high affinity for its own CNS binding site [26]. The affinity of SB-220453 at this CNS binding site correlates well with in vivo activity in the elevation of seizure threshold and inhibition of trigeminal nerve stimulated neurogenic plasma protein extravasation [26]. SB-220453 may therefore have therapeutic potential in conditions associated with neuronal hyperexcitability and/or inflammation. Clearly, the phenomenon of SD represents a propagating region of neuronal hyperexcitability. Previous studies have demonstrated that SB-220453 has a potent and dose dependent inhibitory activity on KCl-induced repetitive SD activity as characterized by changes in d.c. field potential events [27].
We have previously demonstrated that in the anaesthetized cat there is a potent multiphasic release of nitric oxide associated with SD d.c. depolarization. Pre-treatment with an NOS inhibitor was found to uncouple regional cerebral blood flow changes from SD-induced d.c. depolarizations [15]. Conversely, in this study, SB-220453 did not affect coupling of cerebral blood flow and nitric oxide changes from SD-induced d.c. depolarization. SB-220453 therefore appears to act on the metabolic drive of SD, leaving cerebrovascular coupling intact. A similar result has been obtained with the Na+K+2 Cl– cotransporter inhibitor, furosemide. When compared to SB-220453, furosemide only moderately decreased the period of SD activity. However, as with SB-220453, residual SD d.c. depolarizations remained closely coupled to associated cortical nitric oxide release in furosemide pre-treated animals [28].
The involvement of SD in migraine pathogenesis has been strongly implicated in numerous studies. Early regional cerebral blood flow studies proposed the concept of a spreading oligemia occurring during migraine. For example, during a spontaneous migraine attack a bilateral spreading oligemia of the cortex was observed, propagating at a rate consistent with cortical spreading depression [30]. Furthermore, studies in red wine induced headache have also confirmed spreading oligemia originating from the visual cortex with additional changes in flow consistent with activation of nociceptive pathways [31].
Metabolic studies of Cao et al. [8] have further added to a growing body of evidence of SD involvement in migraine. Using functional MRI with blood oxygen level dependent contrast (BOLD) imaging to study occipital cortex function during visually evoked headache in migraine patients, the authors detected a spreading neurovascular event consistent with SD. This BOLD suppression occurred in both migraine with and without aura subjects, and before the onset of headache. Indeed, as with the propagating oligemia [30], the propagation of this BOLD effect was observed to spread over the cortex at a rate between 3 and 6 mm/min. No alteration in BOLD effect was seen in ‘normal’ patients. The authors concluded that the signal change was probably due to a primary neuronal event and followed by a secondary regional cerebral blood flow change.
Therefore, there is a clear rationale that compounds with utility in inhibiting SD, such as SB-220453, may be applicable to the treatment of migraine. Additionally, the evidence implies that SD is not unique to the aura subset of patients, and that SD may precede the headache phase of migraine.
The proposed role of nitric oxide in migraine headache is based upon several pertinent experimental observations. Infusion of GTN into migraineurs induces a reliable and dose dependent immediate headache, followed by a ‘feed-forward’ late migraine attack several hours after termination of GTN infusion [22, 24]. In the clinic, NOS inhibition using L-NG methyl-arginine hydrochloride, has been reported to decrease headache and associated migraine symptoms [25].
It has been suggested by Woolf [32] that SD stimulation of trigeminal afferents may induce a central sensitization which is maintained by further afferent input from sensitized peripheral nerves. Indeed Moskowitz et al. [23] showed that following induction of neocortical spreading depression there was increased expression of c-fos like immunoreactivity in the trigeminal nucleus caudalis (TNC), demonstrating a clear association between SD and nociceptive processing. However, apparently contradictory studies have recently been published [33, 34].
Ingvardsen et al. [33] studied c-fos expression in the TNC following elicitation of SD activity with repeated injections of 1
It is reasonable to suppose given the relative concentrations of cortical nitric oxide produced during SD, when compared to that observed during GTN infusion, that sensitization of nociceptive afferents by nitric oxide released during SD is feasible [15, 17, 29]. The inhibition of NO production per se, may not prove to be a suitable therapeutic approach in the treatment of migraine [25]. Nitric oxide free radicals have a close interaction with superoxide free radicals and react rapidly to form peroxynitrate. Experimental infusions of GTN to anaesthetized rats, have demonstrated that sumatriptan effectively inhibits cortical nitric oxide levels, but concommitantly increases cortical superoxide levels [29]. This is possibly as a consequence of decreased sequestering of superoxide free radicals by nitric oxide to the formation of peroxynitrate. The mechanism of inhibition of cortical nitric oxide by sumatriptan is unknown. Whilst this has not been confirmed for other antimigraine agents, raising superoxide concentrations as a result of inhibiting nitric oxide levels in a patient population with a predisposition to cerebral infarction, may warrant further investigation.
Therefore the inhibition of the metabolic drive of SD and hence the resultant inhibition of the associated cortical nitric oxide release, may be an important aspect of the biological profile of SB-220453. This may prove to be a more preferable approach to decreasing the period and/or concentration of cortical nitric oxide release during the cortical hyperexcitability phase of migraine, than the selective inhibition of nitric oxide production. This is emphasized by the lack of overt vascular activity of SB-220453, common to both NOS inhibitors and sumtriptan. SB-220453 was found to have no effect on diastolic and systolic blood pressure, derived heart rate, or cerebral artery blood flux. In conclusion, this represents a novel profile for an antimigraine compound, potentially treating the overall migraine process rather than providing the symptomatic relief offered by current therapies.