
Abstract
Cortical spreading depression is a pathophysiological excitation wave that occurs during pathophysiological brain conditions such as ischemic brain infarction, migraine aura, and others. Judged from experiments in rodents, the brainstem is thought to be comparatively resistant to the generation of spreading depression. However, because spreading depression can be elicited in the brainstem of rat pups after superfusing the brainstem with solutions enhancing excitability, we reinvestigated spreading depression in the brainstem of the adult rat. Based on theoretical predictions indicating a major role of extracellular potassium in susceptibility to spreading depression, we used conditioning solutions in which chloride ions were replaced by acetate and tetraethylammonium chloride and a small amount of KCI were added. Under these conditions, spreading depression was reproducibly elicited in the brainstem either by topical application of KCI crystals to the brainstem surface or by local microinjection of KCI into the brainstem. The direct current shifts so elicited were accompanied by typical elevation of extracellular potassium ions, propagated in the brainstem, and were prevented by MK-801, an N-methyl D-aspartate blocker. During spreading depression, the regional blood flow in the brainstem was transiently increased. In addition, systemic arterial blood pressure, but not the heart rate, was transiently enhanced. In the nonconditioned brainstem, KCI stimulation neither elicited spreading depression nor induced changes in regional blood flow and blood pressure. These data show that proper conditioning renders the brainstem susceptible to spreading depression, and that spreading depression at this site elicits changes in local circulation and systemic blood pressure.
Introduction
The occurrence of spreading depression (SD) was first described in the cerebral cortex by Leão (1944), (1947). Spreading depression is a propagating, transient negative direct current (DC) potential shift that is paralleled by a rapid and substantial increase in potassium concentration in the extracellular space ([K+]e), water influx into cells, and shrinkage of the extracellular space volume (Kraig and Nicholson, 1978; Gardner-Medwin, 1981; Nicholson, 1993; Somjen, 2001). Spreading depression was shown to occur in pathologic events in the brain, for example, in human cerebral cortex after severe head trauma (Strong et al, 2002) and ischemic stroke after subarachnoid hemorrhage (Dreier et al, 2006), and in cortex and subcortical gray matter of animals in a wide range of conditions associated with acute injury (Nedergaard and Hansen, 1993; Hossmann, 1996; Witte et al, 2000). It is assumed that SDs augment neuronal damage in the penumbra zone by increasing the energy demand and oxygen consumption, which in turn increase the metabolic stress in that region of the brain (Strong and Dardis, 2005; Takano et al, 2007). Furthermore, SD is thought to be involved in the aura phase in human migraine (Richter and Lehmenkühler, 1993; Lauritzen, 1994; Pietrobon and Striessnig, 2003). In animal experiments, SDs can be reliably elicited in the gray matter of the normal brain by pinpricking, and by topical application of KCl or excitatory amino acids to the cortical surface (i.e., glutamate, kainate, or N-methyl
Although in experiments SDs can be easily elicited in the cerebral cortex, the brainstem was thought for a long time to be resistant to SDs. According to Bureš et al (1974), a brain tissue susceptible to SDs should comply with some requirements such as a high density of neurons, a small extracellular space and a low density of myelin, a large enough volume of brain tissue that reacts to the eliciting stimulus, and a relatively homogeneous brain tissue without boundaries that would hinder transition of SDs. The brainstem is an inhomogeneous structure of more or less densely packed neurons together with myelinated boundaries. It should, therefore, be devoid of SDs, and indeed, Somjen et al (1992) listed the brainstem as a structure with low susceptibility to SDs, along with the cerebellar cortex and the spinal cord.
Recently, however, we were able to show that SDs can also be evoked in the brainstem of young rat pups when chloride ions in the extracellular milieu are replaced by acetate ions (this method is frequently used to enhance excitability in SD experiments). Spreading depressions in the brainstem could be elicited by topical application of KCl or NMDA to the brainstem surface, and severe hypoxia and asphyxia conditioned the brainstem for SDs. Interestingly, however, this seemed to be possible only in the immature brainstem at a life period before the cerebral cortex first becomes susceptible to SDs. With maturation, the acetate-conditioned brainstem became resistant to SDs (Richter et al, 2003). For SDs in the immature brainstem, potassium ions were the initiating and propagating force, and a blockade of neither NMDA receptors nor voltage-gated calcium channels inhibited SDs (Richter et al, 2005).
A computational model of cortical SDs in a model neuron (Somjen, 2001) predicted that SD is ignited when persistent inward currents exceed outward currents resulting in a net inward current. In this model, rising [K+]e played an important role by causing a depolarization of the neurons followed by an outflow of K+. The threshold for igniting SDs could be lowered, for example, by elevated [K+]e or by blocking potassium channels, thus enhancing the excitability of neurons. In addition, the K+ distribution is strongly dependent on the Na+/K+-ATPase, that is, blockade of the ATPase enhances neuronal excitability and may even ignite SDs (Balestrino et al, 1999). Based on Somjen's model, we reasoned that SDs in the adult brainstem may be elicited under conditions in which the increase in neuronal excitability is more pronounced than in immature developmental stages, that is, in which [K+]e is elevated in addition to the lowered chloride content in the extracellular milieu.
In the present experiments, we tested the hypothesis that SDs can be elicited in the adult brainstem provided that conventional conditioning of the brainstem is further supported by application of solutions that increase [K+]e. Furthermore, we asked the very important question whether SDs in the brainstem would alter regional blood flow (rCBF) in the brainstem similar to that in the cerebral cortex, and we explored whether SDs in the brainstem affect systemic mean arterial blood pressure (ABP) and heart frequency.
Materials and methods
Animal Preparation
The present study was approved by the Animal Protection Committee and the Regional Government of Thüringen (Reg. No. 02-10/01, 02-040/06). Male Wistar rats (body weight 300 to 450 g) were anesthetized with 100mg/kg sodium thiopentone administered intraperitoneally; supplemental doses (20mg/kg intraperitoneal) were administered as necessary to maintain areflexia. Through the cannulated trachea the animals breathed spontaneously during surgery. A catheter (diameter 0.5 × 0.9 mm; Braun Melsungen, Melsungen, Germany) was inserted into the right femoral artery. In the KCl crystal group, ABP was continuously measured to control depth of anesthesia (pressure monitor BP-1; WPI Instruments, Sarasota FL, USA). In the KCl microinjection group, changes in systemic ABP were measured with the pressure transducer P23Db (Statham Instruments, Puerto Rico, USA). Body temperature was kept at 37°C using a feedback-controlled temperature constanter (L/M-80; List, Darmstadt, Germany). At the end of the experiments, the animals were killed by an overdose of the anesthetic administered intravenously.
The head of the animal was fixed in a stereotaxic frame. The apical and parietal parts of the skull were exposed after a median incision. Over the left hemisphere, the dura mater was exposed in an area of 4 × 4 mm (2 mm posterior to bregma, 1 to 2 mm laterally from the midline) using a mini-drill under artificial cerebrospinal fluid (ACSF) cooling, and kept moist with ACSF (for contents see below) throughout the preparation. The dura mater was opened. Incising the neck muscles and the ligamentum atlantooccipitale exposed the brainstem from the occipital bone to the first cervical vertebra. The dura mater and arachnoidea underneath were removed.
Recording of Direct Current Potentials and of Changes in Extracellular Potassium Concentration
An Ag/AgCl reference electrode containing 2 mol/L KCl was placed on the nasal bone. Intracortical DC potentials were recorded using a glass micropipette filled with 150mmol/L NaCl. This microelectrode was inserted into the somatosensory cortex to a cortical depth of 1,000 to 1,200 μm corresponding to cortical layer V. For DC recordings in the brainstem, three pipettes were glued together with a lateral tip separation of 400 or 1,000 μm, respectively. The electrode assembly was lowered into the brainstem in a region close to the caudal trigeminal nucleus (for one of the electrodes) to a depth of 1,200 to 1,400 μm (Figure 1A, left panel) and allowed the monitoring of SD propagation.
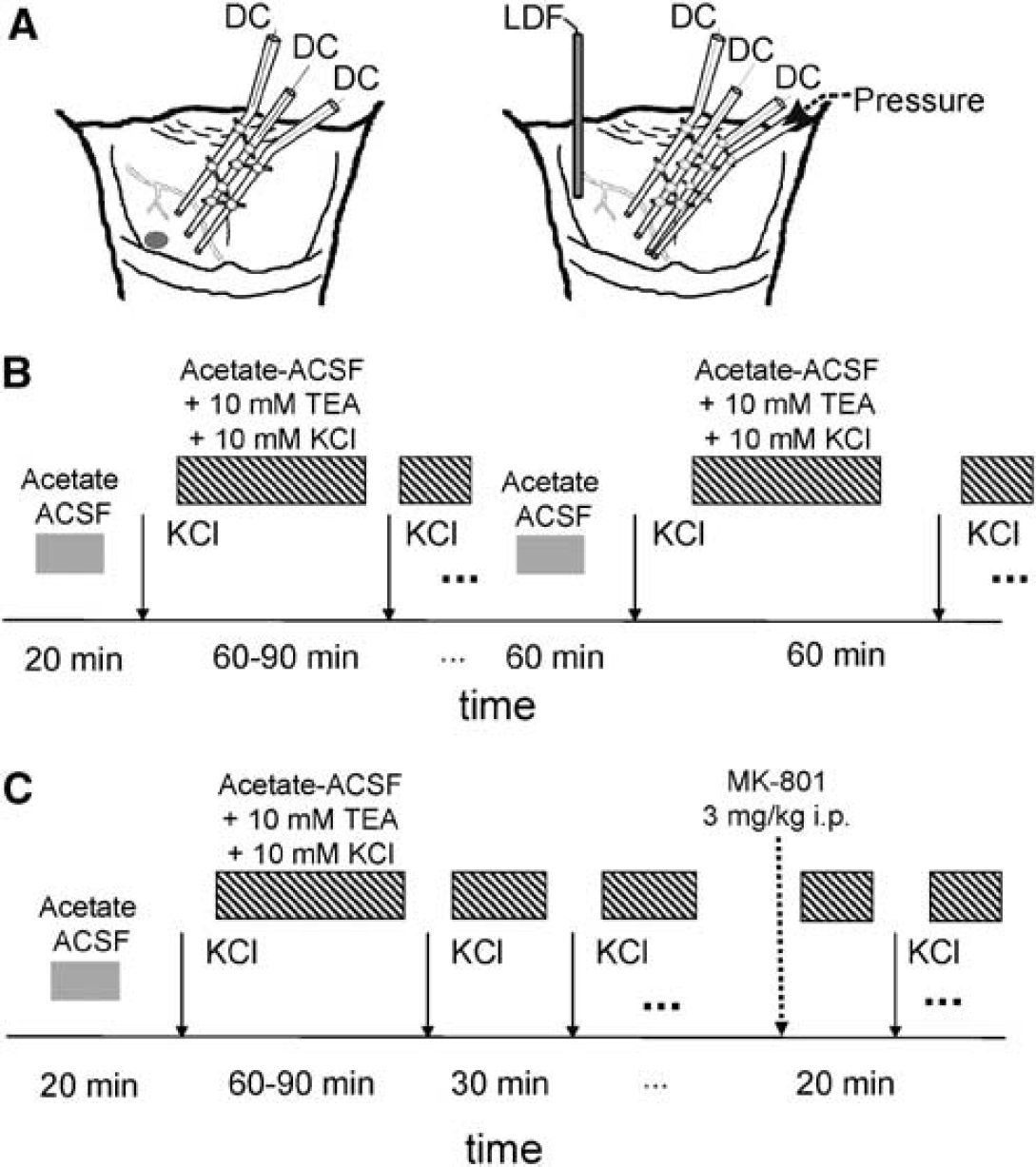
Description of the experimental design and protocols. (
To measure the changes in DC potentials and [K+]e simultaneously, one DC microelectrode filled with 150mmol/L NaCl was glued with a lateral separation of 400 μm to one double-barreled potassium-selective electrode that was made from theta-glass (Kuglstatter, Garching, Germany) and was filled with 100mmol/L KCl. The tip contained potassium ionophore I-cocktail A based on valinomycine (Fluka 60031-1EA; Sigma-Aldrich GmbH, Seelze, Germany). The other barrel contained 150mmol/L NaCl and served as a reference for the potassium-selective electrode. All electrodes were connected by Ag/AgCl wires to a custom-built four-channel high-impedance amplifier (Meyer, Munich, Germany). The signals were stored on a PC. The animals were grounded through an Ag/AgCl electrode below the back skin.
Superfusion of the Brain and Conditioning for Spreading Depression
Overall, 14 rats were included in this study. Exposed brain areas were kept moist with regular ACSF (in mmol/L: NaCl 138.4, KCl 3.0, CaCl2 1.3, MgCl2 0.5, NaH2PO4 0.5, urea 2.2, glucose 3.4, warmed to 37°C and equilibrated with 5% CO2 in O2).
Spreading depressions were elicited with two different methods. In one group of eight adult rats, we applied a small KCl crystal weighing 0.1 to 0.5 mg onto the cortex and the brainstem surface at a distance of approximately 1mm from the electrode. After 20 to 30mins of ACSF superfusion, the first attempt was made to elicit SDs in the cortex and in the brainstem by KCl. Before KCl application, superfusion was stopped and the surface of the cortex and the brainstem was carefully dried with a thread of cotton wool. After having tested for SDs with KCl, the superfusion solution was changed. We then continued superfusing the brainstem with ACSF in which 100 mmol/L of the chloride (75%) was replaced by 100mmol/L acetate, and 10 mmol/L tetraethylammonium chloride (TEA; Sigma, Seelze, Germany) as well as 10 mmol/L KCl was added (acetate-ACSF-TEA-KCl). After 60mins, this superfusion was stopped, the brainstems were carefully dried with cotton wool, and a further attempt was made to elicit SDs. Superfusion was continued either after 5 mins if no DC shift had occurred or after the recovery from a DC shift. In a subset of three animals, after successful elicitation of SDs in the brainstem, the superfusate was changed again into normal ACSF for 1 h, then we again tested for SDs with KCl. After that, we super-fused again with acetate-ACSF-TEA-KCl for 1 h and then we tested for SDs (Figure 1B). During the whole superfusion time, the animals were artificially ventilated with room air and paralyzed with pancuronium bromide (Pancuronium Organon; Organon GmbH, Oberschleißheim, Germany; 1mL/kg intravenous).
In another group of six adult rats, we used the same superfusion schedule, but we injected an amount of approximately 0.5 μL of 1mol/L KCl solution according to Moskowitz et al (1993) with a pressure of 100 kPa for 1 sec using a microinjector (pico-injector PLI-100; Harvard Apparatus, Holliston, MA, USA). After having tested the efficacy of injection time and injection pressure in the first rat, these parameters were kept constant in all animals. To monitor the KCl injection and SD propagation, a double-barreled microelectrode (tip diameter 5 μm), containing in one shank 1 mol/L KCl solution for injection and in the other 150 mmol/L NaCl for DC recording, was glued with a lateral distance of 1,000 μm each to two microelectrodes filled with 150 mmol/L NaCl for DC recording. This assembly was lowered into the same brainstem areas as in the KCl crystal group. Superfusion with acetate-ACSF-TEA-KCl was stopped before each microinjection and continued either 5 mins afterward (if no DC deflection occurred) or after repolarization of the negative DC deflection (if a propagating SD was observed).
Regional blood flow in the brainstem, systemic ABP, and heart rate (HR) were measured in five animals of the latter group (KCl microinjection). An electrocardiogram was recorded from standard limb leads using stainless-steel needle electrodes. Local cerebral blood flow (CBF) was measured continuously using a laser-Doppler flowmeter (MBF3D; Moor Instruments, Exminster, UK) with a single fiber probe (fiber separation was 225 μm) and a maximal possible sampling rate of 40 sec−1. The optic probe was placed on the brainstem surface of a region devoid of large vessels (>100 μm) and approximately 1 mm apart from the injection point. After stable baseline recordings had been obtained, the probe was left for the duration of the experiment. A schematic drawing of this assembly is shown in Figure 1A (right panel).
In three rats of the KCl crystal group and three rats of the KCl microinjection group, after repeated elicitation of SDs, we injected the NMDA receptor blocker MK-801 (Sigma, Seelze, Germany) (3 mg per kg body weight, intraperitoneal) and made another attempt to elicit SDs 20 mins after injection. In intervals of 20 mins, attempts to elicit SDs were repeated. In the mean time, superfusion with acetate-ACSF-TEA-KCl was continued and only stopped immediately before each SD elicitation (Figure 1C).
Data Acquisition and Evaluation
Data were recorded on PC by using custom signal acquisition programs. In accordance with Somjen et al (1992), DC shifts were classified as SD if their amplitudes were larger than 5 mV and if peaks showed a time difference at the horizontally separated electrodes in the brainstem indicating propagation. The SDs were analyzed regarding occurrence, amplitude, SD half-amplitude duration (time interval between half of the amplitude during depolarization and half of the amplitude during repolarization phases), and shape of the DC deflection.
For HR calculations, the R-R intervals were estimated using the R wave peak as the trigger point. The reciprocal of the R-R interval series represented the instantaneous HR (in bpm). All changes in CBF as well as ABP and HR were calculated as percent of the baseline value immediately preceding the SD release. The laser-Doppler flowmetry monitor displays blood flow readings in arbitrary units that do not allow for measurement of CBF in terms of absolute values, but the method is valid in determining relative changes in CBF during moderate flow increases (Fabricius and Lauritzen, 1996).
Results
Characterization of Spreading Depression Elicitability in the Brain Tissue before Conditioning
In all rats tested, application of a KCl crystal to the cerebral cortex reproducibly elicited 4 to 8 repetitive, self-regenerating cortical SD waves in a period of 20 mins. In none of the rats, the cortical SDs caused a related DC shift in the brainstem. In agreement with our previous findings in immature rats (Richter et al, 2003), the same stimuli failed to evoke SDs in the native brainstem. We performed 13 attempts to elicit SDs in the adult brainstem by a KCl crystal (n = 8 rats) and 6 attempts by microinjection of 1 mol/L KCl solution (n = 6 rats), and none of them was able to elicit an SD-related DC deflection that propagated within the brainstem.
Spreading Depression in the Adult Brainstem after Superfusion with Acetate-ACSF-TEA-KCL
Spreading depressions could be elicited in the adult brainstem after a particular conditioning protocol. We used a solution in which we replaced 75% of the chloride ions by acetate, added 10mmol/L KCl, and blocked potassium channels by 10mmol/L TEA. During superfusion with this conditioning solution, [K+]e continuously increased by 3–6mmol/L per h at a depth of 1,200 μm in the brainstem. Notably, even the longest continuous superfusion lasting 3.5 h did not influence systemic ABP or HR. However, the brainstem became susceptible to SDs after a superfusion time of approximately 60 to 75 mins.
In 8 rats, we applied 26 times a KCl crystal to the brainstem surface and observed 22 propagating SDs fulfilling the criteria of cortical SD (start of the SD-related DC shift after the application of the KCl crystal, amplitude larger than 5mV, well-defined propagation). In 13 SDs, we recorded the concomitant transient elevation of [K+]e. As in the cortical gray matter, [K+]e increased very steeply and recovered within the time course of the DC shift. A typical example of an SD induced by KCl crystal is shown in Figure 2. The parameters of these SDs are summarized in Table 1. The increase in susceptibility to SD was reversible (Figure 3). After successful testing for SD in three rats, the superfusate was changed back to normal ACSF for 1 h, and after this superfusion, KCl did not induce anymore SDs in the brainstem. Switching back again to acetate-ACSF-TEA-KCl superfusion restored the elicitability of brainstem SDs in all the three animals tested (Figure 3).
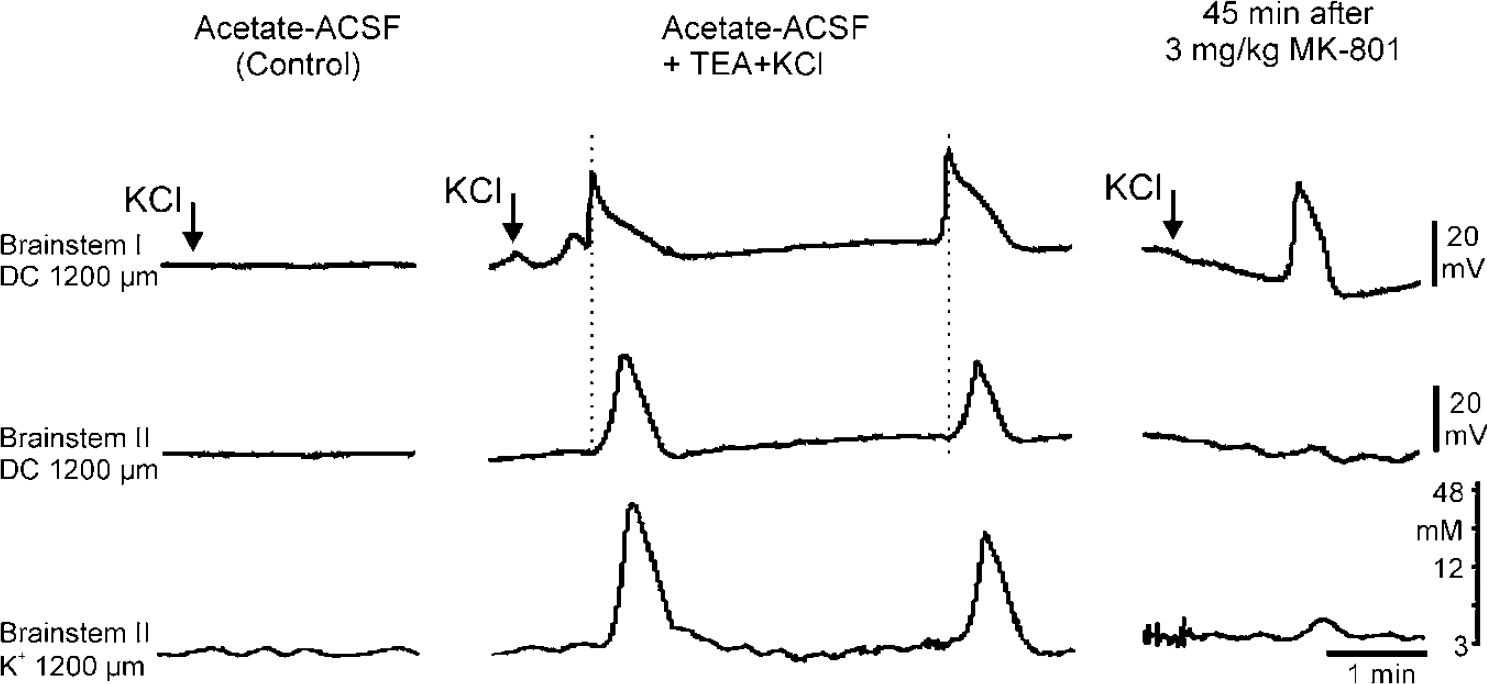
Simultaneous recordings of both DC potentials and changes in [K+]e in the adult brainstem at a depth of 1,200 μm. The horizontal distance between the electrodes was 400 μm; electrode II is a double-barreled one measuring DC potentials and [K+]e. The panels from left to right show the schedule of one experiment. During the control phase, a KCl crystal was unable to elicit SD in the brainstem (left panel). After superfusion with acetate-ACSF-TEA-KCl, a KCl crystal elicited propagating SDs in the brainstem. Note the different times when the peak amplitudes were reached accentuated by thin dotted lines. Each DC deflection is accompanied by an increase in [K+]e up to 45 mmol/L (middle panel). Systemic application of the NMDA receptor blocker MK-801 prevented elicitation and propagation of SD by NMDA. A KCl crystal still elicited a local depolarization that did not propagate and therefore did not fulfill the criteria of SD (right panel).
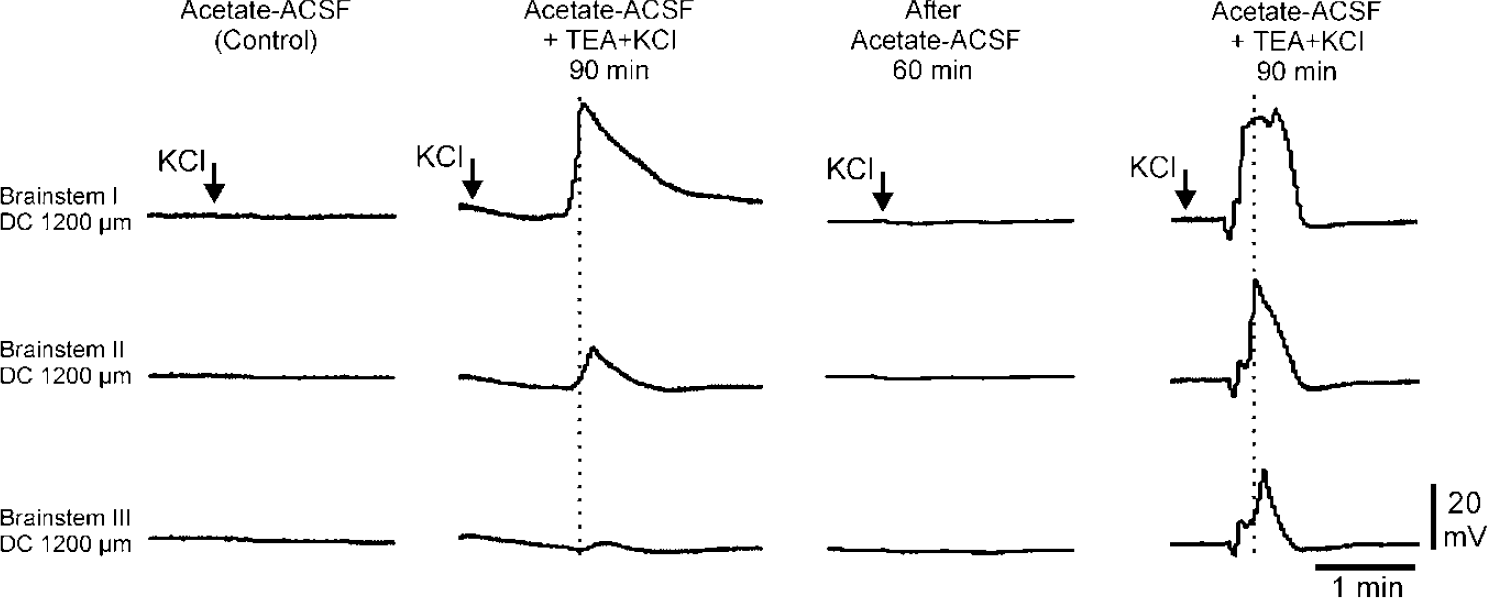
Simultaneous recordings of DC potentials in the adult brainstem at a depth of 1,200 μm with three DC microelectrodes at horizontal distances of 400 μm each. The panels from left to right show the schedule of one experiment. In the unconditioned brainstem, a KCl crystal was unable to elicit SD. Superfusion with acetate-ACSF-TEA-KCl rendered the adult brainstem susceptible to SD evoked by a KCl crystal with a negative DC shift that fulfilled the criteria of an SD. The thin dotted lines accentuate the different times when the peak amplitudes were reached. After a washout for 60mins with regular acetate-ACSF, the capability to generate SD disappeared, but could be restored by another 90-min superfusion with acetate-ACSF-TEA-KCl. Spreading depressions could still be elicited after a total superfusion time of 240mins.
Comparison of parameters of SDs (mean ± s.d.) in the brainstem of adult rats elicited either by application of a KCl crystal to the surface of the brainstem or microinjection of 1 mol/L KCl solution into the brainstem at a depth of 1,200 μm

Abbreviations: DC, direct current; SD, spreading depression.
After application of a KCl crystal, we observed in two out of eight rats repetitive negative DC deflections amounting between 5 and 45 mV every 60 to 120 sees that lasted up to half an hour without any change in amplitude. As in the typical SD, we found signs of propagation, that is, the peaks showed a time difference at the different recording sites, and every DC shift was accompanied by a transient elevation of [K+]e. The simultaneous DC recordings in the cerebral cortex did not show any related DC deflections (Figure 4).
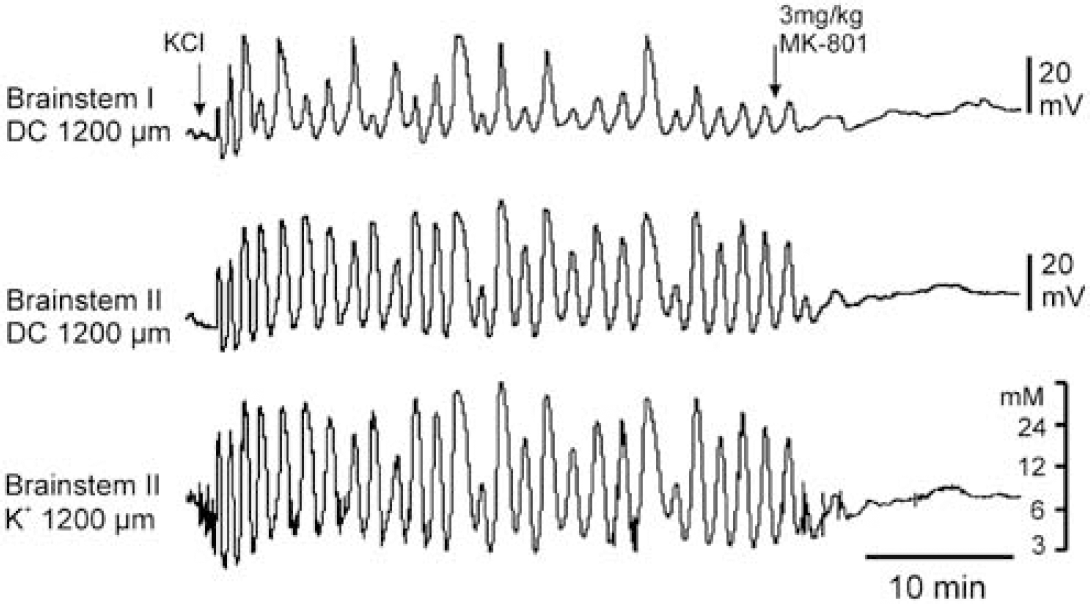
Specimen of a simultaneous recording of both DC potentials and of changes in [K+]e in the adult brainstem at a depth of 1,200μm. The horizontal distance between the electrodes is 400μm; electrode II is a double-barreled one. The application of a KCl crystal to the surface of the brainstem initiated a series of subsequent following SDs that lasted more than 30mins without a decrease in amplitudes. Note the different amplitudes and time courses of the DC potentials comparing the two recording sites, and note the close amplitude correlation between DC potentials and the accompanying changes in [K+]e at electrode II. The systemic application of the NMDA receptor blocker MK-801 abolished repetitive DC deflections.
In six rats, we used KCl microinjection at sites deep into the brainstem to elicit SDs. After 60 mins of conditioning with acetate-ACSF-TEA-KCl, we made 28 attempts to elicit SDs with the same injection parameters. All microinjections caused a DC shift that fulfilled the criteria of SD. At the site of microinjection, only the injection artifact was observed in most cases. This was caused by the elevation of potassium and chloride ions that were picked up by the NaCl-filled microelectrode. The injection artifacts lasted between 3 and 11mins. At the remote microelectrode glued in a horizontal distance of 1 mm, the DC potential increased with a clearly visible delay indicating propagation of the DC shift. Then a DC shift followed under the microelectrode 2 mm away from the site of microinjection (Figure 5A). Negative DC shifts were only classified as SDs if the DC shifts at the injection site and the remote site exhibited a time interval of at least 10 sees. In 23 of the attempts, we observed ‘full blown’ propagating SDs, and in five attempts, we observed propagating DC shifts that did not reach the amplitude of 5mV. The SDs at the remote electrodes had a uniform shape and time course (Figure 5B). The parameters of microinjection-induced SDs are summarized in Table 1. In three of the rats, we observed spontaneously occurring SDs in the brainstem. They were either repetitively ignited at the injection site by the long-lasting elevation of [K+]e, or they traveled back from the farthest microelectrode.
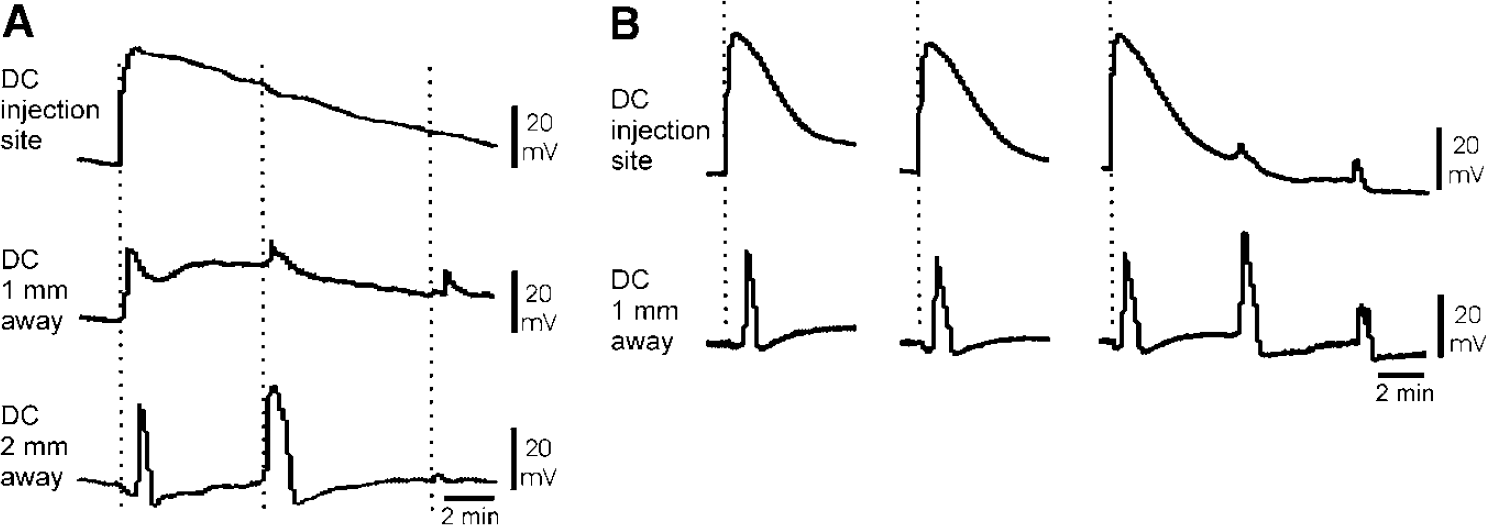
Representative samples of SDs elicited by KCl microinjection into the rat brainstem. (
In four of the rats of the microinjection group, microinjection of KCl elicited repetitive negative DC deflections similar to those depicted in Figure 4. These DC shifts reached amplitudes of 10 to 35 mV, and repetitions were observed every 50 to 150 sees.
Effect of MK-801 on Brainstem Spreading Depressions
MK-801 inhibited brainstem SDs elicited by topical application of KCl. Injection of this NMDA receptor blocker inhibited the propagation of SDs in all three conditioned animals in which SDs were elicited by topical application of KCl crystals. After MK-801 treatment, no propagating SDs could be elicited throughout a period of 60mins. In some cases, only a nonpropagating, local negative DC shift was observed (Figure 2, right panels). Spreading depressions caused by microinjection of KCl were also blocked by MK-801. This was tested in four rats. In two rats, we could still elicit an SD 20mins after MK-801 treatment but not after 40mins. In the other two rats, SDs could be elicited neither at 20 nor at 40mins after MK-801 treatment. MK-801 also inhibited repetitive SDs that were elicited in the brainstem by KCl crystals (Figure 4) or by KCl microinjection.
Effect of Brainstem Spreading Depression on Regional Blood Flow, Mean Arterial Pressure, and Heart Rate
Because application of KCl crystals to the brainstem occasionally evoked changes in blood pressure parallel to brainstem SDs, we systematically investigated the impact of brainstem SDs on rCBF, ABP, and HR in five animals. In these experiments, we used microinjection of KCl because administration of KCl crystals to the brainstem surface disturbed stability of blood flow measurement with the laser-Doppler flowmetry probe. First we tested whether KCl injection itself elicited changes in CBF, ABP, and HR. Figure 6A shows an experiment in which KCl was microinjected without the SD conditioning protocol. Notably, the injection of KCl into the native brainstem neither evoked an SD nor any changes in CBF or ABR In contrast, in the conditioned brainstem, KCl microinjection caused SDs, and during each SD, we observed increases in CBF and ABP (see typical example in Figure 6B). On average, during SDs, CBF increased by 29% ± 14% of baseline and persisted for 50.7 ± 18.8 sees. We did not observe a spreading oligemia after the increase in CBF. Simultaneous ABP elevations showed considerable variations in magnitude (42% ± 44% of baseline) and duration (19.3 ± 16.6 sees) (Figure 6B). Whereas 30% of ABP responses showed a maximum increase of < 10% of baseline, the same percentage of ABP responses elicited a peak elevation of >80% of baseline. In contrast, SD-associated HR responses occurred in only 5 of 23 SDs, with an HR decrease of < 7% of baseline. The magnitude of the CBF response was not correlated to the amplitude of the SD (Figure 6C) and did not depend on the magnitude of ABP responses (Figure 6D).
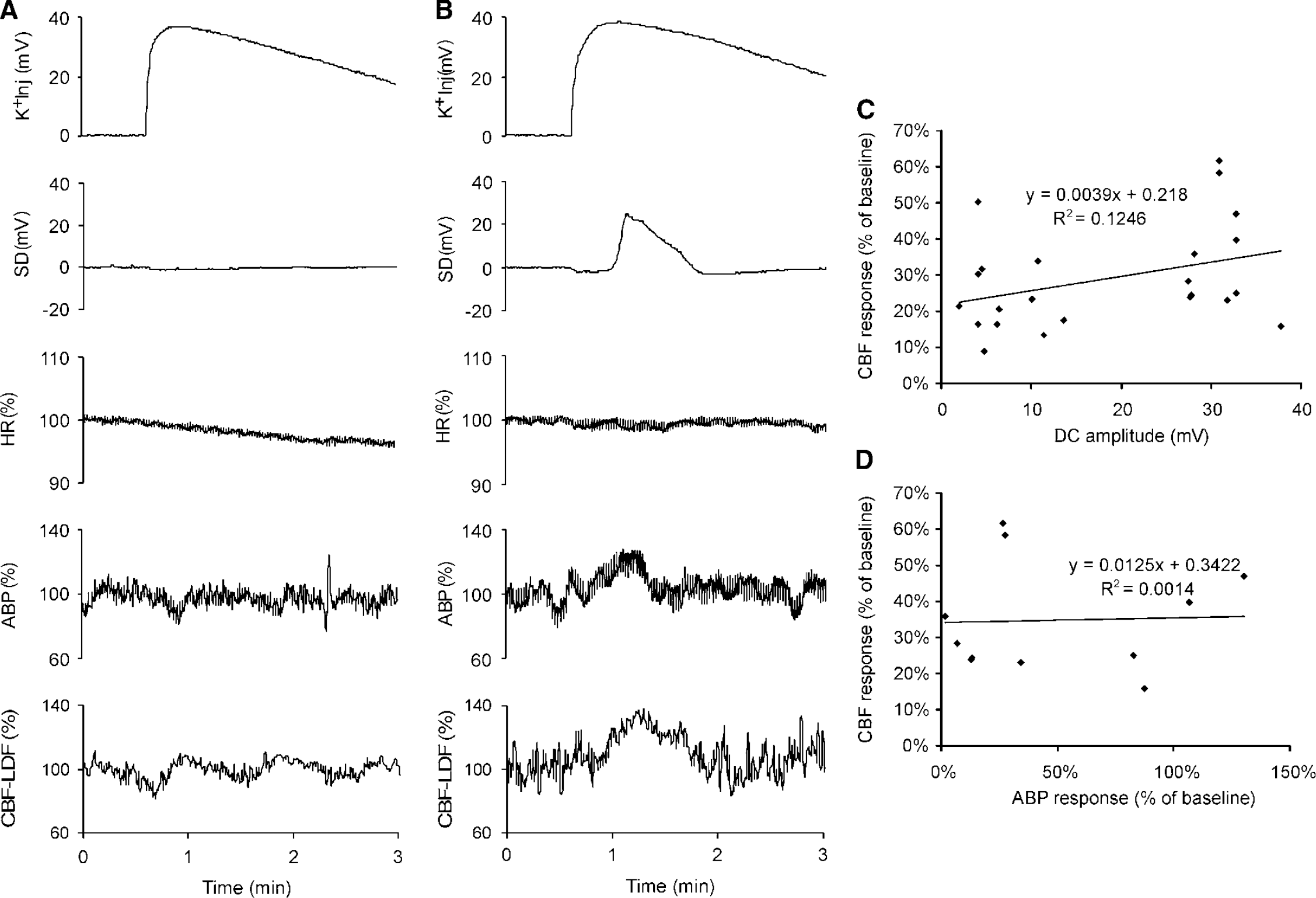
Simultaneous recordings of DC potentials at the microinjection site (K+Inj), and at a horizontal distance of 1 mm, of SD, HR, ABR and brainstem CBF). (
Discussion
In the present study, we have shown for the first time that the brainstem of the adult rat is not completely resistant to SD. Under suitable conditions, negative DC deflections could be elicited that were accompanied by elevations of [K+]e and migrated within the brainstem tissue at least over distances of 1 to 2 mm. The blockade of NMDA receptors prevented the generation and propagation of negative DC deflections, indicating that glutamate is involved in the propagation process. Furthermore, negative DC deflections in the brainstem were regularly accompanied by transient increases in rCBF and ABP.
In accordance with Somjen (2001), we consider the negative DC deflections in the brainstem as SDs, since (i) they depolarized the brainstem tissue, (ii) they had amplitudes and time courses similar to SDs in the cerebral cortex, (iii) they were accompanied by transient increases in [K+]e, and (iv) they propagated within the brainstem tissue at a similar speed as SDs in the cerebral cortex (Richter et al, 2003). The change in [K+]e during brainstem SDs was closely correlated with the amplitude of the DC shift. The average increase in [K+]e during brainstem SDs, however, was slightly lower than the increase in [K+]e during cortical SDs (Richter and Lehmenkühler, 1993). This difference might be because of the blockade of TEA-sensitive potassium channels, as a similar reduction in the magnitude of [K+]e was observed in hypoxia-induced hippocampal SDs after TEA treatment (Aitken et al, 1991).
To the best of our knowledge, so far only Takahashi et al (1981) attempted to monitor changes in [K+]e in the native adult rat brainstem. They induced an acute closed head injury by a falling Bakelite column to the skull and found that this impact caused increases in [K+]e in the brainstem ranging from 10 to 50mmol/L and more, and these changes were accompanied by transient increases in ABP. However, they only observed the cortical electroencephalogram and did not explore whether the increase in [K+]e in the brainstem was related to a DC deflection in the brainstem. In their work, cortical SDs evoked by KCl did not trigger changes in [K+]e in the brainstem.
The present experiments confirm that the adult brainstem is resistant to negative DC deflections in the native state (Bureš et al, 1974). Why this is the case has not been clarified yet. According to the criteria of Bureš, SDs are prevented by the inhomogeneous structure of the native brainstem where nuclei and myelinated zones form boundaries, thus protecting the normal function of cardiovascular and respiratory centers located nearby. Furthermore, according to Bureš et al (1974), susceptibility to SD is indirectly proportional to the amount of extracellular space and myelin per unit volume brain tissue. However, the conclusions of Bureš are derived from experiments on normal brains. More recent observations, for example, in animals with channelopathies, revealed that SD principally can occur in most brain areas including the cerebellum (Ebner and Chen, 2003). Recent data on cortical SDs in man (Strong et al, 2007a) clearly demonstrated that SDs occur spontaneously in ischemic human neocortex, and they may even reemerge, thus probably increasing the volume of damaged brain areas. It is likely, therefore, that resistance to SD is not completely explained by histomorphological data. Probably susceptibility or resistance to SD is also controlled by functional parameters such as cellular permeability for ions and the metabolic state of the cell.
As reported before, even the replacement of extracellular chloride ions by acetate alone was insufficient to render the brainstem susceptible to SDs (Richter et al, 2003). The major step toward increased susceptibility to SDs in the present study was the increase in [K+]e, which was achieved by blocking K+ channels with TEA and by adding KCl to the solution. This is in accordance with theoretical models suggesting that significant depolarization of neuronal membranes is a prerequisite to ignite an SD wave (Somjen, 1979, 2001; Somjen et al, 1992) and that a continuous supply of K+ could provide sufficient depolarization (Kager et al, 2002). Tetraethylammonium chloride was successfully used for conditioning of frog retina for SD (Fujimoto and Yanase, 1991). Tetraethylammonium chloride affects a number of different potassium channel types such as delayed rectifier potassium channels, the M potassium channels that are inactivated by acetylcholine, and inward rectifier potassium channels (Aronson, 1992), whereas background (leak) potassium channels are relatively insensitive to TEA (Lesage, 2003). Obviously, the sum of these effects slightly increases excitability without causing sustained depolarizations. The addition of potassium ions successfully conditioned the brainstem of rats aged 14 to 21 days for SD ignition by KCl in previous experiments. In the present study, the superfusion of the brainstem with acetate-ACSF-TEA-KCl caused an increase in [K+]e up to 6mmol/L per h and most likely partly depolarized the neurons and increased their excitability to the SD stimulus (KCl). Importantly, both topical application of a KCl crystal to the brainstem surface (a conventional method we previously used in immature rats; Richter et al, 2003, 2005) and microinjection of KCl deep into the brainstem generated similar SDs, showing that SDs can be ignited at different depths in the brainstem. Microinjection of KCl caused shorter SDs of a more uniform shape, which is probably related to the different application mode.
Notably, this conditioning process and elevation of [K+]e was not toxic because (i) the elevated susceptibility against SD was not associated with sustained depolarization, (ii) the elevated susceptibility against SD was reversed by washing out the conditioning solution, (iii) in the same rat susceptibility against SD could be restituted by superfusion with acetate-ACSF-TEA-KCl, and (iv) animals survived this conditioning procedure over 3 to 4 h without changes in HR or blood pressure indicating that cardiovascular regulatory centers in the brainstem were not significantly affected.
In the cortex, NMDA receptors are significantly involved in SD generation (Marrannes et al, 1988). It is assumed that the initial depolarization by an increase in [K+]e triggers the release of glutamate, which then activates NMDA receptors, and that this mechanism is able to further increase [K+]e and induce migration of SDs (Somjen, 2001). N-methyl
Effect of Brainstem Spreading Depression on Regional Blood Flow and Arterial Blood Pressure
Cortical SDs are typically accompanied by changes in rCBF but the sequence of changes may show different patterns depending on the conditions of the brain. During migraine in patients, Lauritzen et al (1983) observed a hyperemia followed by an oligemia, and supposed that these rCBF changes are related to cortical SDs. By contrast, during experimental SD in ischemic tissue, an ‘inverse coupling’ was observed, namely, first a spreading oligemia, which was then followed by a spreading hyperemia (Strong et al, 2007b, Shin et al, 2006). In the normal brain of cats (Piper et al, 1991) and rats (Lehmenkühler et al, 1976; Ueda et al, 1997; Brennan et al, 2007), cortical SDs usually caused a temporary increase in rCBF. However, an inverse coupling of cortical blood flow was observed in rat neocortex when [K+]e was elevated and nitric oxide synthase was inhibited (Dreier et al, 1998), and elevation of [K+]e alone caused a short initial hypoperfusion accompanying a cortical SD (Dreier et al, 2000). In general, the mechanisms of blood flow changes are not entirely clear. Sanchez-del-Rio and Reuter (2004) supposed that hyperperfusion during cortical SD is partly mediated by the release of trigeminal and parasympathetic neurotransmitters from perivascular nerve fibers. Endothelium-derived NO obviously did not contribute to the vasodilatation in the normal cerebral cortex (Shimizu et al, 2002), but might play a role in a state of hypercapnia (Scheckenbach et al, 2006).
In the present experiments, we monitored blood flow in vessels of brainstem tissue because the dura and arachnoid above the brainstem were removed before measurements. It was a consistent finding that rCBF in the brainstem transiently increased during SD. The changes in blood flow were tightly linked to SDs, suggesting that SDs were a cause for blood flow changes. Indeed, increases in rCBF started after a short delay after initiating the SD by microinjection, and the time course paralleled the time course of SD. The strongest evidence for a causal role of SD in the blood flow change is the fact that the application of KCl to the native brainstem did not cause any change in rCBF. Only when the brainstem was conditioned for SDs and when KCl application elicited SDs, changes in rCBF were observed. It should be noted, however, that the magnitude of rCBF changes did not depend on the SD amplitude. Furthermore, unlike in the cortex (Lauritzen et al, 1983), SD-induced hyperemia in the brainstem was not followed by spreading oligemia. It may be that in thiopental-anesthetized animals, the increases in rCBF are too small to evoke an oligemia after hyperemia (Wada et al, 1996).
During brainstem SDs, we also noted increases in the ABP. We assume that SD waves might have reached cardiovascular regulatory areas in the brainstem and that activation of these centers caused transient blood pressure changes. It is thus possible that rCBF changes were caused by increases in systemic ABP. However, the magnitude of CBF changes was not correlated to the magnitude of blood pressure changes. This indicates that local factors in the brainstem were at least partly responsible for increases in brainstem blood flow changes. The HR was influenced by brainstem SDs only in few rats. This weak effect may be because of barbiturate anesthesia. This anesthetic agent causes no change in basal tonic sympathetic nerve activity, but markedly diminishes the baroreceptor reflex control of HR and sympathetic nerve activity (Barringer and Bunag, 1990; Shimokawa et al, 1998).
In conclusion, the present study shows that the brainstem of the adult rat is not generally resistant to generation of SDs. However, the brainstem is much more protected against SDs than the cerebral cortex, and the present study provides evidence that particular conditioning factors are necessary in this respect. When elicited, cortical and brainstem SDs share many common features, such as modes of ignition and propagation and the influence on rCBF. Under which conditions brainstem SDs can occur in human and whether they are functionally significant is unknown yet. In immature rat brainstem, periods of hypoxia or a transient asphyxia of 2 to 3 mins was able to condition this brain area for SDs (Richter et al, 2003). It can be speculated that SDs in adult human brainstem is involved in brainstem pathophysiology, such as brainstem ischemia, provided conditioning factors lower threshold for elicitation of SDs. In this context, it may be speculated that brainstem SDs are involved, for example, in basilar-type migraine. During this migraine subtype, patients show typical brainstem symptoms in the aura phase (Kirchmann et al, 2006). Furthermore, in magnetic resonance imaging investigations, migraineurs exhibit more ‘hyperin-tensities’ in the brainstem than healthy control subjects (Kruit et al, 2006). It may be speculated that these changes are caused by ischemic processes in the brainstem, and that brainstem SDs are involved in the generation of such changes.
Disclosure/Conflict of Interest
No competing interests are disclosed by the authors of this study.
Footnotes
Acknowledgements
We thank Mrs Konstanze Ernst and Mrs Gabriele Cuny for excellent technical assistance.