
Abstract
Deficiency of phenylalanine hydroxylase activity in phenylketonuria (PKU) causes an excess of phenylalanine (Phe) throughout the body, predicting impaired synthesis of catecholamines in the brain. To test this hypothesis, we used positron emission tomography (PET) to measure the utilization of 6-[18F]fluoro-
Keywords
Introduction
Phenylketonuria (PKU) is caused by hereditary deficiency of the enzyme phenylalanine hydroxylase, or deficient production of the cofactor tetrahydrobiopterin, both conditions resulting in elevated levels of phenylalanine (Phe) and its metabolites throughout the body. When untreated, elevated Phe in the plasma of PKU patients can competitively inhibit the brain uptake of tyrosine via the common carrier for large neutral amino acids (LNAAs) (Choi and Pardridge, 1986). Magnetic resonance spectroscopy has shown that the Phe concentration in normal human brain is elevated after oral Phe loading (Moller et al, 1998), predicting a decline in the availability of tyrosine in brain; this phenomenon has been conclusively shown in a mouse model of PKU (Puglisi-Allegra et al, 2000).
It is commonly accepted that PKU patients require a Phe-restricted diet from birth until adolescence, and require oral tyrosine supplements to prevent mental retardation. Although some expert panels recommend lifelong continuation of the PKU diet (NIH Consensus Development Panel, 2001), it remains a matter of debate as to which blood level of Phe is acceptable in adult PKU patients (Department of Health, 1991; Przyrembel, 1996). Even adults with documented adequate diet during childhood and adult Phe levels less than 1 mmol/L (<17 mg/dL) may show reduced performance in executive functioning, attention, verbal memory, expressive naming, and verbal fluency tests. Adults with Phe levels above this threshold have even lower performance in tests for focused attention, verbal fluency, reaction time, verbal recognition memory, visual memory, and naming (Brumm et al, 2004). In addition to these cognitive deficits, motor dysfunction characterized by increased muscular tone, ataxia, and tremor can occur in adult PKU; comorbidity with parkinsonism has also been reported in rare cases (Evans et al, 2004). Furthermore, reduced concentrations of the main dopamine metabolite, homovanillic acid, and the serotonin metabolite, 5-hydroxy-indoleacetic (Lykkelund et al, 1988), have been reported in the cerebrospinal fluid of PKU patients, suggesting a central impairment of monoamine neurotransmission. This raises the possibility that certain aspects of neurophysiological impairment in PKU patients tests may be mediated by impaired dopamine transmission, affecting the dorsolateral prefrontal cortex, which subserves the aspects of executive function performance (Channon et al, 2005; Ullrich et al., 1996), or indeed the striatum itself.
There have been few functional imaging studies of human PKU; a pilot study with [18F]fluoro-ethyl-spiperone did not reveal major changes in dopamine D2 receptor availability, but did indicate reduced [11C]tyrosine uptake throughout the brain (Paans et al, 1996). Radioactive tyrosine is a tracer for LNAA transport and cerebral protein synthesis, but only a tiny fraction is incorporated into brain catecholamines (Cumming et al, 1998). In contrast, the positron-emitting levodopa analogue 6-[18F]fluoro-
Materials and Methods
Subjects and Methods
Seven patients with PKU (two males, five females; age: 21 to 27 years) were included in the study. To avoid confounding effects of infantile brain damage, great care was taken to select patients with no impairment in intellectual or neurologic function; all patients underwent extensive neuropsychological testing and neurologic examination. Psychometric testing was performed before the PET scan with the LPS (Leistungsprüfungssystem, a performance testing system), TMT (trail making test), tower of Hanoi, RWT (Regensburger word fluency test), HAWIE (Hamburg—Wechsler intelligence test for adults), and WCST (Wisconsin card sorting test). The patients were neurologically completely unremarkable and performed at average to above average in all tests of cognitive performance. A reference group of seven age-matched, healthy male volunteers (age: 23 to 34 years) was recruited for FDOPA/PET imaging under identical conditions. As there is no gender difference of FDOPA influx in striatum, gender matching seemed unnecessary, and would, in any event, have been problematic with respect to regulatory issues concerning the recruitment of healthy female volunteers of childbearing age. All subjects gave written, informed consent.
The PKU patients imaged had adhered to a Phe-restricted diet until the age of 15 to 18 years, and were without strict diet from 4 to 11 years. MRI (magnetic resonance imaging) (MP-RAGE (magnetization prepared rapid gradient echo) 1 mm) was performed for each subject to get three-dimensional images of individual brain anatomies; no subject had to be excluded due to brain morphologic abnormalities. Immediately before PET, blood samples were obtained for assay of Phe, tyrosine, and other LNAAs. Serum amino-acid levels were measured by ion-exchange chromatography and ninhydrin postcolumn derivatization (amino acid analyzer LC 3000) (Schulpis et al, 2002).
Positron Emission Tomography
All the subjects were administered carbidopa (2 mg/kg body weight) orally, 60 mins before scanning, to block DOPA decarboxylase activity outside the brain. To correct for tissue attenuation, transmission scans using three rotating 68Ge line sources were acquired before injecting FDOPA. Emission data were acquired dynamically (30 time frames: 3 × 20 secs, 3 × 1 mins, 3 × 2 mins, 3 × 3 mins, 15 × 5 mins, and 3 × 10 mins) with an ECAT EXACT PET in three-dimensional mode on intravenous administration of FDOPA (185±12 MBq).
Dynamic time—radioactivity curves were obtained for the cerebellum, the bilateral caudate nucleus, and the putamen ROIs (region of interests) from each subject, as described below. The fractional rate constant for the striatal utilization of FDOPA was calculated by linear graphical analysis (Gjedde, 1982; Patlak and Blasberg, 1985), modified for the case of a reference tissue analysis (Hartvig et al, 1991). Basically, to evade the need for an arterial plasma sampling, the individual cerebellum time—activity curve is used as a reference tissue input function. After modification of the Patlak—Blasberg plot (Patlak and Blasberg, 1985) for determination of striatal rate constants using a reference region, the equation for the graphical analysis can be described as R(T)=R0+k3R0((∫T0Mr(t)dt)/Mr(t)), where R is the ratio of activity concentration in the striatum to the reference tissue and Mr(t) the activity content of a reference region. The slope (k3R0) of the linear regression in this analysis correlates with the relative activity of DOPA decarboxylase (k3S, also k3ref, ki; min−1), whereas the ordinate intercept (R0) corresponds to the equilibrium distribution volume of FDOPA in the cerebellum reference tissue (for further details see Hoshi et al, 1993). We used recording frames 12 to 22 (30 to 90 mins) for control subjects. Because of the delayed kinetics of FDOPA, we used recording frames 17 to 25 (60 to 120 mins) for the linear regression analysis in PKU patients.
In addition, parametric maps for the voxelwise calculation of k3S were made for each subject, and mean parametric maps by group were calculated after spatial normalization of the individual maps to a common coordinate system, using a ligand-specific template. As a surrogate for the unidirectional blood—brain clearance of FDOPA (K1D), we made average maps of the summed uptake from the first till the sixth minute of the recording, scaled to the amount injected and to body weight. The fraction of untransformed FDOPA in venous plasma was measured in two representative patients (patient A male and patient B female) and all healthy volunteers by high-performance liquid chromatography at selected time points, as described previously (Heinz et al, 2004).
Region of Interest Analysis
Region of interests for caudate nucleus, putamen, and cerebellum were drawn on individual MRI images. For summation, images of early PET emission frames (2 to 8 mins) were coregistered to the individual MRIs, and dynamic time—radioactivity curves were extracted from the entire emission recordings in native space (for details see Siessmeier et al, 2005).
Statistical Analysis
An ROI-based categorical comparison of the magnitude of the estimates of k3S in striatum (and caudate and putamen separately) in PKU patients and controls was performed using SPSS. We tested the hypotheses that PKU patients show reduced FDOPA uptake in the striatum, and that plasma Phe levels correlated with striatal FDOPA utilization. After testing for normal distribution with the Kolmogorov—Smirnov and Shapiro—Wilk tests (P<0.05), we performed a two-sided Student's t-test (P<0.05) and a correlation analysis (Pearson's, P<0.05).
Results
Blood plasma levels of Phe in PKU patients ranged from 8.5 to 21.5 mg/dL (mean 21 mg/dL, normal mean <2 mg/dL). Blood tyrosine levels ranged from 0.31 to 2.4 mg/dL (mean 0.9 mg/dL, normal mean 0.6 to 1.6 mg/dL). The time—activity curves for FDOPA showed significantly delayed maxima in the cerebellum of PKU patients relative to controls (30±5 versus 12±2 mins; P<0.001), and likewise in the striatum (71±8 versus 43±4 mins; P>0.001) (Figure 1, upper row). Additionally, the normalized maxima in whole striatum were of lower magnitude in the PKU patients (mean 0.0015% of injected FDOPA) than in controls (mean 0.0028% of injected FDOPA; P<0.001). Average maps of the summed uptake from the first till the sixth minute of the recording, scaled to the amount injected and to body weight, are illustrated in Figure 1 (upper row).
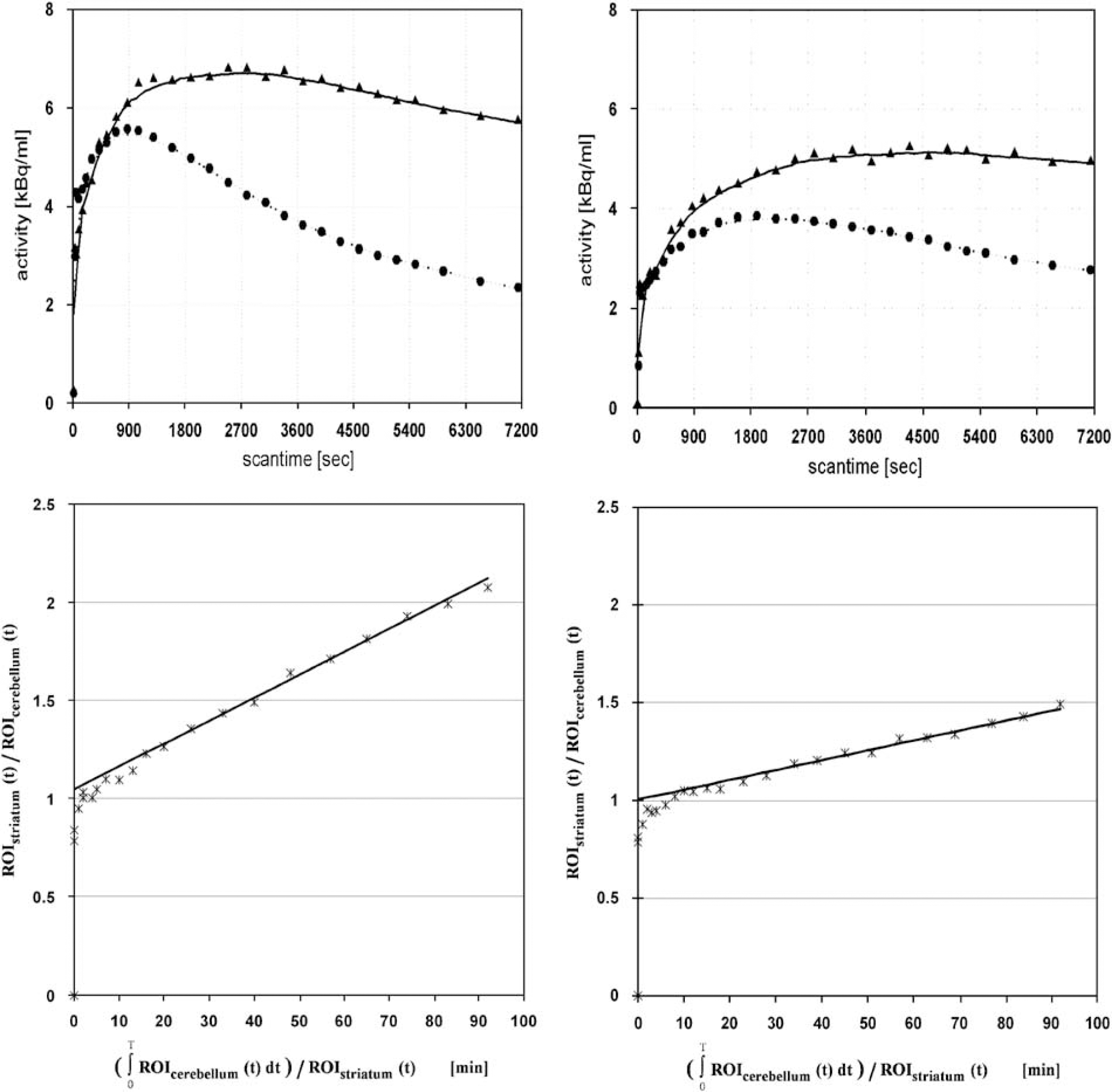
(Upper row) Time activity curves of a typical control subject (left image) and a PKU patient (right image). The triangles represent the striatal time—activity curve and the circuits the cerebellar time—activity curve. (Lower row) Reference tissue linear graphical analysis of striatal population mean results of controls (left image) and PKU patients (right image).
The fraction of untransformed FDOPA in venous plasma was consistently higher in the two representative patients compared with controls. Sixty minutes after injection, this fraction was 48% in patient A and 54% in patient B, compared with a mean fraction of 28±5% in the control subjects. After adjustment for body weight and injected activity, the concentration of activity of unmetabolized FDOPA in plasma at 90 mins was 0.376 and 0.324 kBq/mL in the two patients compared with 0.284±0.122 kBq/mL in the control group.
The mean magnitude of k3S in striatum was 0.0088±0.0009 min−1 (caudate 0.0089±0.0010, putamen 0.0086±0.0010) in the control group and 0.0052±0.0004 min−1 (caudate 0.0056±0.0008, putamen 0.0048±0.0005) in the PKU group (P<0.001). Figure 1 (lower row) shows the mean striatal reference tissue linear graphical analysis of controls and PKU patients. Relative reductions in the patients ranged from 33% to 46% (mean 41%). The ordinate intercept of the plots, corresponding to the partition volume of FDOPA, was close to 1 mL/g in both groups (Figure 1, lower row). The spatially normalized mean parametric images in Figure 2 (lower row) show the severely reduced k3S of FDOPA in PKU patients (right image) compared to controls (left image). Contrary to expectation, neither did the plasma concentration of Phe correlate significantly with the magnitude of striatal k3S in the PKU patients nor was there any correlation with the sum of LNAAs in plasma or with the Phe/tyrosine ratio.
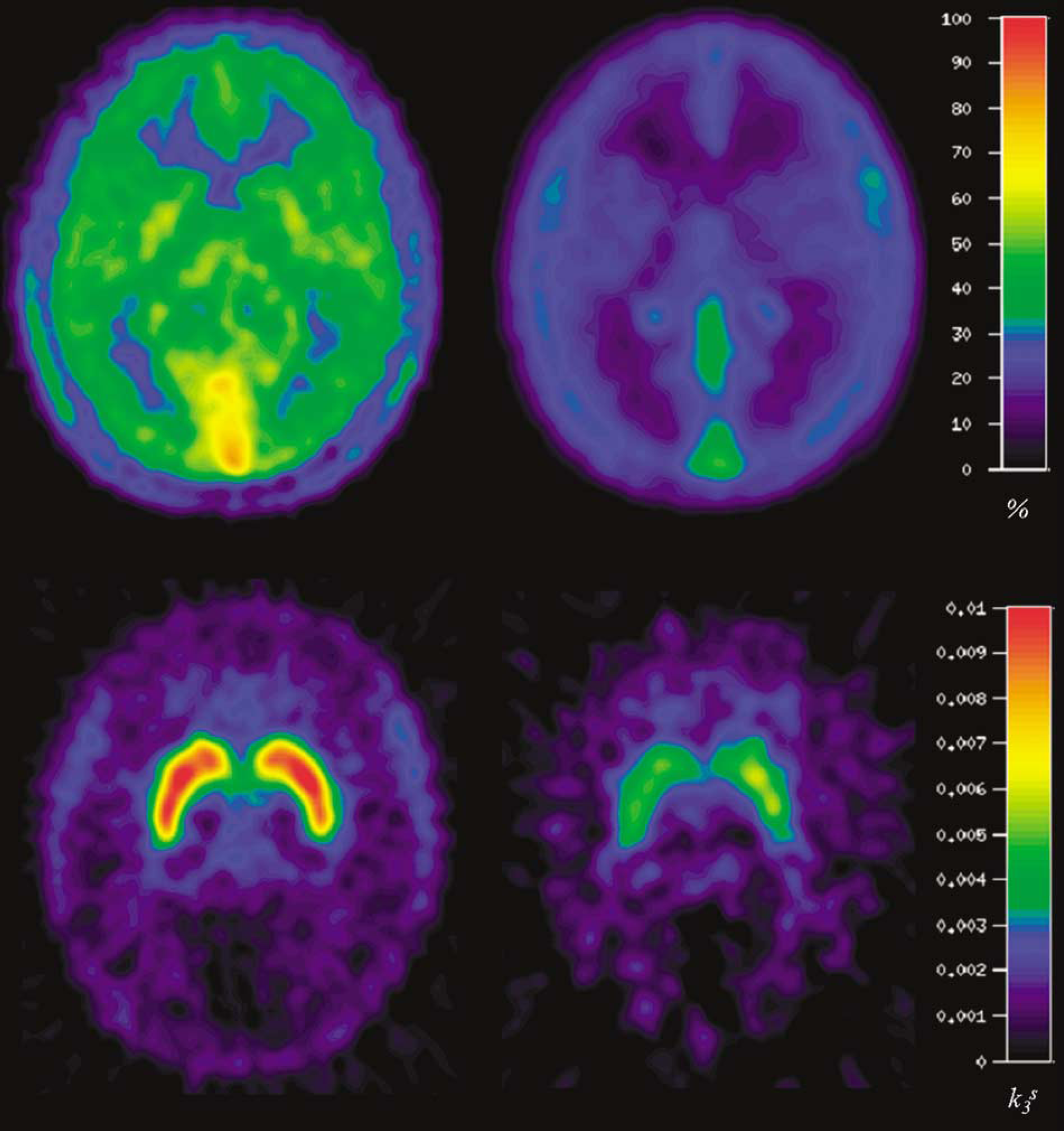
(Upper row) Early summed emission images (1–6 mins after injection, relative uptake: standardized uptake value and stereotactical normalized) of healthy control subjects (left image) and PKU patients (right image). Low cortical uptake compared with f.e. to the skull. (Lower row) k3S images (stereotactical normalized, scale showing k3S values) of healthy control subjects (left image) and PKU patients (right image).
The neuropsychological performance of all our PKU patients was within the normal range and did not differ from mean scores in the present control group. The mean percentile scores were LPS word fluency 75%; RWT: p-words 50%, s-words 43%, prenames 40%, animals 35%; HAWIE: arithmetic 10 points, similarities 29 points, pictures completion 14 points; numbers symbol test 12 points; WCST: total errors 61%, perseverative answers 60%, perseverative errors 68%, nonperseverative errors 53%, conceptual level answers 71%, categories completed 6, tries to complete 1 category 11, failure to keep set 1.4, learning to learn 2.4; TMT A 24 secs, TMT B 47 secs; tower of Hanoi moves in 4+5.34. Furthermore, we found no significant negative correlation between the performance of any task with k3S in the caudate nuclei, putamen, or whole striatum, nor was there any significant correlation with plasma Phe or tyrosine levels (P>0.05).
Discussion
The main quantitative finding of the present investigation is a 41% reduction in the rate of utilization of FDOPA in the striatum of adult PKU patients. This kinetic finding was associated with a discernible delay and attenuation of the peak radioactivity concentrations seen in cerebellum and striatum. The initial clearance of FDOPA across the blood—brain barrier (BBB) (K1D, mL per g per min) is determined by the local cerebral blood flow and the permeability—surface area product of the capillary endothelium, reduced by competition from other substrates for the LNAA carrier; this clearance is nearly the same in striatum and cerebellum (Cumming and Gjedde, 1998; Stout et al, 2000; Shoghi-Jadid et al, 2000). Owing to the lack of arterial input curves, we were unable to quantify the magnitude of K1D in this study. However, using the early summation images, normalized for body weight and injected dose as a surrogate for K1D, it is readily apparent that the initial clearance of FDOPA is substantially reduced in the PKU group (Figure 2, upper row).
In the two PKU subjects with high-performance liquid chromatography analysis, the fractions of unmetabolized FDOPA were considerably higher than in the control group, suggesting an impairment in the hepatic activity of catechol-O-methyl transferase. We are unaware of earlier reports of such a phenomenon, but note that low levels of hepatic copper and zinc and elevated hepatic iron were present in the liver of PKU mice (Gropper et al, 2004). If hepatic magnesium was likewise abnormal in the present group, this could alter the kinetics of catechol-O-methyl transferase, thus increasing the bioavailability of FDOPA. However it was obtained, the present finding of greater bioavailability of plasma FDOPA in the PKU patients would suggest that the distribution of the tracer across the BBB must be even more abnormal than is suggested from the delay to peak brain concentrations of tracer.
The delayed and attenuated cerebral influx of FDOPA in the PKU group is to be anticipated, as Phe has a higher affinity for the LNAA transporter than other substrates (Hargreaves and Pardridge, 1988). Indeed, the LNAA transporter is nearly saturated by normal levels of its several substrates (Smith et al, 1987). Consequently, elevated Phe levels will attenuate the influx of the endogenous competitors (Pardridge, 1998) and FDOPA. In this study, the extra burden of plasma Phe in the PKU patients, corresponding to 1,100 μmol/L, has to be compared with the sum of the concentrations of the other LNAAs, which was 530±281 μmol/L. Given the hyperbolic relationship between LNNA transport and the sum of the substrate concentrations in plasma (Stout et al, 1998), we attribute the present finding of apparently reduced unidirectional clearance of FDOPA to increased competition from Phe. This finding is entirely in line with the earlier report of decreased [11C]tyrosine transport in PKU patients (Paans et al, 1996). This scenario likewise predicts the occurrence of substantial increases in the brain concentration of free Phe, which accounts for the apparently decreased washout of FDOPA. This phenomenon can be construed from the similar ordinate intercepts in the linear graphical plots from the control group and in PKU patients. Given that the magnitude of K1D is reduced in the patients, the magnitude of k2D must likewise be reduced, so as to maintain a constant ratio of the two parameters.
The present reference tissue method provides an estimate of the relative activity of DOPA decarboxylase with respect to FDOPA (Hartvig et al, 1991; Hoshi et al, 1993). The time—radioactivity curve in the reference region, assumed to be nearly devoid of DOPA decarboxylase activity, serves as a surrogate input for the formation of [18F]fluorodopamine elsewhere in the brain. As such, the magnitude of the present rate constant k3S should be robust to the impaired influx of FDOPA across the BBB of PKU patients. This finding may thus indicate competitive inhibition of DOPA decarboxylase by Phe. However, we did not detect a significant correlation between individual plasma Phe and the magnitude of k3S in the striatum of the PKU group. This might be due to the small sample size and the rather limited spread in the individual estimates of the measured clinical parameters.
In agreement with earlier reports (Hanley et al, 2000; Cabalska et al, 2000), plasma tyrosine was slightly reduced in the present PKU patients. Under physiologic conditions, there is only limited conversion from Phe to tyrosine in the brain by phenylalanine hydroxylase (Lichter-Konecki et al, 1999) or tyrosine hydroxylase (Katz et al, 1976). It is not to be expected that the increased Phe can compensate for a presumed deficiency of the precursor tyrosine for levodopa synthesis in the brain of PKU patients. However, the rate of dopamine synthesis cannot be directly calculated from the present assay of DOPA decarboxylase activity alone, so it cannot be ascertained that the observed 41% reduction in k3S corresponds to an equal reduction of dopamine synthesis. Although the magnitude of k3S in the present PKU group is almost as low as is typical of Parkinson's disease patients (Cumming and Gjedde, 1998), PKU patients seldom develop parkinsonism. Other factors may intervene so as to maintain sufficient dopamine synthesis under conditions of reduced DOPA decarboxylase activity. In particular, it has been argued that a considerable fraction of levodopa is exported from the brain rather than being transformed into dopamine (Cumming et al, 1997, 1998, 2001). Elevated brain levels of Phe in the PKU patients may serve to rectify dopamine synthesis by impeding the facilitated diffusion of levodopa (and FDOPA) from brain into circulation, as is suggested by the lack of effect on the magnitude of the ratio K1D/k2D, occurring in the presence of reduced K1D, discussed above.
The above arguments suggest that competing effects of brain Phe on DOPA decarboxylase and BBB efflux may fortuitously cancel, such that adequate dopamine synthesis is maintained in the present PKU patients. Nonetheless, reduced dopamine metabolite levels are reported in the cerebrospinal fluid of PKU patients (Lykkelund et al, 1988), suggesting that cerebral dopamine synthesis is actually impaired, but to an extent falling below a threshold for emergence of motor symptoms. An additional compensating mechanism could be an upregulation of postsynaptic dopamine receptors, as occurs in patients suffering from untreated Parkinson's disease.
That we found no significant negative correlation between the performance of any task, nor any significant correlation with plasma Phe or tyrosine levels might be explained by the selection bias of our PKU patients, who were selected as performing unremarkably in neuropsychological testing, or by the discussed mechanisms compensating for the possible lack of dopamine.
Conclusion
The results show a severely qualitatively impaired influx and distribution of FDOPA throughout the brain of adult patients suffering from PKU, which we attribute to competitive inhibition of the LNAA transporter by plasma Phe. The reference tissue graphical analysis shows a 41% reduction in the rate of decarboxylation of FDOPA in brain, which may also be attributed to increased competition from Phe. As the patients did not show symptoms of Parkinsonism, we speculate that impaired export of brain levodopa across the BBB may compensate for reduced activity of DOPA decarboxylase.
Footnotes
Acknowledgements
We thank our technicians Mrs Heike Armbrust and Mr Stefan Maus for assistance and Dr Ralf Schirrmacher for radiopharmaceutical preparation and metabolite analysis.
None of the authors has any financial or otherwise involvement that might bias his work.