
Abstract
The brain's adaptive response to ischemic preconditioning (IPC) is mediated in part via hypoxia inducible factor (HIF)-responsive genes. We previously showed that IPC induces cytochrome P450 2C11 expression in the brain, associated with protection from stroke. Cytochrome P450 2C11 is an arachidonic acid (AA) epoxygenase expressed in astrocytes, which metabolizes AA to epoxyeicosatrienoic acids (EETs). We tested the hypotheses that hypoxic preconditioning (HPC) induces 2C11 expression in astrocytes via HIF-1α, and that the P450 epoxygenase pathway contributes to enhanced astrocyte tolerance to ischemia-like injury induced by oxygen-glucose deprivation (OGD). Primary cultured astrocytes were incubated under normoxic or hypoxic conditions for 1, 3, 6, 24, or 48 h, and protein levels of P450 2C11 and HIF-1α were measured by Western blotting. Additionally, 2C11 mRNA was measured by Northern blotting, and binding of HIF-1α to 2C11 promoter was evaluated using electrophoretic mobility shift assay (EMSA) with 2C11 promoter DNA containing putative HIF-binding sites. Levels of 2C11 mRNA and protein were significantly increased starting at 3 and 6 h of hypoxia, respectively. The increase in 2C11 expression was preceded by an increase in HIF-1α protein at 1 h of hypoxia, and EMSA showed a specific and direct interaction between 2C11 promoter DNA and HIF-1α in nuclear extracts from astrocytes. HPC and EETs reduced astrocyte cell death, and P450 epoxygenase inhibition prevented protection by HPC. We conclude that HPC induces tolerance in astrocytes, at least in part, via HIF-1α-linked upregulation of P450 2C11.
Keywords
Introduction
Brief exposure to ischemia renders the brain more resistant to subsequent prolonged periods of cerebral ischemia. This phenomenon, referred to as ischemic preconditioning (IPC), represents a fundamental adaptive response to environmental stress, whereby cells adapt to stress by upregulating defense mechanisms and switching to a more protected phenotype. Several mechanisms involved in preconditioning-acquired ischemic tolerance are similar to a highly conserved genomic response to hypoxia mediated via the transcription factor hypoxia-inducible factor-1 (HIF-1) (Dirnagl et al, 2003). Hypoxia-inducible factor-1 is a heterodimer composed of hypoxia-sensitive HIF-1α, and constitutively expressed HIF-1β, also known as the aryl hydrocarbon receptor nuclear translocator (ARNT). Hypoxia inducible factor-1α has been shown to be induced by hypoxic preconditioning (Semenza, 2000; Bergeron et al, 2000; Jones and Bergeron, 2001), where it is thought to associate with ARNT to transactivate several hypoxia-regulated genes, including glucose transporter-1 GLUT1, erythropoeitin (EPO), vascular endothelial growth factor (VEGF), insulin-like growth factor-2 (IGF-2), and IGF-binding protein (IGF-BP) (Semenza, 2003), all of which have been proposed to play a role in mediating ischemic tolerance in the brain (Ruscher et al, 2002; Bernaudin et al, 2002b; Prass et al, 2003; Wang et al, 2004). We previously showed that ischemic preconditioning induces cytochrome P450 2C11 expression, associated with protection from ischemic brain injury (Alkayed et al, 2002). P450 2C11 is an arachidonic acid (AA) epoxygenase expressed in astrocytes, which metabolizes AA to epoxyeicosatrienoic acids (EETs), which have been shown to protect against ischemia-reperfusion injury in the intact heart (Wu et al, 1997), and to directly inhibit cell death in culture induced by hypoxia-reoxygenation (Yang et al, 2001), hydrogen peroxide (H2O2) and serum deprivation (Chen et al, 2001). Many P450 genes are regulated via the aryl hydrocarbon receptor (AhR), which dimerizes with ARNT (HIF-1β) to drive P450 gene transcription (Park, 1999; Nie et al, 2001), and ARNT knockout leads to loss of target gene induction by AhR and HIF-1α (Tomita et al, 2000). Recent evidence also suggests that some P450 genes are inducible by hypoxia via HIF-1α (Fradette and du Souich, 2003; Mastyugin et al, 2004). However, it is not known if cytochrome P450 2C11 is regulated by hypoxia, and whether HIF-1α is a regulator of 2C11 expression. We examined cytochrome P450 2C11 promoter sequence, and found at least two potential HIF-binding sites. Therefore, in this study, we set out to determine if hypoxic preconditioning increases P450 2C11 expression in primary cultured astrocytes via HIF-1α, and whether the P450 epoxygenase pathway mediates, at least in part, tolerance to ischemia-like injury induced by oxygen-glucose deprivation (OGD) in preconditioned astrocytes.
MATERIALS AND METHODS
Animals
Timed-pregnant Sprague-Dawley rats (16 to 18 days of gestation) were purchased (Charles River Wilmington, MA, USA) and housed at the Johns Hopkins University animal facilities until delivery. Primary cultured astrocytes were prepared from 1- to 3-day-old Sprague-Dawley rat brains in accordance with the National Institutes of Health guidelines, and experimental protocols were approved by the Johns Hopkins University Animal Care and Use Committee.
Chemicals
All four regioisomers of EETs (5,6-, 8,9-, 11,12-, and 14,15-EET) were purchased from BIOMOL (Plymouth Meeting, PA, USA), and added individually at a final concentration of 10 μmol/L, as previously described (Chen et al, 2001; Peng et al, 2002, 2004; Lu et al, 2002). Epoxygenase inhibitor N-methylsulfonyl-6-(2-propargyloxyphenyl) hexanamide (MS-PPOH) was synthesized by Dr John R Falk at the University of Texas Southwestern Medical Center (Dallas, TX, USA) and was kindly provided to us by Dr David Harder at the Medical College of Wisconsin (Milwaukee, WI, USA). MS-PPOH was used at a final concentration of 20 μmol/L, as previously described (Lu et al, 2002; Peng et al, 2002, Peng et al, 2004).
Cell Culture
Primary cultured astrocytes were prepared from the cerebral cortex and hippocampus separately, as previously described (Alkayed et al, 1996; Park, 1999). Briefly, under aseptic conditions, brain tissue was dissected free of meninges, cut into small pieces, and digested in 2 mg/mL trypsin (Sigma, St Louis, MO, USA) in the presence of 0.006% DNase (Sigma) at 37°C for 5 mins. Digestion was stopped by rinsing twice with HEPES-buffered salt solution (HBSS, Invitrogen, Grand Island, NY, USA) and by adding 1 mg/mL trypsin inhibitor (Invitrogen) and placing the tube on ice for 10 mins. Tissue pieces were then washed two times in HBSS, followed by mechanical dissociation with a pipette. Cell suspension was diluted with a feeding medium consisting of 10% fetal bovine serum (FBS, HyClone, Logan, UT, USA) in DMEM (Invitrogen) with 1% penicillin-streptomycin solution (Sigma), seeded in 75-cm2 culture flasks (Coring Incorporate, Corning, NY, USA) and incubated at 37°C in a 95%/5% mixture of atmospheric air and CO2. Feeding medium was changed after 2 days and subsequently twice a week. For all experiments reported here, we used confluent monolayers (10 to 14 days in vitro, div) of primary cortical and hippocampal cultures.
Immunocytochemistry
Cells were identified by indirect immunofluorescence using antibodies against astrocyte-specific marker glial fibrillary acidic protein (GFAP, Chemicon, Temecula, CA, USA), oligodendrocyte-specific marker galactocerebroside (Gal-C, Sigma), and the microglial marker integrin CD11b (OX-42, Serotec, Raleigh, NC, USA), with FITC-conjugated goat anti-mouse IgG (Chemicon) as the secondary antibody. Cells were visualized by costaining with universal nucleic acid stain 4',6-diamidino-2-phenylindole (DAPI, Molecular Probes, Eugene, OR, USA). Cells were plated on 22 × 22-mm coverslips (VWR, West Chester, PA, USA) and incubated at 37°C in 95% air/5% CO2 for 10–14 days. Cells were then rinsed with phosphate-buffered saline (PBS) containing Ca2+ and Mg2+ (Invitrogen), fixed in cold methanol for 5 mins at –20°C, permeabilized in 0.25% Triton X-100 in PBS for 2 mins at room temperature, blocked with 3% normal goat serum in PBS for 30 mins at room temperature, and incubated with 100 μL of 5 μg/mL (1:4 dilution in 0.1% normal goat serum) of the primary antibody for 1 h at room temperature and with 100 μL of 2.68 μg/mL (1:500 dilution in 0.5% bovine serum albumin/PBS) of the secondary antibody for 1 h at room temperature in the dark. Coverslips were then washed with PBS and mounted on 25 × 75-mm slides (VWR) with a drop of Gel-Mount (Biomedical Corp., Norwalk, CT, USA). Slides were visualized with a Nikon TE200 prism inverted microscope equipped for epifluorescence.
Northern Blot Analysis
Expression of 2C11 mRNA in astrocytes was examined by Northern blotting. Total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. Confluent monolayers were studied at 3 or 6 h after hypoxia (94% N2/5% CO2/1% O2) or normoxia (74% N2/5% CO2/21% O2). Briefly, the culture medium was discarded and cells were washed three times with ice-cold PBS, and then lysed by adding the TRIzol reagent directly into the dish. Cells were further disrupted and homogenized by passing the lysate several times through a syringe needle. The aqueous phase was separated using chloroform, and the RNA was precipitated from the aqueous phase with isopropanol. The RNA pellet was washed with 75% ethanol and redissolved in RNase-free water. Northern blot hybridization was performed using a 296-bp, PCR-amplified rat 2C11 cDNA probe, as previously described (Alkayed et al, 2002). Total RNA (20 μg) was fractionated by electrophoresis through 1% agarose gel containing 0.7 mol/L formaldehyde and transferred to a Nytran Plus membrane (Schleicher & Schuell). The membrane was then air-dried, and the RNA was immobilized by UV cross-linking (Stratalinker, Stratagene, La Jolla, CA, USA). Blots were prehybridized for 1 h at 68°C in ExpressHyb hybridization solution (Clontech Laboratories, Inc., Palo Alto, CA, USA), and incubated for 2 h at 68°C with 2C11 cDNA probe labeled with [α-32P]dATP using Prime-It II random primer labeling kit (Stratagene, La Jolla, CA, USA). Blots were washed twice at room temperature for 15 mins each with 2 × sodium chloride/sodium citrate (SSC)/0.05% sodium dodecyl sulfate (SDS) and one time in 0.1 × SSC/0.1% SDS for 30 mins at 50°C with continuous shaking. The blots were immediately covered with plastic wrap to prevent drying, mounted on a filter paper for support, and exposed to an X-ray film at –80°C overnight.
Western Blot Analysis
To study the effect of hypoxia on HIF-1α and 2C11 protein expression, sister cultures were incubated under normoxic (74% N2/5% CO2/21% O2) or hypoxic (94% N2/5% CO2/1% O2) conditions for 1, 3, 6, 24, or 48 h. At each time point, cells were washed three times with PBS and scraped for preparation of microsomes as previously described (Alkayed et al, 2002). Briefly, cells were homogenized in 250 mmol/L sucrose, 1 mmol/L EDTA, and 10 mmol/L KPO4, 0.1 mmol/L phenylmethylsulfonyl fluoride (PMSF, pH 7.7) in the presence of protease inhibitors (2 μg/mL aprotinin, 2 mg/mL leupeptin, 1 μg/mL pepstatin), and the pellet was resuspended in 100 mmol/L KPO4, 1 mmol/L EDTA, 1 mmol/L dithiothreitol (DTT), and 30% glycerol (pH 7.25). The homogenate was centrifuged for 15 mins at 10,000 g at 4°C, and the supernatant was further centrifuged at 100,000 g for 60 mins at 4°C. The protein concentration was measured by Bradford protein assay using bovine serum albumin as a standard. Microsomal proteins (30 μg) were then boiled in Laemmli buffer for 5 mins, electrophoresed on a 10% SDS-polyacrylamide gel, and blotted onto polyvinylidene fluoride (PVDF) membrane (Bio-Rad, Hercules, CA, USA). Blots were blocked in PBS containing 5% nonfat dry milk and 0.05% Tween for 1 h at room temperature, and incubated overnight at 4°C with a polyclonal rabbit antirat 2C11 antibody (1:2,500, obtained from Dr David Harder at the Medical College of Wisconsin, Milwaukee, WI, USA) (Alkayed et al, 1996). Membranes were then washed with PBS/T, incubated at room temperature for 1 h with peroxidase-coupled goat anti-rabbit IgG (1:5,000), and overlaid with enhanced chemiluminescence (ECL) detection reagent (Amersham, Piscataway, NJ, USA) for 1 min. Blots were visualized by autoradiography (Hyperfilm ECL, Amersham) and films were photographed with a digital camera, and analyzed with ImageQuant. Blots were stripped (30 mins at 50°C in 62.5 mmol/L Tris base, 2% SDS, 100 mmol/L β-mercaptoethanol) and reprobed with a monoclonal β-actin antibody (1:2,500) (Sigma) as an internal control. Optical density of 2C11 bands was divided by that of β-actin within each lane and expressed relative to control cells after adjusting for background. Hypoxia-inducible factor-1α (HIF-1α) protein was examined in nuclear extracts prepared as described below with a mouse HIF-1α monoclonal antibody 2 mg/mL (OSA-601; Stressgen Biotechnologies Corp. Inc., Victoria, BC, Canada), and peroxidase-coupled goat anti-mouse IgG (1:5,000).
Nuclear Protein Extract
Hypoxia-inducible factor-1α Western blot and EMSA were performed on nuclear protein extracts prepared from primary cultured astrocytes as previously described (Bernaudin et al, 2002b). Briefly, at 1, 3, or 6 h of hypoxia, astrocytes were washed and lysed in the following buffer: 20 mmol/L HEPES, 10 mmol/L KCI, 1 mmol/L EDTA, 1 mmol/L dl-DTT, 0.2% NP40, 10% glycerol, 1 mmol/L PMSF and 1% protease inhibitor cocktail (Sigma) in distilled water. Lysates were incubated for 5 mins on ice and then centrifuged for 10 mins at 13,000g at 4°C. The supernatant, containing the cytosolic fraction, was stored at –80°C, and the pellets, containing the nuclei, were resuspended in 50 μL of a resuspension buffer containing 350 mmol/L NaCI, 20% glycerol, 20 mmol/L HEPES, 10 mmol/L KCI, 1 mmol/L PMSF, and 1% protease inhibitor cocktail (Sigma) in distilled water. Suspension was mixed vigorously with a pipette tip and incubated on ice for 30 mins. Samples were then centrifuged for 10 mins at 13,000g at 4°C and supernatants (nuclear extracts) stored at –80°C.
Electrophoretic Mobility Shift Assay
Hypoxia-inducible factor-1α binding to P450 2C11 promoter DNA was examined using EMSA. Two independent sequences containing putative hypoxia response elements (HRE) were derived from published 2C11 promoter sequence (Strom et al, 1994). Double-stranded oligonucleotides with the following sequences were synthesized at Johns Hopkins Molecular Core Facility: 2C11-HRE1: 5'-TTC CGG GGC CA
Hypoxic Preconditioning
To determine time course of 2C11 and HIF-1α induction by hypoxia, confluent monolayers of primary cultured astrocytes were incubated under hypoxia (94% N2/5% CO2/1% O2) for 1, 3, 6, 24, or 48 h in a humidified, three-gas incubator (Forma, Marietta, OH, USA). To determine if hypoxic preconditioning (HPC) induces tolerance to ischemia-like injury, astrocytes were incubated for 3 h under hypoxia, returned to normoxic environment (74% N2/5% CO2/21% O2) for 24 h, and then subjected to 6 h of OGD, as described below. Cell death was then measured at 24 h by propidium iodide (PI) staining and compared with the effect of OGD alone without prior preconditioning, as described below.
Oxygen-Glucose Deprivation
To simulate ischemia/reperfusion, astrocytes were incubated under hypoxic conditions in glucose-free medium for 6 h, followed by 24-h incubation under normoxic conditions in glucose-containing feeding medium. Briefly, at 10 to 14 div, culture medium was exchanged with glucose-free feeding medium, and cells incubated under hypoxic atmosphere (94% N2/5% CO2/1%O2) for 6 h. Immediately after OGD, the medium was replaced with fresh feeding medium containing glucose, and cells were incubated under normoxic atmosphere (74% N2/5% CO2/21% O2) for 24 h. Cell death was assessed by PI (Sigma) staining, as described below. Synthetic EETs (10 μmol/L) and MS-PPOH (20 μmol/L) were added 1 h before OGD, and supplemented as necessary during the switch to glucose-free medium and back to normal medium to maintain the drug presence throughout OGD and reoxygenation.
Cell Death Assay
Effects of HPC, OGD, and combined HPC plus OGD, as well as various pharmacologic manipulations were assessed by dividing the number of dead cells by the total number of dead and surviving cells. Cell death was identified using PI staining (Sigma), a red-fluorescent nuclear dye that stains only dead cells because the dye is excluded from live cells with intact cell membranes. Surviving cells are identified by calcein-AM (Molecular Probes), which is retained by live cells with intact membranes.
Statistical Analysis
Number of experiments per group (n) refers to the number of separate experiments performed on separate cultures from different litters. To obtain sufficient material for expression and gel shift studies, cells were grown in 100-mm culture dishes and 75-cm2 flasks. For cell death assay, astrocytes were grown in 24-well plates, and each experiment was performed in triplicate (three wells). Cell death in each well was measured by counting PI- (dead) and calcein-positive (live) cells in at least 6 to 10 different visual fields under fluorescent microscope (x 40 objective, × 10 eye piece). The average of 6 to 10 counts of three replicates was considered, n = 1. Differences among groups in densitometric measurements and in percent cell death were analyzed with one-way ANOVA and post hoc Newman-Keuls test. The criterion for statistical significance was P<0.05. Values are presented as mean±s.e.m.
RESULTS
As illustrated in Figure 1, immunofluorescent staining of primary cultures revealed that the majority of cells used in the current studies were GFAP positive (98.4±0.5% of all DAPI-positive cells, n = 5 separate cultures, upper panels), with no detectable staining for Gal-C (n = 3, lower panels) or CD11b (OX-42, n = 4, middle panels), indicating that cultures predominantly consisted of astrocytes, with no microglia or oligodendrocytes.
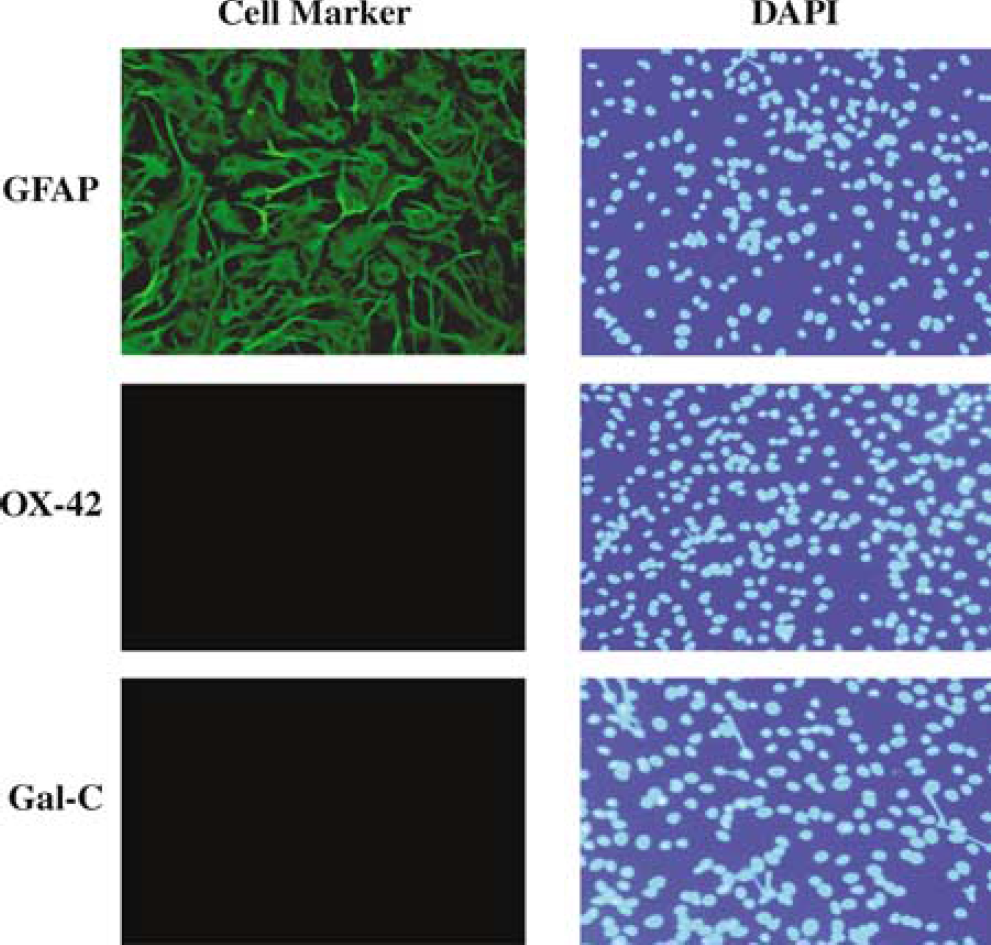
Immunofluorescent identification of rat brain primary astroglial culture. The majority of cells (98.4±0.5%, n = 5 separate cultures) were positive for astrocyte-specific marker GFAP. No staining was observed for either oligodendrocyte-specific marker Gal-C (n = 3) or microglial marker CD11b (OX-42, n = 4). Right panels show cell nuclear staining with DAPI.
To determine if hypoxia induces 2C11 protein expression, sister cultures were incubated under hypoxic and normoxic conditions, and cells collected for Western blot analysis using anti-2C11 antibody. Figure 2A is representative Western blot illustrating marked 2C11 protein induction by hypoxia in astrocytes. Figure 2B summarizes the time course of 2C11 protein induction by hypoxia in astrocytes. We observed differences in the timing of 2C11 induction by hypoxia between cortical and hippocampal astrocytes. In cortical astrocytes, 2C11 protein was significantly increased in hypoxia-treated cells compared with cells incubated under normoxia (N) starting at 6 h (256±36% versus 100±16%, respectively, n = 4, P<0.05), and a similar increase was maintained on days 1 and 2 (207±20% and 235±25%, respectively, n = 4 per group at each time point, P<0.05 versus N). In hippocampal cells, the level of 2C11 protein at 6 h was not different in hypoxia-treated cells compared with normoxic cells (110±13% versus 100±13%, n = 4, P>0.05). However, 2C11 protein expression in hippocampal astrocytes was significantly increased by hypoxia after 1 and 2 days (252±15% and 305±20%, respectively, n = 4 each time point, P<0.05 versus N). To determine if 2C11 upregulation by hypoxia is observed at the mRNA level, we measured 2C11 mRNA expression by Northern blot with 2C11 cDNA probe. Figure 3 is representative Northern blot demonstrating 2C11 mRNA induction at 3 and 6 h of hypoxia in primary cultured cortical and hippocampal astrocytes.
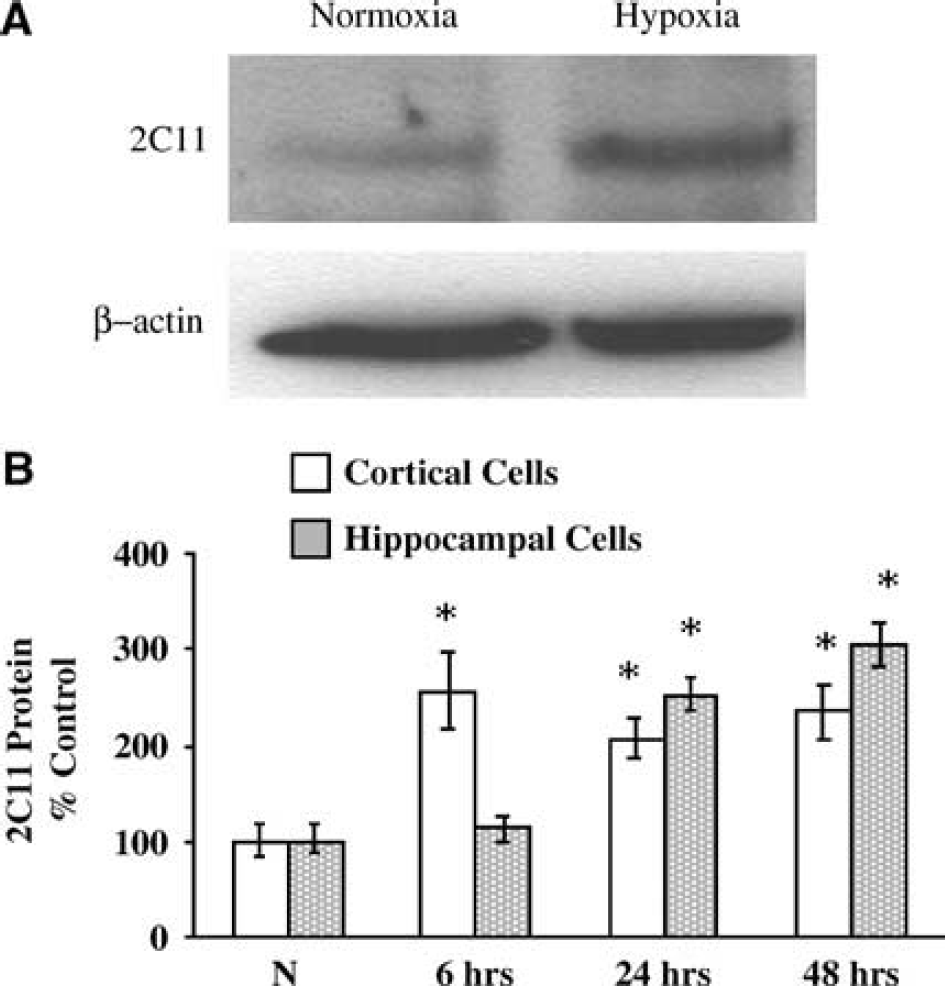
Hypoxia induces 2C11 protein expression in astrocytes. (
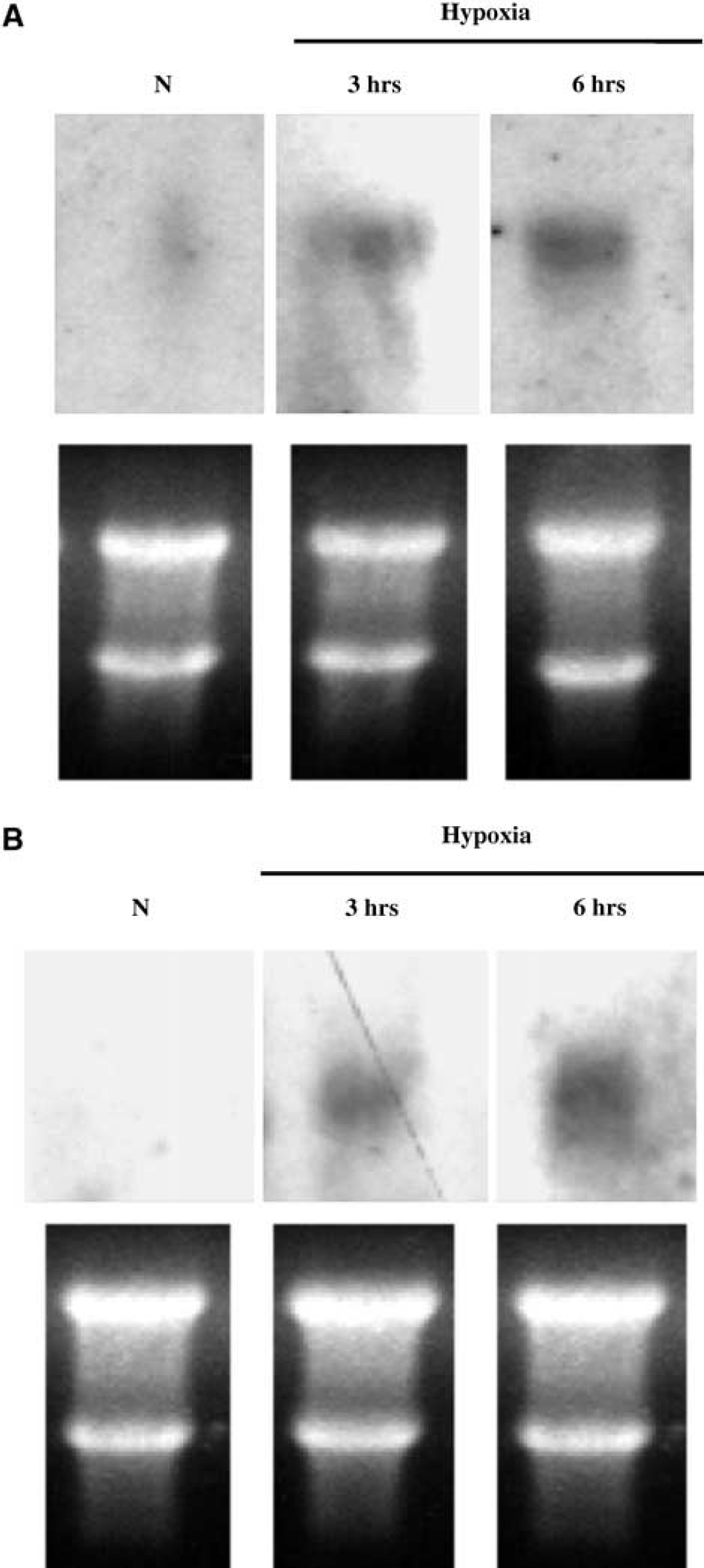
Hypoxia induces 2C11 mRNA expression in astrocytes. Representative Northern blot analysis showing 2C11 mRNA induction in cortical (
We then determined if 2C11 upregulation by hypoxia is accompanied by an increase in HIF-1α protein. Astrocytes were incubated under hypoxia for 1, 3, and 6 h, and cells collected and homogenized, and nuclear extract probed with anti-HIF-1α antibody. Figure 4 shows that HIF-1α protein level is increased by hypoxia in nuclear extract from cortical astrocytes as early as 1 h, and that increased level is maintained for 3 and 6 h. To determine if 2C11 induction by hypoxia is linked to HIF-1α, we searched 2C11 promoter sequence (Strom et al, 1994) and found at least two HIF-binding core sites (Semenza, 1999). Figure 5 shows that HIF-1α protein interacts with an oligonucleotide sequence derived from 2C11 promoter sequence that contains an HIF-binding site, and this hypoxia increases HIF-1α protein-2C11 promoter DNA interaction in primary cultured cortical astrocytes. Figure 6 shows the specificity of the interaction between HIF-1α and 2C11 DNA. Two independent sequences containing putative hypoxia response elements (2C11-HRE1 and 2C11-HRE2) derived from 2C11 promoter sequence (Strom et al, 1994) were shifted upward by incubation with astrocytic nuclear extracts. Bands were shifted to the same position as HIF-containing probe from the human erythropoietin gene promoter (Semenza and Wang, 1992; Strom et al, 1994). Furthermore, shifted bands disappeared when coincubated with 100-fold excess unlabeled wild-type probes (cold +), but not probes with mutated HIF binding sites (mutant +).
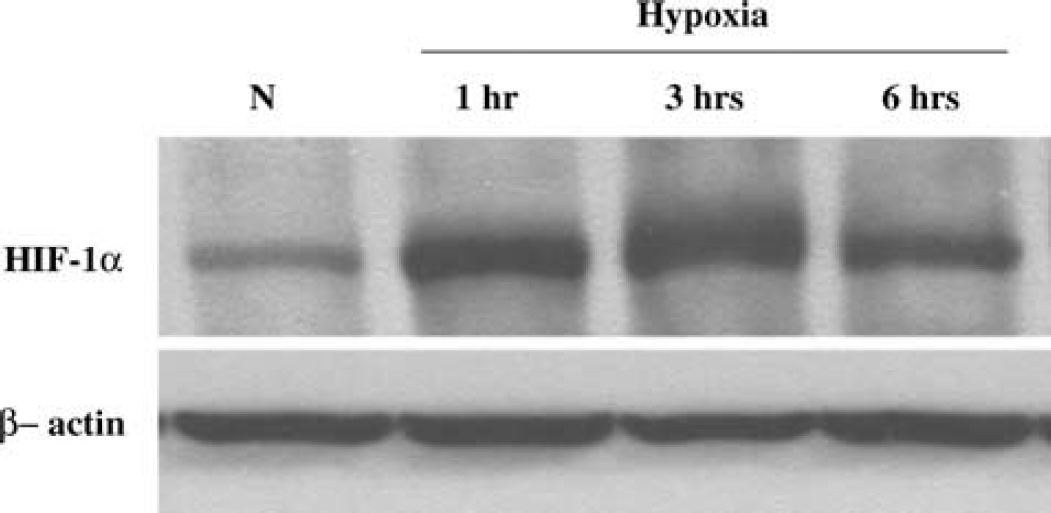
Hypoxia increases HIF-1α protein in astrocytes. Western blot analysis of HIF-1a shows that the level of HIF- 1α protein is increased at 1, 3, and 6 h of hypoxia in primary cultured astrocytes
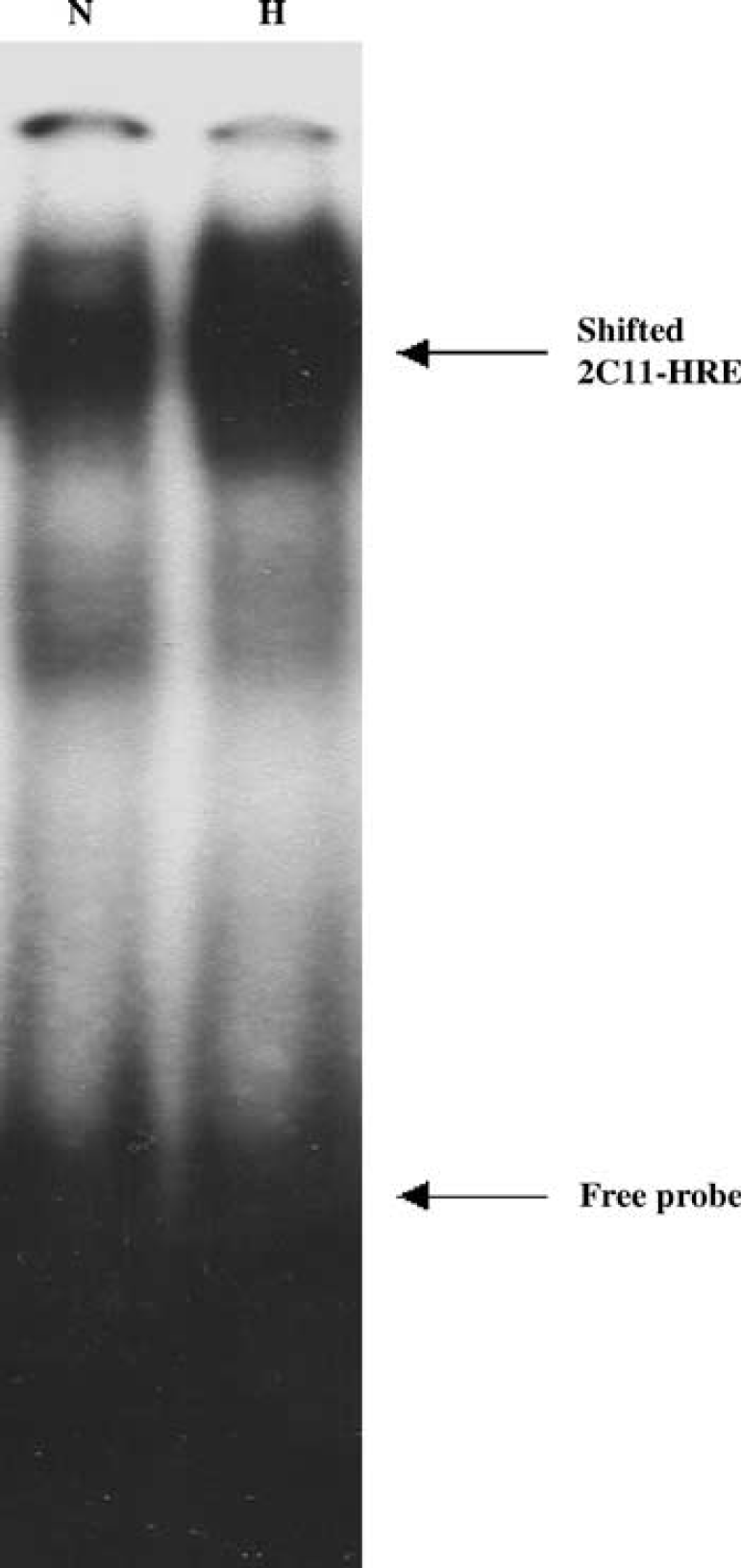
Hypoxia increases DNA-protein complex formation between HIF response element (HRE) and nuclear protein extract from primary cultured astrocytes.
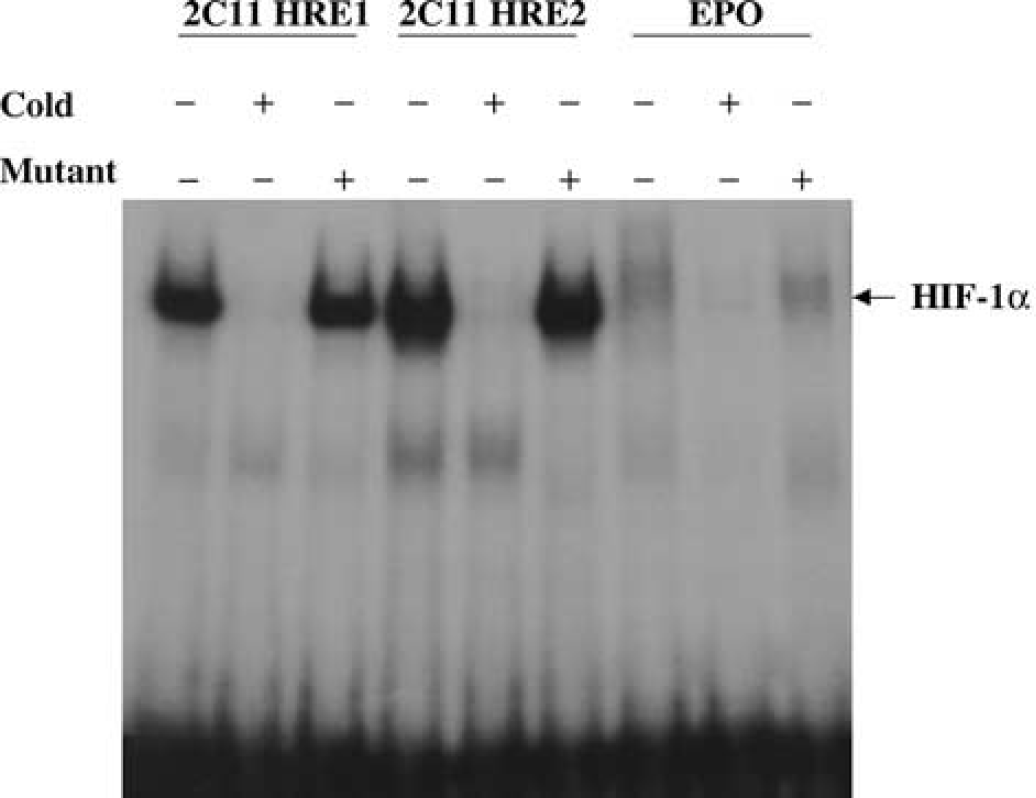
Hypoxia inducible factor-1α interaction with 2C11 promoter DNA in primary cultured astrocytes. Two independent 2C11 probes containing HIF-binding site (HRE1 and 2) were shifted upward in the presence of astrocyte nuclear extracts. Bands were shifted to the same position as HIF-containing probe from human EPO gene promoter. Shifted bands disappeared when coincubated with 100-fold excess unlabelled probes containing HIF binding sites (cold, +), but not probes with mutated HIF binding sites (mutant, +).
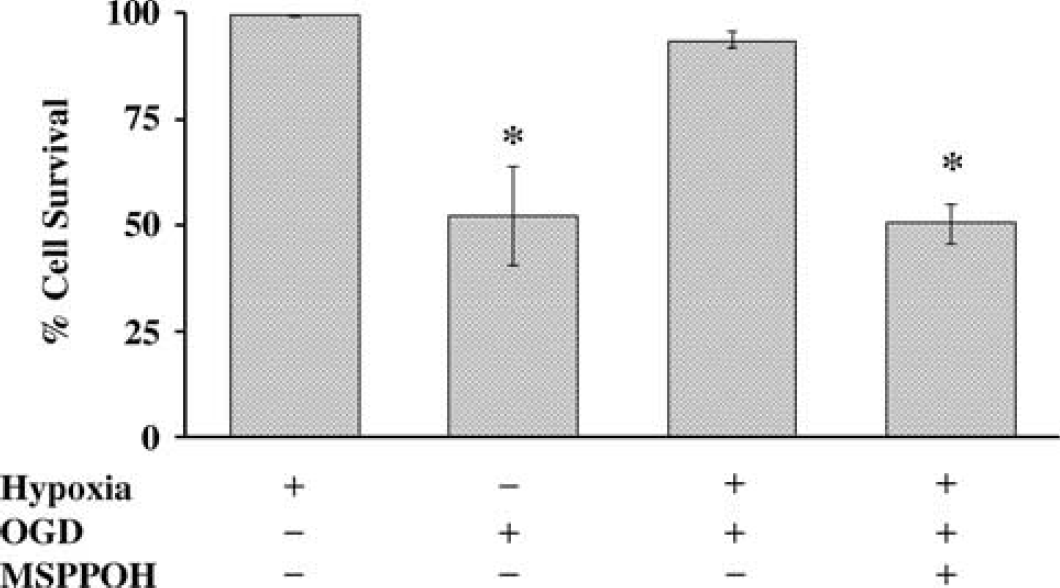
Hypoxic preconditioning reduces OGD-induced cell death in primary cultured astrocytes via P450 epoxygenase pathway. The protection by HPC was prevented by epoxygenase inhibitor MS-PPOH (20 μmol/L) (P<0.05, n = 5). *Different from hypoxia.
Finally, we examined the functional significance of 2C11 upregulation by hypoxia in astrocytes. To determine if HPC is protective in astrocytes via the P450 epoxygenase pathway, we subjected primary cultured cortical astrocytes to 6 h of OGD with and without prior preconditioning using a level and duration of hypoxia shown above to induce 2C11 expression. Figure 7 shows that 3-h hypoxia alone does not induce cell death (hypoxia +, 99.4±0.2% survival), while 6-h OGD induces 47.7±11.8% (OGD+, n = 9) cell death in primary cultured cortical astrocytes (in hippocampal astrocytes, 6-h OGD induced 37.9±9.1%, n = 6, not included in Figure 7). However, preconditioning with 3-h hypoxia 24 h earlier (Hypoxia +, OGD +) significantly reduces OGD-induced cell death (6.3±1.5%, n = 5, P>0.05 versus OGD alone), and pretreatment with epoxygenase inhibitor MS-PPOH (20 μmol/L) abolishes protection by HPC (49.5±4.7%, n = 5). MS-PPOH alone does not induce cell death in astrocytes (46.3±1.8%, n = 5). To determine if EETs are protective against OGD-induced cell death, we applied EETs (10 μmol/L) 1 h before 6-h OGD, and cell death was assayed at 24 h after OGD. Figure 8 illustrates that OGD induces 46.3±1.8% cell death in vehicle-treated cells. Pretreatment with EETs reduces cell death by 50%, from 46.3±1.8% in vehicle-treated cells to 23±3% (n = 5) in EET-treated cells. We observed no differences among the 4 regioisomers of EETs (5,6-EET; 8,9-EET; 11,12-EET and 14,15-EET). Therefore, the effect of EETs in Figure 8 represents an average of all 4 isoforms.
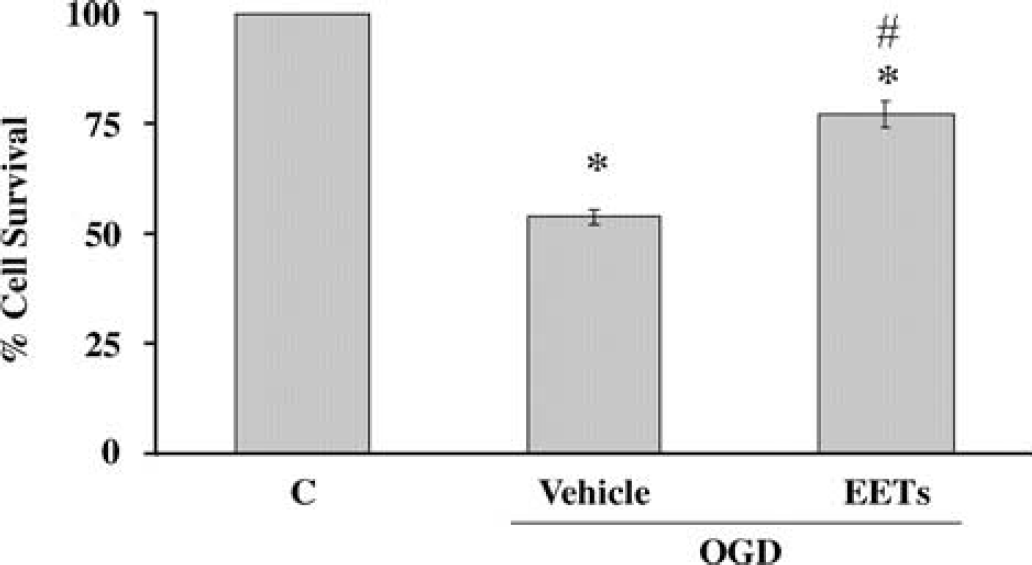
Epoxyeicosatrienoic acids (EETs, 10 μmol/L) reduce OGD-induced cell death in primary cultured astrocytes (P<0.05, n = 5). No differences were observed among the four regioisomers of EETs. *Denotes a difference from nomoxia, and #denotes a difference from OGD.
DISCUSSION
The principal findings of this study are as follows: (1) hypoxia induces P450 2C11 expression in astrocytes, (2) hypoxia increases HIF-1α, which binds HIF-response elements on 2C11 promoter, (3) HPC protects against OGD-induced cell death in part via the P450 epoxygenase, and (4) EETs are protective against OGD-induced cell death in astrocytes. These findings suggest that preconditioning-acquired ischemic tolerance is mediated, at least in part, via HIF1α-induced upregulation of 2C11 in astrocytes, and the protective effect of EETs.
Mild ischemia or hypoxia is protective against subsequent lethal ischemia. This adaptive response, referred to as preconditioning (Dirnagl et al, 2003), is in part because of alterations in gene expression (Stenzel-Poore et al, 2003). A key transcription factor that mediates the brain's genomic response to hypoxia is HIF-1 (Sharp and Bernaudin, 2004). A functional HIF-1 is a heterodimer that consists of a constitutive HIF-1β, known as the ARNT, and a regulated HIF-1α. Under normoxia, HIF-1α is continually degraded via oxygen-dependent ubiquination. Hypoxia stabilizes HIF-1α, leading to induction of hypoxia-responsive genes that carry out important adaptive functions, including angiogenesis, erythropoiesis, anaerobic metabolism, and cell survival (Sharp and Bernaudin, 2004).
Hypoxia-inducible factor-1α has specifically been implicated in the protection by preconditioning. Hypoxia-inducible factor-1α induction is associated with protection from hypoxic-ischemic brain injury in newborn rats (Bergeron et al, 2000) and permanent focal cerebral ischemia in adult mice (Bernaudin et al, 2002a). A number of HIF-responsive genes have been proposed to play a role in the protection by preconditioning, including EPO (Prass et al, 2003), IGF-2, IGF-BP (Semenza, 2003), and GLUT-1 (Jones and Bergeron, 2001).
We previously determined that ischemic preconditioning induces P450 2C11 expression in the brain, which was associated with infarct size reduction after MCA occlusion in the rat (Alkayed et al, 2002). Cytochrome P450 2C11 is an AA epoxygenase expressed in astrocytes (Alkayed et al, 1996), which metabolizes AA to EETs. Therefore, this study was designed to determine if 2C11 upregulation by HPC in astrocytes is mediated via HIF-1α. We found that HPC increases 2C11 mRNA and protein in astrocytes. The time course of 2C11 induction was different in cortical versus hippocampal astrocytes, which may account for the differential sensitivity of hippocampal versus cortical astrocytes to injury observed in this and other studies (Zhao and Flavin, 2000; Xu et al, 2001).
To determine if 2C11 upregulation by hypoxia is mediated via HIF-1α, we first searched published 2C11 promoter and found two HIF-binding sites. We then determined that hypoxia increases HIF-1α expression and binding to 2C11 HRE. The increase in 2C11 expression likely contributes to increased tolerance to OGD, because the P450 epoxygenase inhibitor MS-PPOH abolished the protection by HPC. MS-PPOH is a terminal acetylenic fatty acid analog (Wang et al, 1998). It selectively inhibits P450 AA metabolism by acting as a substrate, binding and irreversibly inactivating P450 enzyme (Brand-Schieber et al, 2000). MS-PPOH specificity has been examined in vitro and in vivo. In a study by Wang et al (1998), MS-PPOH dose-dependently inhibited EET formation, but had no effect on the formation of 20-hydroxyeicosatetraenoic acid (20-HETE), a product of the P450 omega-hydroxylase. In a study by Brand-Schieber et al (2000), MS-PPOH was administered in vivo and shown to inhibit AA epoxygenase activity in rat renal cortical microsomes, without affecting 20-HETE formation. Finally, as we had previously shown, MS-PPOH reduces CBF response to whisker activation (Peng et al, 2002), forepaw stimulation (Peng et al, 2004), and N-methyl-d-aspartate, without affecting nitric oxide synthase (NOS) activity (Bhardwaj et al, 2000).
In agreement with our observation that 2C11 upregulation is associated with protection, and that the preconditioning effect was attenuated by MS-PPOH, a recent report showed that transgenic mice with overexpression of another epoxygenase, P450 2J2, in cardiac myocytes improves postischemic recovery in heart function, which is abolished by MS-PPOH (Seubert et al, 2004). To examine the protective effect of EETs directly, we subjected astrocytes to OGD in the presence of EETs or vehicle. We found that astrocyte cell death was significantly reduced by EETs, with no observed differences among EETs regioisomers. This is in agreement with previous reports in non-neural tissue showing that EETs protect against cell death induced by hypoxia-reoxygenation (Yang et al, 2001), H2O2, etoposide, and serum withdrawal (Chen et al, 2001).
The mechanism of astrocyte protection by preconditioning and EETs is unknown. However, EETs and preconditioning share common mechanisms of protection. Overexpression of P450 epoxygenase is associated with less superoxide (Yang et al, 2001), and preconditioning increases antioxidant enzyme activity (Arthur et al, 2004). Similarly, EETs inhibit NF-κB signaling, which induces preconditioning (Jones et al, 2003). Furthermore, EETs regulate intracellular calcium (Rzigalinski et al, 1999), stimulate ATP-sensitive K+ channel (Lu et al, 2002), activate phosphatidylinositide-3'-OH kinase/AKT pathway (Potente et al, 2003), and increase cAMP (Sun et al, 2002), all of which have been implicated in the preconditioning effect (Heurteaux et al, 1995; Ohta et al, 1996; Yano et al, 2001). Our finding that hypoxia induces P450 2C11 epoxygenase is consistent with other reports showing that the epoxygenase pathway plays an important role in mediating the response to hypoxia in a variety of tissues. P450 epoxygenase inhibition impairs hypoxic dilation of skeletal muscle arteries (Frisbee et al, 2001), and chronic hypoxia increases P450 2C9 expression and EETs, which may contribute to enhanced K-channel activity and attenuated vasoconstriction in mesenteric arteries (Earley et al, 2003; Michaelis et al, 2003). Hypoxia also increases P450 4B1 in the corneal epithelium (Vafeas et al, 1998) via HIF-1α, AP-1 and NF-κB (Mastyugin et al, 2004), which may play a role in corneal new vessel formation via VEGF (Mastyugin et al, 2001). Finally, in agreement with our finding that 2C11 upregulation by hypoxia is linked to HIF-1α, P450 3A6 upregulation by hypoxia in hepatocytes has also been linked to HIF-1α (Fradette and du Souich, 2003), which may play a role in altered hepatic drug metabolism in patients with chronic pulmonary disease and heart failure with hypoxemia.
In summary, we provided evidence that HPC is protective against ischemic cell death in astrocytes, at least in part, via HIF1α-induced upregulation of P450 2C11 in astrocytes and the protective action of EETs. This is the first demonstration of a P450 epoxygenase upregulation by hypoxia, and the first evidence of brain cell protection by EETs. In this study, we did not examine the effect of EETs on neuronal cell death. However, 2C11 upregulation by hypoxia in astrocytes may impact neuronal cell death directly via EETs release and action on surrounding neurons, or indirectly by protecting and stabilizing astrocytes, which provide vital metabolic, antioxidant, and trophic support to neurons (Chen and Swanson, 2003; Takuma et al, 2004; Ouyang and Giffard, 2004). The findings of this study suggest that the P450 epoxygenase pathway in astrocytes is an important endogenous mechanism of protection against ischemic injury. Enhancing EET synthesis or preventing its degradation may serve as a novel neuroprotective strategy against ischemic brain injury.