
Abstract
Ion-selective microelectrodes were used to study acute effects of N-methyl-
Keywords
Traumatic brain injury (TBI) produces a range of disturbances in tissue functions. Some of these changes (i.e., ischemia, seizures, hemorrhages, disturbances in ion homeostasis and metabolism, edema) are potential mechanisms of secondary injury to cells that have survived the initial insult. A better understanding of these mechanisms is essential for improvements in treatment of TBI.
CNS trauma results in large transient increases in interstitial (IS) levels of excitatory amino acids (EAAs) (Faden et al., 1989; Katayama et al., 1990; Nilsson et al., 1990) and disturbances in Ca2+ homeostasis (Young et al., 1982; Young and Koreh, 1986; Fineman et al., 1993; Nilsson et al., 1993). Treatment with N-methyl-
The present study was undertaken to determine whether blockade of NMDA or α-amino-3-hydroxy-5-methyl-4-isoxazole (AMPA) receptors affects acute Ca2+disturbances seen after cerebral cortical compression contusion and to determine whether delayed changes of regional Ca2+homeostasis correlate with tissue damage.
MATERIALS AND METHODS
Thirty-eight male Sprague Dawley rats (302–490 g) from Møllegaard (Copenhagen, Denmark) were used. They had free access to pellets and water. Anesthesia was induced by placing the rats in a gas mixture of 3% halothane and O2:N2O (1:1). After induction, they were intubated and artifically ventilated in a small animal ventilator with 1.5% halothane and O2:N2O (1:3). Arterial and venous catheters were implanted in the tail vessels. Mean arterial blood pressure (MABP) and body temperature were continually measured and arterial blood gases were checked throughout the experiment. After catheter preparation the animals were placed in a stereotaxic frame and lidocaine was administered subcutaneously over the galea. Halothane was replaced with isoflurane (1.4–1.6%). The rationale for using two anesthetics was twofold: (i) we find it easier to intubate the animals when induced with halothane, and (ii) we wanted to reduce isofluranecosts. Succinylcholine (5 mg/ml, 0.2 ml) was given every 15–20 min henceforth. A craniotomy was made over the right parietal cortex (diameter, 6 × 9 mm) with its center 3 mm posterior to the bregma and 2.5 mm lateral to the midline. This is 1.5 mm posterior to the location employed in our previous studies (Nilsson et al., 1990, 1993). Trauma was produced (with maintained general anesthesia) by dropping a 21-g weight from 35 cm onto a piston (diameter, 4.5 mm) resting on the dura. The piston was constructed to allow a maximum compression of 1.5 or 2 mm (Feeney et al., 1981).
IS [Ca2+] measurements
Twenty animals were divided into four groups for [Ca2+]e studies: Group 1 (four animals), 1.5-mm trauma; Group 2 (six animals), 2-mm trauma; Group 3 (five animals), 2-mm trauma and a 3 mg/kg intravenous (i.v.) bolus dose of dizocilpine maleate (MK-801) given 20 min pretrauma; and Group 4 (five animals), 2-mm trauma and a 30 mg/kg i.v. bolus dose of 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[F] quinoxaline (NBQX) given 10 min pretrauma.
Double-barreled ion-sensitive microelectrodes were used to register the [Ca2+]e and direct current (DC) potential as described by Nilsson et al. (1993). The liquid ion-exchanger for the [Ca2+]e measurements was based on a calcium ionophore (ETH 1001). The electrodes were positioned by micromanipulators in the shear stress zone (SSZ) and in the central region (CR) (Fig. 1), 1 mm below the surface of the cortex, through a small slit in the dura. A baseline level was established and then the electrodes were removed. Immediately after trauma, ≤1 min, the electrodes were replaced into exactly the same position and registration was carried out up to 120 min. The electrodes were calibrated before and after the experiment in ion solutions of known concentrations. The electrodes were connected to high-impedance differential amplifiers and the signals displayed on a pen recorder. The DC potential was recorded between the reference barrel and a grounded Ag/AgCl electrode placed in the hindpaw (see Nilsson et al., 1993).
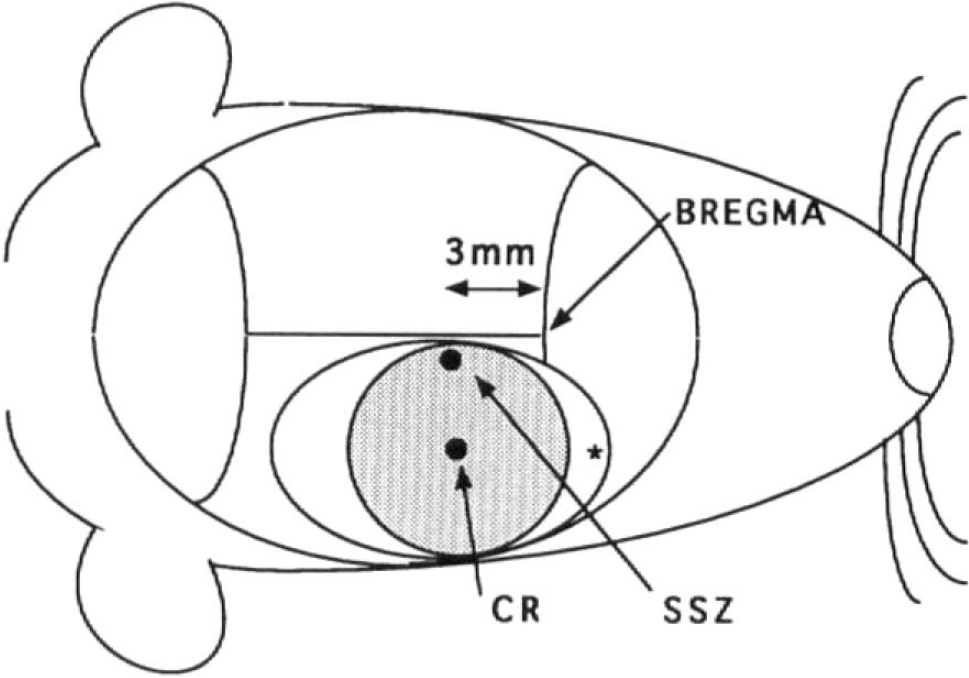
Schematic drawing of the rat head. The shaded circle represents the area under the piston and the oval outside this area shows the boundaries for the craniotomy. (*) Area where needle prick for CSD was done. CR, Central region; SSZ, shear stress zone.
To test the functional integrity of the tissue, cortical spreading depression (CSD) (Leão, 1944) was produced at different time intervals postinjury by pin pricks to the noninjured cortex along the anterior edges of the craniotomy (Fig. 1). A lack of ionic changes (decrease in [Ca2+]e to ∼0.1 mM) and a DC shift (>10 mV) typical for CSD (Hansen and Zeuthen, 1981), recorded in the SSZ or CR, were taken as a sign of functional disturbance of the tissue (see Nilsson et al., 1993).
The experiments were terminated by giving the animals an i.v. bolus of lidocaine that resulted in immediate cardiac arrest.
Morphology
A group of nine rats was used to define the gross and light microscopic changes. The group was subdivided into controls (n = 3), MK-801-treated (3 mg/kg i.p. given 30 min posttrauma) (n = 3), and NBQX-treated (30 mg/kg i.p. given 30, 60, and 90 min posttrauma) (n = 3) animals. Two-millimeter trauma was produced in the same manner as described above with the exception that no slits were made in the dura and the bone flap was replaced (without fixating it to the intact skull) prior to closure of the scalp. The animals were extubated ∼20 min posttrauma and then kept under observation in their cages.
The rats were reanesthetized with mebumal (50 mg/kg i.p.) after 3 days. The thorax was rapidly opened and the brain perfusion fixated by infusion of 250 ml of physiological saline followed by 250 ml of a 4% formaldehyde solution through the heart's left ventricle, using the opened right atrium as an outlet. Four-micrometer-thick paraffin coronal sections were stained with hematoxylin and eosin (H&E) or Klüver-Barrera or immunohistochemically for glial fibrillary acid protein (GFAP) and serum proteins. The GFAP was detected by a rabbit anti-cow GFAP antibody (DAKO Z 334) diluted 1:1,600. Serum proteins were detected by a rabbit anti-rat whole-serum antibody (DAKO Z 178) diluted 1:1,600. All antibodies were used in a peroxidase antiperoxidase (PAP) procedure. The tissue was incubated with a primary antibody overnight at 4°C. Swine anti-rabbit IgG diluted 1:40 was used as the secondary antibody for 30 min and rabbit PAP complex diluted 1:100 was used for another 30 min. The peroxidase activity was demonstrated by staining with 3-amino-9-ethylcarbazol (AEC; Sigma A-5754). Between each step the tissue was washed three times with phosphate-buffered saline at pH 7.4. As negative controls the primary antibody was omitted in the staining procedure. Endogenous peroxidase was blocked by pretreatment of the tissue with 3% hydrogen peroxide in water.
45Ca2+ autoradiography
Two-millimeter trauma was produced in nine animals in the same manner as for the morphology group. Prior to sacrifice the rats were reintubated and a catheter was placed in the left femoral vein. We used the procedure described by Dienel (1984), in which 250 μCi of 45Ca2+(Amersham) was given as an i.v. bolus at 3, 19, and 67 h postinjury. The rats were extubated and kept under observation until 5 h had elapsed. The preparation was planned so that the three groups (n = 3) could be sacrificed at 8, 24, and 72 h after injury, respectively. Mebumal (50 mg/kg i.p.) was given and the animals were decapitated. The brains were carefully removed, submerged in −70°C isopenthane, and stored at −70°C until sectioned. Ten-micrometer-thick frozen sections were stained with H&E or Klüver-Barrera or immunohistochemically for GFAP and serum proteins. For autoradiography 20-μm-thick frozen sections were placed on films (Hyperfilm MP; Amersham) and exposed for 3 weeks at 4°C. The sections were evaluated by comparing the traumatized side with the contralateral side.
Statistical analysis
For statistical analysis Ca2+ data were evaluated with a repeated measures analysis of variance (ANOVA) followed by Dunnett's test at each time point (SuperA-NOVA; Abacus, CA, U.S.A.). Differences with a p value <0.05 were considered to be statistically significant.
Ethics
The experimental protocol was approved by the local ethics committee for animal research.
RESULTS
Physiological parameters
Physiological parameters were maintained at the following levels: rectal temperature, 37.5°C; Pco2, between 35 and 41 mm Hg (4.7–5.5 kPa); Po2, 83–135 mm Hg (11–18 kPa); pH, 7.35–7.45; and MABP, ≥100 mm Hg.
Morphology and histochemistry
At 3 days tissue changes closely resembled earlier findings with a 1.5-mm trauma (Nilsson et al., 1993). In the SSZ, H&E staining showed spongiosis, distorted neurons, numerous eosinophilic neurons, polymorphonuclear leukocytes, and phagocytic cells (Fig. 2). Intense GFAP activity was also seen. Serum proteins were present IS, as well as in neurons with either distorted outlines and pyknotic nuclei or with a preserved structure (Fig. 3).
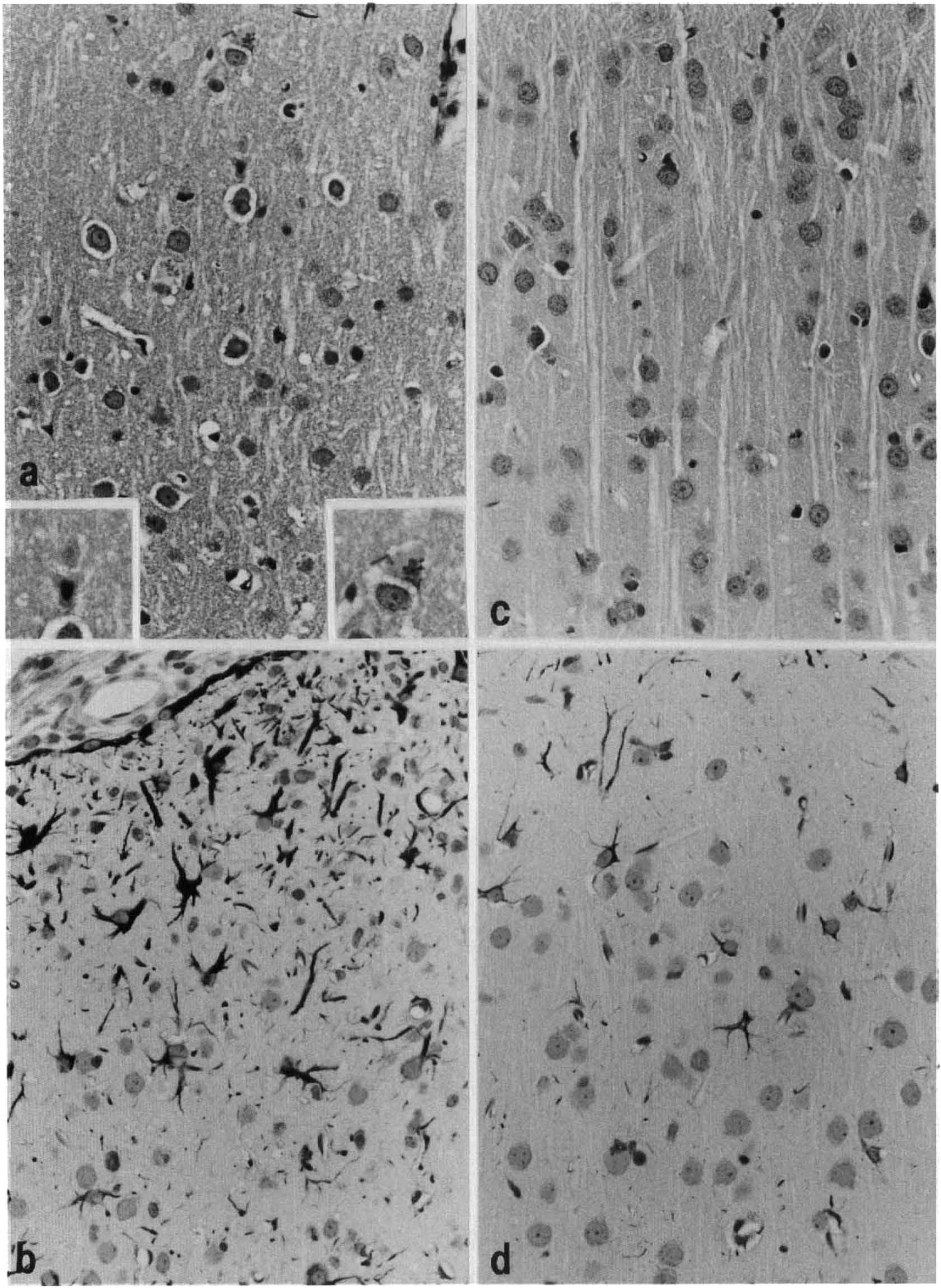
Histology at 3 days after 2-mm trauma,
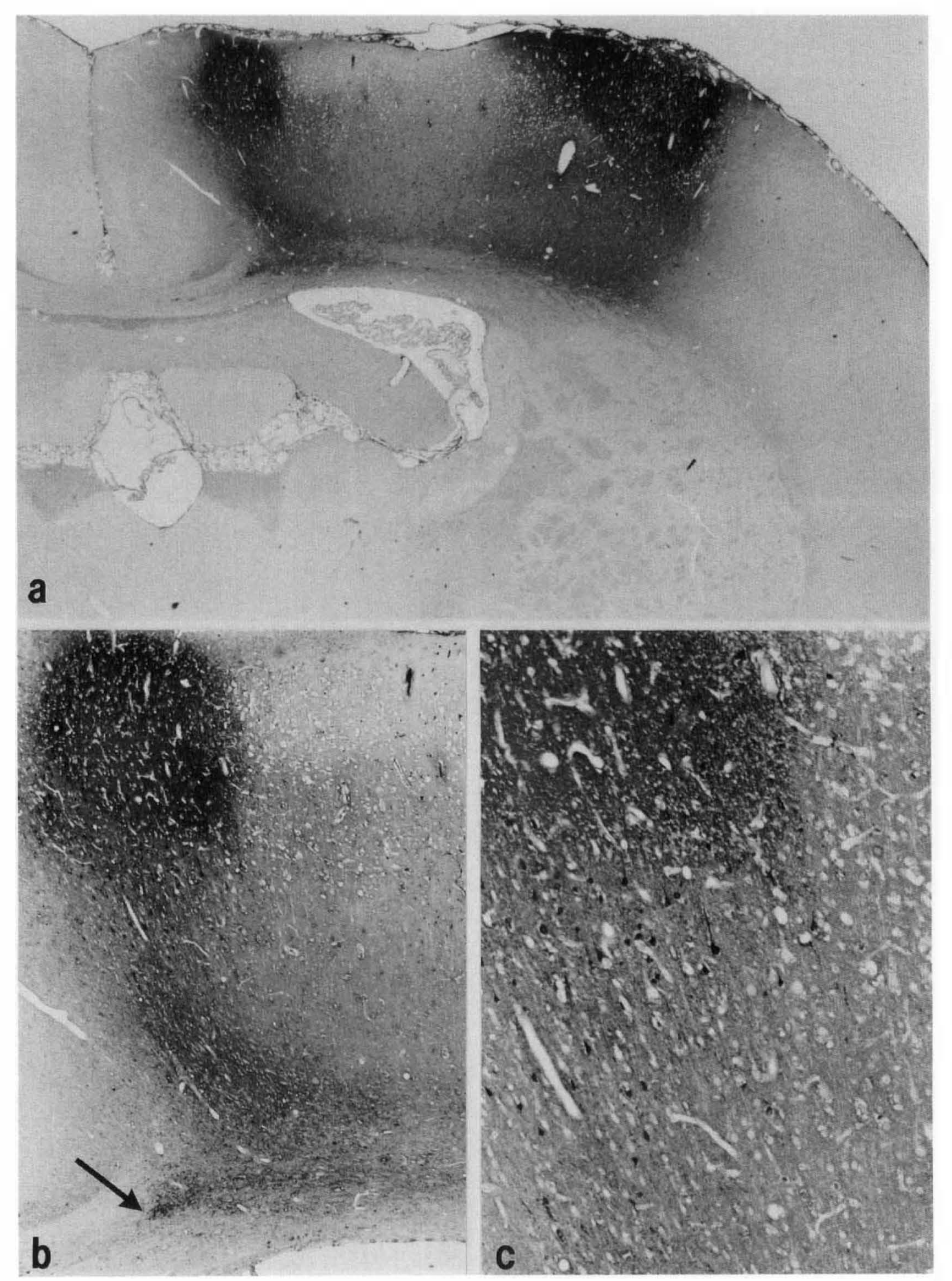
Immunohistochemical staining for serum proteins 3 days after 2-mm trauma,
In the CR the only pathological changes were occasional eosinophilic neurons. Staining for serum proteins and GFAP was not different from that of controls.
Pretreatment with MK-801 or NBQX did not have any obvious effect on the histological pattern.
Frozen sections: Histochemistry and 4SCa2+ autoradiography
These rats showed a progression of changes over time in the SSZ. At 8 h posttrauma there were spongiosis, distorted neurons, and an increase in serum proteins. At 24 h eosinophilic neurons had become numerous, and the presence of serum proteins was more pronounced and partly located IC. Polymorphonuclear leukocytes were also seen. At 72 h the above-mentioned changes were even more pronounced. Macrophages and reactive astrocytes, as shown by GFAP staining, were present.
Autoradiography showed an accumulation of 45Ca2+ in the SSZ at 8 h (Fig. 4). The density of this area increased at 24 and 72 h, signifying further 45Ca2+ accumulation. Inspection of Figs. 3 and 4 reveals that the area of 45Ca2+accumulation corresponded to the presence of serum proteins but exceeded by far the distribution of neuronal necrosis. The correlation between 45Ca2+and serum protein accumulation was further stressed by the localization of both 45Ca2+ and serum proteins in non-blood–brain barrier (BBB) areas such as the pituitary gland (Fig. 4).
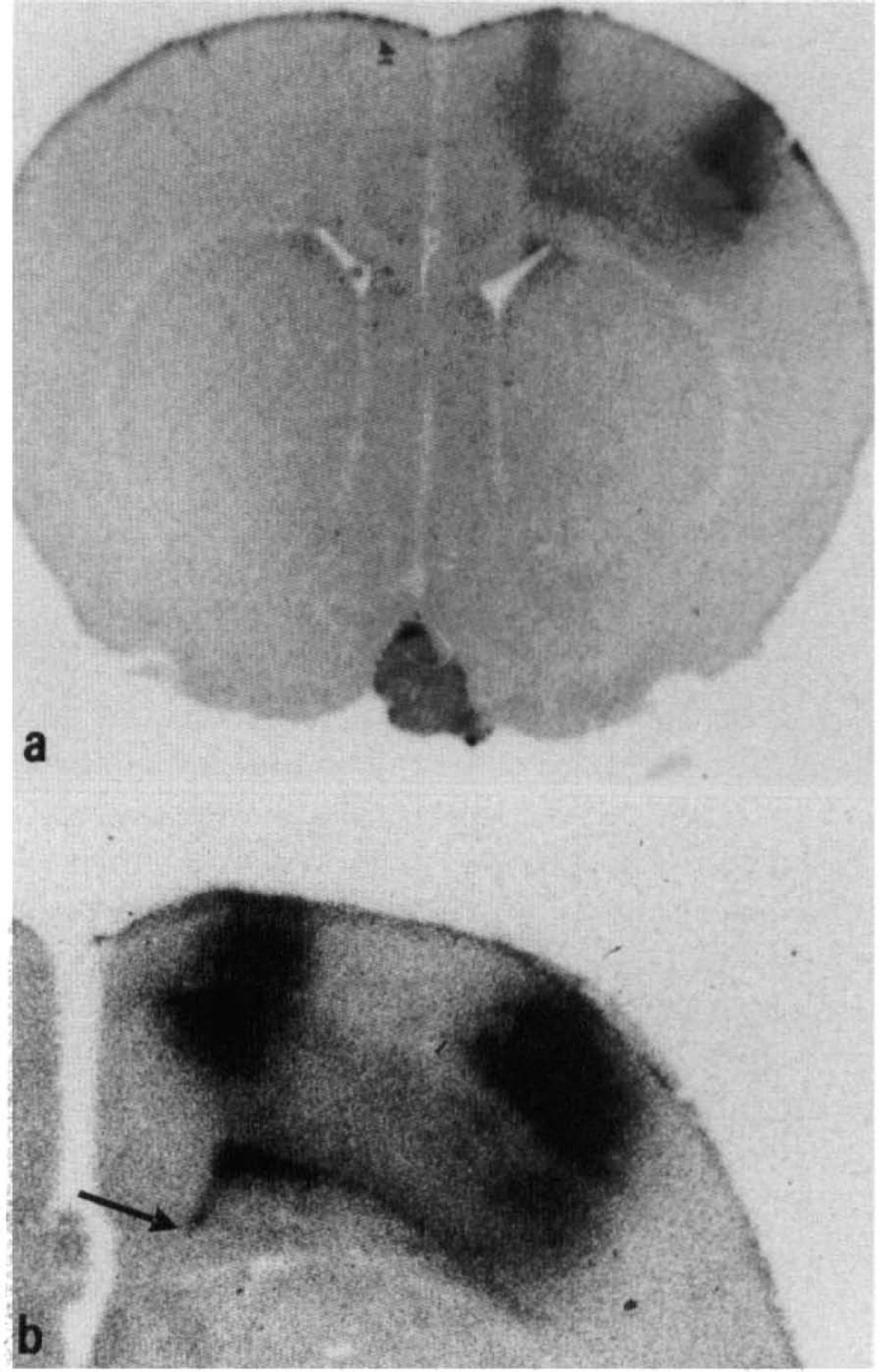
45Ca2+ autoradiograms after 2-mm trauma,
In the CR there were no pathological changes present other than occasional eosinophilic neurons. There was no apparent 45Ca2+accumulation in the CR compared to the contralateral side.
Measurements of [Ca2+]e
Ionic changes in response to 1.5-mm trauma differed between the SSZ and the CR (Fig. 1, Table 1) in accordance with Nilsson et al. (1993). Immediately after trauma the [Ca2+e decreased dramatically, from a basal level of 1.1 to 0.1 mM in the SSZ and from 0.9 to 0.3 mM in the CR. Recovery of [Ca2+e in the SSZ (50 min) was significantly delayed compared with that in the CR (10 min).
Trauma-induced changes of interstitial Ca2+ levels
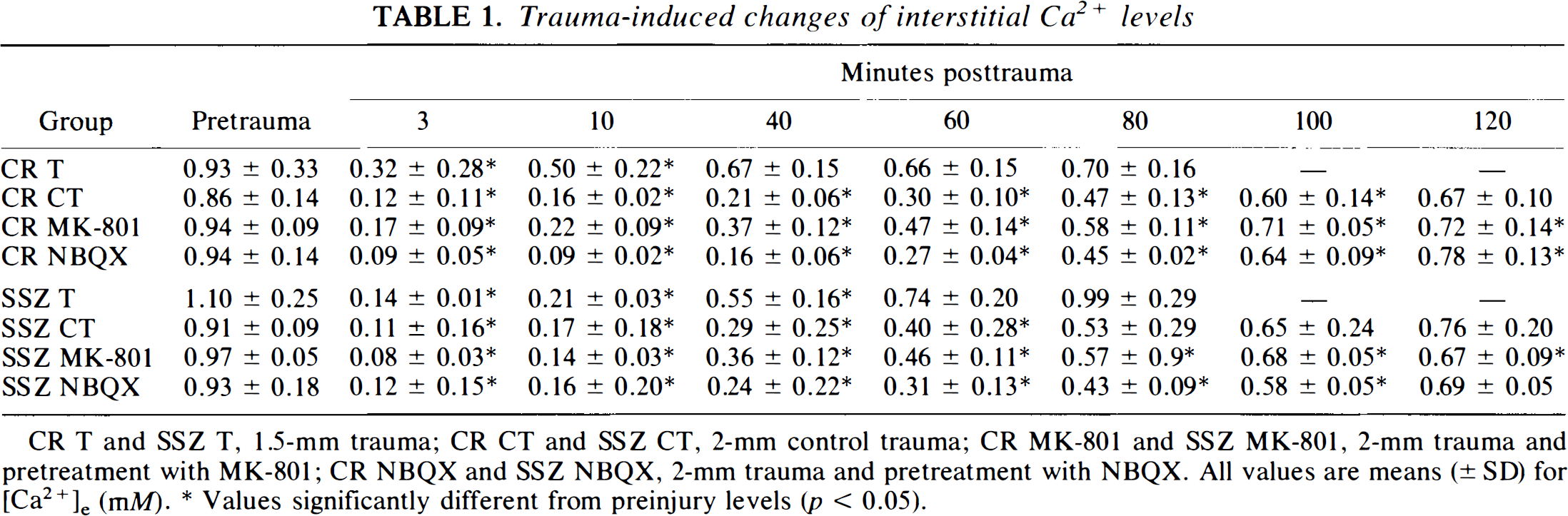
CR T and SSZ T, 1.5-mm trauma; CR CT and SSZ CT, 2-mm control trauma; CR MK-801 and SSZ MK-801, 2-mm trauma and pretreatment with MK-801; CR NBQX and SSZ NBQX, 2-mm trauma and pretreatment with NBQX. All values are means (± SD) for [Ca2+e, (mM).
Values significantly different from preinjury levels (p < 0.05)
In the SSZ, CSD could not be detected after the trauma, while the CR regained the ability during the 70-min observation period. In one of the four rats, however, no CSD could be elicited, perhaps because the animal required a higher isoflurane concentration (2%).
In the 2-mm control trauma group both the SSZ and the CR regions exhibited similar decreases and recovery rates for [Ca2+e (Table 1, Fig. 5). CSD was not seen in the SSZ following trauma in five of six animals, while the CR regained its ability to react to CSD at between 35 and 120 min in five of the six animals (two of these reacted with DC shifts between 4 and 6 mV).
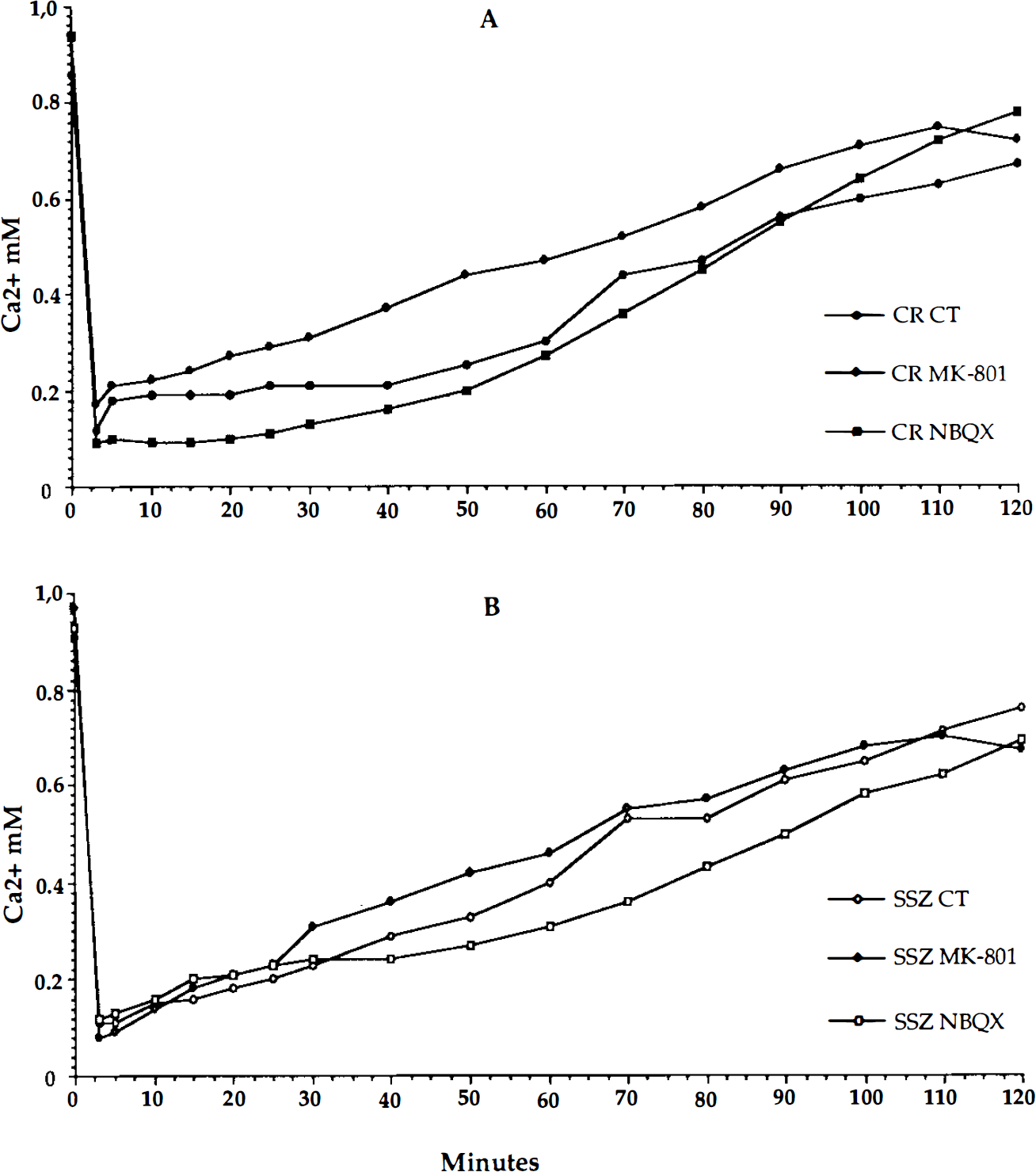
Pretreatment with MK-801 (3 mg/kg i.v.) did not result in any differences between the regions with regard to decrease or recovery rate for [Ca2+e following trauma (Table 1, Fig. 5). As expected (Gill et al., 1992; Nellgård and Wieloch, 1992), CSD reactions were not seen in MK-801-treated animals.
Pretreatment with NBQX (30 mg/kg i.v.) did not influence the decrease or rates of recovery for [Ca2+e following trauma (Table 1, Fig. 5). In the SSZ, CSD could not be evoked in any of the five animals during the 120-min period. In the CR, CSD were seen in three of the animals (two of these were incomplete with DC shifts between 2 and 6 mV). The two animals that did not regain CSD required a slightly higher isoflurane concentration (2%).
Comparison of means from control, MK-801-pretreated, and NBQX-pretreated animals showed no difference in [Ca2+e changes in either the SSZ or the CR (Table 1, Fig. 5). Recovery rates were also the same for the different groups and regions.
DISCUSSION
Severity of trauma
Previous work has shown that 1.5-mm trauma resulted in a dramatic increase in IS levels of EAAs in the SSZ (Nilsson et al., 1990, 1994). The EAA increase was accompanied by an acute lowering of [Ca2+e in the cerebral cortex, which was prolonged in the SSZ, while the CR recovered within 10 min (Nilsson et al., 1993). We assumed that the decrease in [Ca2+e reflected an IC accumulation of Ca2+and proposed that the ensuing neuronal loss, observed only in the SSZ, was related to the prolonged Ca2+ disturbance in this region. This hypothesis was tested in the present study.
The severity of the trauma was increased in this study by using a 2-mm compression depth instead of 1.5 mm. We found that the changes in [Ca2+e were now similar in the CR and SSZ but that the histological changes were still confined exclusively to the SSZ. In addition, the functional state of the cells in the CR appeared to be better preserved since the ability to propagate CSD returned during the 120-min observation period, albeit at a later time point than after 1.5-mm trauma (Nilsson et al., 1993). The SSZ did not regain this ability. Our results show that regions with comparable acute changes of [Ca2+e have different functional capacities and exhibit different degrees of neuronal damage. Other studies have also shown that disturbances in [Ca2+e, as indirect evidence of IC overload, are not necessarily associated with neuronal death. Repeated CSD, in which transient Ca2+ influxes are observed, did not produce histological damage (Hansen and Nedergaard, 1988). Furthermore, even prolonged Ca2+ disturbances were not damaging as shown in fluid percussion injury to the cerebral cortex (Fineman et al., 1993) and in spinal cord injury (Young et al., 1982). These results seem to question a pivotal role of calcium in trauma-induced cell damage. However, data on IC calcium accumulation and perhaps regional IC calcium levels are needed to clarify this issue.
Ca2+ measurements and glutamate receptor blockade
Since IC Ca2+ accumulation is thought to be mediated partly by agonist (i.e., EAA)-operated Ca2+channels and since EAA receptor antagonists are protective in TBI, we studied the effects of MK-801 and NBQX. Our results showed that pretraumatic NMDA or AMPA receptor blockade did not influence [Ca2+e changes in either region and that post-treatment did not have any profound effects on neuronal necrosis in the SSZ. Hence, the acute [Ca2+e decrease caused by trauma was not directly caused by glutamate acting on these receptors but, rather, mediated by other mechanisms. There is other evidence supporting this view. The IS changes of EAAs in the CR after 1.5-mm trauma (unpublished) were similar to those seen in the SSZ (Nilsson et al., 1990, 1994), while the [Ca2+e disturbances in the two regions were different (Nilsson et al., 1993).
What are the mechanisms behind the decreases in [Ca2+e seen after trauma? A regional lowering of [Ca2+e is the result of several processes: net uptake into brain cells, opposed by diffusion of calcium from unaffected regions and from the blood.
Uptake of Ca2+ into cells is probably the most important process and takes place by several routes: by unselective cationic channels operated by transmitters or by other means [e.g., stretch-activated channels (Sachs, 1991)], voltage-activated Ca2+channels, and carrier-mediated transport (e.g., reversal of the Na+/Ca2+ antiporter).
In our earlier study short-lived K+ and Ca2+changes in the CR were very similar to those during CSD (Nilsson et al., 1993). However, in the SSZ Ca2+ changes were prolonged and we concluded that they were caused by the combined effects of EAA receptor activation, stretch-activated channels, and buffering by mechanically damaged neurons (Nilsson et al., 1993). When trauma was increased to 2 mm the Ca2+ disturbances in the CR became prolonged and corresponded with changes seen in the SSZ. Since MK-801 and NBQX pretreatment did not influence [Ca2+e changes in either region, the role of EAAs in mediating these movements is questionable. It is possible that the trauma allows massive movements of Ca2+ into neurons and glia in both regions via stretch-activated channels. Buffering by damaged cells and serum proteins could also explain the prolonged Ca2+ disturbances in the SSZ, which exhibits acute histological changes, but not the Ca2+ disturbances in the histologically intact CR (Young et al., 1982). In this region the movements are more likely caused by disturbances of membrane functions including reversal of Na+/Ca2+ exchange or by activation of voltage-sensitive calcium channels. The fact that the tissue in the CR recovers the ability to support CSD also suggests intact function. Since [Ca2+e recovery in the SSZ is not coupled with a return of CSD activity, it is more likely that it is caused by diffusion of Ca2+ in the IS from surrounding, uninjured tissue (Young and Koreh, 1986) or from plasma via a damaged BBB (Cortez et al., 1989; Tanno et al., 1992a, b ).
45Ca2+ autoradiography
It is believed that accumulation of Ca2+ is the cause or sign of imminent cell death (Dienel, 1984). As shown in Fig. 4, 45Ca2+ accumulation seems to be confined to the SSZ where cell damage is seen. However, we found that 45Ca2+accumulation was evenly distributed within and outside the SSZ and it correlated directly with the distribution of serum proteins. Entry of serum proteins occurs via a defect BBB. Since these proteins (i.e., mainly albumin) are able to bind Ca2+, the accumulation of 45Ca2+is a result not only of cell damage but also of the presence of serum proteins.
In the CR region 45Ca2+ accumulation was not different from that in the contralateral site and no increase was observed at 8, 24, and 72 h. This regional pattern of 45Ca2+accumulation differs markedly from that seen following fluid percussion injury, where the entire ipsilateral hemisphere is significantly affected (Fineman et al., 1993). This probably reflects differences in the severity of the injury with respect to BBB damage.
In conclusion, the present results fail to support the hypothesis that cell loss in the SSZ is caused by acute intracellular Ca2+ overload mediated by EAAs in our TBI model. The acute [Ca2+e changes were not affected by blocking NMDA or AMPA receptors, and posttraumatic treatment with MK-801 or NBQX had no obvious effect on neuronal injury. The autoradiographic results show that 45Ca2+ accumulation is related mainly to the presence of serum proteins after BBB breakdown.
Footnotes
Acknowledgment:
The authors are grateful to Ms. Anne Krag Vinther and Ms. Ann Meisler for excellent technical help. The study was supported by the Swedish Association of Persons Disabled by Accidents or Polio, Swedish Society for Medical Research, Swedish Society of Medicine, Swedish Medical Research Council, Laerdal Foundation for Acute Medicine, and 1987 Foundation for Stroke Research.