
Abstract
Cell death after traumatic brain injury (TBI) evolves over days to weeks. Despite advances in understanding biochemical mechanisms that contribute to posttraumatic brain cell death, the time course of cell injury, death, and removal remains incompletely characterized in experimental TBI models. In a mouse controlled cortical impact (CCI) model, plasmalemma permeability to propidium iodide (PI) was an early and persistent feature of posttraumatic cellular injury in cortex and hippocampus. In cortical and hippocampal brain regions known to be vulnerable to traumatic cell death, the number of PI + cells peaked early after CCI, and increased with increasing injury severity in hippocampus but not cortex (P < 0.05). Propidium iodide labeling correlated strongly with hematoxylin and eosin staining in injured cells (r = 0.99, P < 0.001), suggesting that plasmalemma damage portends fatal cellular injury. Using PI pulse labeling to identify and follow the fate of a cohort of injured cells, we found that many PI+ cells recovered plasmalemma integrity by 24 h and were terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling negative, but nonetheless disappeared from injured brain by 7 days. Propidium iodide-positive cells in dentate gyrus showed significant ultrastructural damage, including plasmalemma and nuclear membrane damage or overt membrane loss, in all cells when examined by laser capture microdissection and transmission electron microscopy 1 to 24 h after CCI. The data suggest that plasmalemma damage is a fundamental marker of cellular injury after CCI; some injured cells might have an extended window for potential rescue by neuroprotective strategies.
Introduction
Traumatic brain injury (TBI) induces cell death that occurs during a period of days to months after experimental and human TBI (Dixon et al, 1999; Graham et al, 2000; Raghupathi, 2004; Smith et al, 1997; Zhang et al, 2002). Although a growing number of studies provide insight into molecular mechanisms that contribute to posttraumatic brain cell death (Raghupathi, 2004; Zhang et al, 2005), fundamental questions regarding the fate of injured cells, such as reversibility of acute cellular injury and time course of individual cell death, remain to be explored.
One direct approach to examining these questions in brain involves labeling injured cells with membrane-impermeable fluorescent dyes and monitoring their fate with other markers of cell death such as terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) and hematoxylin and eosin (H&E) staining. Using a model of fluid percussion TBI, Singleton and Povlishock (2004) showed altered membrane permeability to fluorescent tracers in scattered neurons, many of which showed ultrastructural features of early necrosis and some that had little or no ultrastructural damage 2 h after injury. The finding that some labeled cells showed no apparent injury was interpreted as evidence that some cells might repair their membrane damage and survive diffuse TBI.
Propidium iodide (PI) is a 668 Da membrane-impermeable nucleic acid stain that emits bright red fluorescence when bound to RNA or DNA. Acute excitotoxic, ischemic, or traumatic cellular injury may induce plasmalemma permeability to PI, although the exact mechanism of PI-permeability is unknown (Matsumoto et al, 2004; McKinney et al, 1996; Unal-Cevik et al, 2004). Propidium iodide and TUNEL histochemistry have been used extensively to distinguish between apoptosis and necrosis after acute injury in cultured cells (Hamabe et al, 2003; Kristensen et al, 2003), and PI labeling has been used to examine cell death phenotypes in ischemic kidney and brain (Kelly et al, 2003; Unal Cevik and Dalkara, 2003). These studies indicate that PI may be a useful marker for loss of membrane integrity in vivo.
Biochemical studies pointing to a complex interplay among numerous distinct mechanisms and modes of traumatic brain cell death have left unanswered a number of fundamental questions regarding the fate of injured cells after TBI. The length of time injured cells remain in brain, and whether they are functional after injury, is not known, and their capacity for membrane repair after initial plasmalemma damage has only been investigated in one short-term study using a mild, diffuse TBI model (Farkas et al, 2006). To begin to address some of these issues relevant to cell death after more severe focal TBI, we employed an in vivo PI pulse-labeling protocol to follow the fate of injured cells after controlled cortical impact (CCI) in mice. We found that plasmalemma permeability to PI occurs early after CCI in brain regions selectively vulnerable to acute cell death, that some initially damaged cells restore their plasmalemma integrity, and that PI-positive cells have a much longer clearance time than previously anticipated. The data suggest that plasmalemma damage assessed by the methodology herein may portend a fatal outcome, but that some injured cells may have an extended therapeutic window for rescue by neuroprotective strategies.
Methods
Mouse Controlled Cortical Impact Model
The trauma protocol was approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee and complies with the NIH Guide for the Care and Use of Laboratory Animals. Anesthesia was induced in a plexiglass chamber with 4% isoflurane, balance N2O/O2 (2:1), and maintained with 2% isoflurane balance N2O/O2 (2:1). Anesthetized mice were positioned in a stereotaxic frame and a midline scalp incision was performed. A 5-mm craniotomy was made using a portable drill and a trephine over the left parieto-temporal cortex. The bone flap was removed and mice were subjected to CCI using a pneumatic cylinder with a 3-mm flat-tip impounder, velocity 6 mins/sec, and set impact depth of depth of 0.6 mm. In addition, some studies comparing the effect of impact depth on PI labeling employed 0.1 mm depth. The 0.6 mm depth produces an injury severity that results in significant hippocampal tissue loss by 3 weeks after CCI, whereas the depth of 0.1 mm results in a lesion, in which cortical tissue is lost but the hippocampus remains intact. After CCI, the scalp was sutured closed, anesthesia was discontinued, and mice allowed to recovered in room air in their cages. Sham-operated mice were subjected to all aspects of the protocol except for cortical impact.
Administration of Propidium Iodide
Propidium iodide (10 mg/mL; Sigma, St Louis, MO, USA) was diluted in 0.9% NaCl and 1 mg/kg was administered by intraperitoneal injection in a total volume of not more than 100 μL. In some experiments, PI (50 μg/mL, 2μL) was administered into the ipsilateral cerebral ventricle (coordinates 0.1 mm posterior, 1 mm lateral, and 2.5 mm deep to bregma) before CCI (see below for specific protocols).
In Vivo Propidium Iodide Labeling Protocols
Three experimental protocols were used to label injured brain cells with PI. In Protocol 1 (n = 82 mice), PI was administered at various times after CCI intraperitoneally 1 h before killing. This protocol was used to label cells that became permeable to PI at both early and later times after injury.
Protocol 2 (n = 6 mice) was used to label cells within the first 5 mins after CCI. This protocol was used to minimize any potential delay in PI labeling owing to kinetics of intraperitoneal absorption into blood and passage of PI across the blood—brain barrier. Mice were administered PI into the ipsilateral cerebral ventricle 1 h before CCI and intraperitoneally 20 mins before CCI. Mice were killed at 5 mins after CCI for examination of PI-positive cells. This protocol ensured adequate blood and cerebrospinal fluid PI levels to label injured brain cells at 5 mins after CCI.
Protocol 3 (n = 72 mice, PI pulse label) was used to label injured cells that became permeable to PI within the first 2 h after CCI. Pulse-labeling studies were used to determine the time course of elimination of PI-positive cells from traumatic mouse brain, to determine the percentage of early PI-labeled cells that developed histological evidence of cell death over time, and to assess the progression of ultrastructural changes in PI+ cells from 1 to 24 h after CCI.
We first determined the bioavailability of PI, administered intraperitoneally, to the injured brain. This was done by administering PI to mice (n = 3 to 8/time point) at various times (0 to 4 h) before CCI and examining for PI-positive cells at 1 h after CCI, a time when PI labeling is robust when PI is administered at the time of injury. These pilot studies showed that the maximum time that PI could be administered before CCI and still produce cellular labeling was 1 h. Thus, in Protocol 3, PI was administered at the time of CCI to pulse label a discrete cohort of cells injured within 0 to 2 h after CCI.
Preparation of Brain Tissue for Histochemistry
For evaluation of PI labeling in nonperfused, frozen brain sections, mice were deeply anesthetized with isoflurane and decapitated at various times after CCI. Brains were removed taking care to keep the contusion region intact and then snap-frozen in liquid nitrogen vapor and stored at −80°C. Brains were cut in the coronal plane on a cryostat. A series of sections (10 μm thick) was collected (150 to 200 μm apart) from anterior to posterior hippocampus from each brain (bregma −1.90 to −3.00). Cryostat sections were placed on poly-
For evaluation of PI staining using fixed tissue, anesthetized mice were transcardially perfused with 4% paraformaldehyde and the brains were removed and embedded in paraffin. Serial brain sections (5 μm) were cut on a microtome and placed on poly-
Multiple Label Histochemistry
Cryostat sections of the brain were fixed in 100% ethanol for 10 mins at room temperature and then allowed to air dry completely. For TUNEL staining, sections were incubated with terminal deoxynucleotidyl transferase buffer (30 mmol/L Tris, 140 mmol/L sodium cacodylate, 1 mmol/L cobalt chloride, pH 7.2) containing terminal deoxynucleotidyl transferase (0.5 U/mL) and fluorescein-12-dUTP (2‘-deoxyuridine 5‘-triphosphate) (0.04 mmol/L) (all reagents from Boehringer-Mannheim, Indianapolis, IN, USA) in a total volume of 30 μL per slide, for 1 h at 37°C. The TUNEL reaction was terminated by immersing the slide in 100% ethanol, and slides were coverslipped in Permount.
For immunohistochemical detection of PI-labeled neurons, astrocytes, and vascular endothelium, cryostat sections of the brain were fixed in 100% ethanol for 10 mins at room temperature, allowed to dry, and reacted for 5 mins with either anti-mouse NeuN (1:300; Chemicon, Temecula, CA, USA) conjugated to Cy2 (Amersham Biosciences, Piscataway, NJ, USA) or fluorescein-labeled Griffonia simplicifolia lectin (1:200; Vector Laboratories, Burlingame, CA, USA) to label vascular endothelium. To label astrocytes, sections were reacted for 30 mins with rabbit anti-mouse glial fibrillary acidic protein (1:100 in phosphate-buffered saline; Sigma), washed briefly in phosphate-buffered saline, and then reacted for 5 mins with goat anti-rabbit-FITC (1:200; Vector Laboratories). Sections were rinsed in 100% ethanol, air dried, and coverslipped with Permount. Propidium iodide labeling was photographed using emission and excitation wavelengths of 568 and 585 nm, respectively. Fluorescein labeling was detected using excitation and emission filters of 490 and 520 nm, respectively.
Assessment of Propidium Iodide and Hematoxylin and Eosin Staining
To evaluate PI and H&E staining within injured tissue, mice were subjected to CCI and administered PI at 5, 23, or 47 h, and then killed at 6, 24, or 48 h, respectively (n = 1 to 2/time point). Frozen brain sections cut on a cryostat (10 μm) were collected at 150 to 200 μm intervals and placed on poly-
Quantitation of Propidium Iodide- and Terminal Deoxynucleotidyl Transferase-Mediated Biotinylated UTP Nick End Labeling-Positive Cells (Protocol 1) and Pulse-Labeled Propidium Iodide-Positive Cells (Protocol 3)
Cells labeled with PI in time-course experiments (Protocols 1 and 3) were counted in predetermined brain regions of interest in all animals from bregma −1.70 to −3.00. Cortical brain regions were chosen from all × 400 cortical fields from within contused cortex. Hippocampal regions were medial and mid-dentate gyrus, medial CA1, and the lateral most region of CA3, based on the spatial distribution of PI+ cells after CCI (Figure 2). From the brains of each mouse, 5 to 8 × 400 (450 × 450 μm) cortical fields from 5 to 8 brain sections separated by at least 150 μm were chosen for analysis using a random number generator. One cortical field per brain section was analyzed. For hippocampal cell counts, two adjacent × 400 dentate gyrus fields were counted in three different brain sections separated by at least 150 μm (six 400 fields per animal total); for CA1 and CA3 regions, one × 400 field encompassing approximately half of the entire anatomic region of interest was examined in three different brain sections, for a total of three × 400 fields per mouse. The mean number of positive cells for a given time point was calculated by summing the cell count data from all the brain sections counted and dividing by the number of × 400 fields analyzed.
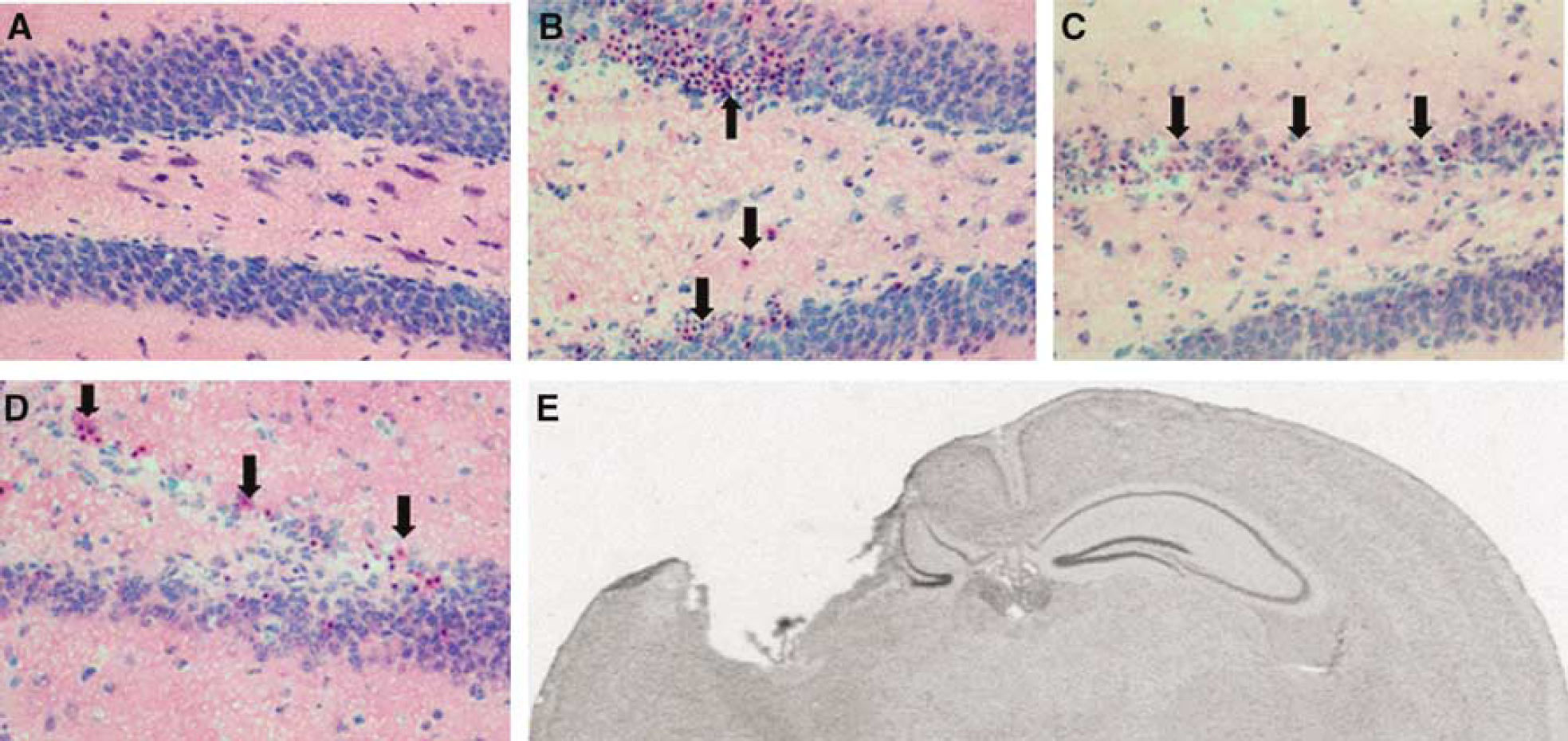
Temporal progression of cellular degeneration and loss in vulnerable brain regions after controlled cortical impact (CCI). (
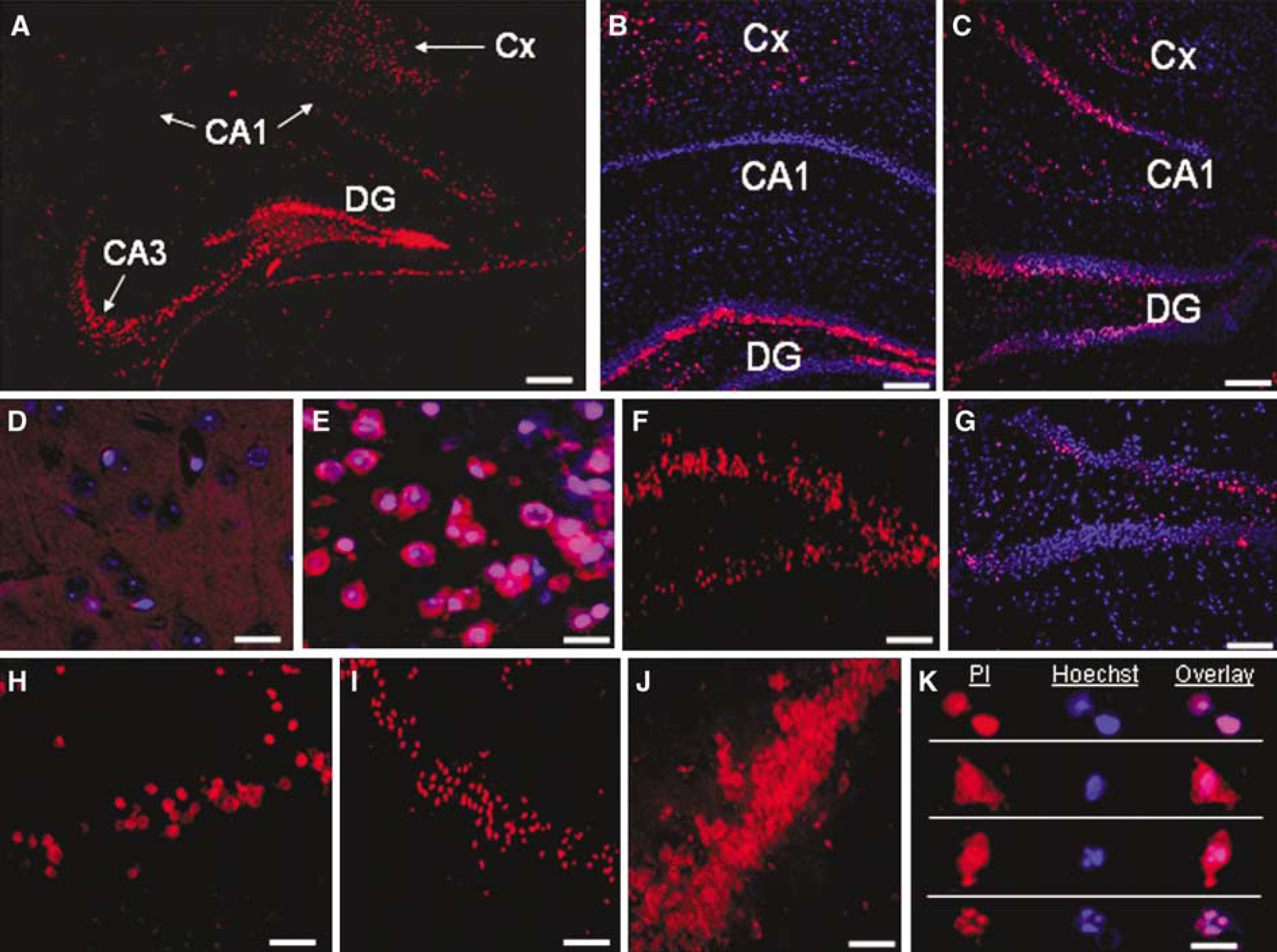
Propidium iodide (PI) labels injured cells in selectively vulnerable brain regions after controlled cortical impact (CCI). Propidium iodide was administered to mice at various times after CCI (Protocols 1 and 2, Methods), and paraformaldehyde-fixed (
For experiments in which PI + cells were colabeled with TUNEL ex vivo, cortical regions of interest were randomly chosen and photographed as described above. Propidium iodide labeling was photographed using emission and excitation wavelengths of 568 and 585 nm, respectively, and fluorescein labeling was detected using excitation and emission filters of 490 and 520 nm, respectively. For multiple label experiments, photomicrographs were imported into PowerPoint and pseudocolored using maximal or near-maximal saturation levels for red, blue, and green wavelengths by adjusting the brightness and contrast of all wavelengths concomitantly. The mean number of PI- or TUNEL-positive cells for a given time point was calculated by summing the cell count data from all the brain sections counted and dividing by the number of × 400 fields analyzed.
Determination of Plasmalemma Resealing Using Propidium Iodide and YOYO-1
Mice were subjected to CCI and PI labeling Protocol 3 (PI pulse label). At 23 h after CCI, mice were administered YOYO-1 intracerebroventricularly (4 μL of 10 μg/mL YOYO-1 in phosphate-buffered saline; Molecular Probes, Eugene, OR, USA). Mice were killed at 24 h after CCI and brain sections were analyzed by fluorescence microscopy using excitation and emission wavelengths for YOYO-1 identical to those used for fluorescein. Propidium iodide-positive cells that underwent membrane resealing by 24 h after CCI were defined as PI +/YOYO-1–, whereas injured cells with persistent plasmalemma permeability were PI+/YOYO-1+. Cells that were initially intact early after CCI that later developed membrane permeability were defined as PI–/YOYO-1+.
Electron Microscopy
To prepare brain tissue for transmission electron microscopy, mice were deeply anesthetized and transcardially perfused with a solution containing 2% paraformaldehyde and 2% glutaraldehyde (electron microscopy grade; Sigma) in phosphate-buffered saline (pH 7.4). Brains were postfixed overnight in that solution and then cryoprotected in 12.5% sucrose for 24 h followed by 25% sucrose for 24 h. Brains were frozen in dry ice crystals and sectioned on a cryostat (8 μm) and transferred to RNase-free membrane-mounted metal frame slides (Molecular Machines and Industries (MMI), Glattsbrugg, Switzerland) for laser capture microdissection. Propidium iodide-positive cells were identified on a Nikon TE 2000 inverted microscope connected to CellCut laser capture microscopy equipment (MMI) and dissected with UVCut software (MMI). Brain tissue was transferred to mini-isolation caps and postfixed in 1% glutaraldehyde. In some experiments, alternate tissue sections were transferred to poly-
For electron microscopy, brain tissue on the capture tube caps was postfixed with a mixture of 1% osmium tetroxide (OsO4) + 1.5% potassium ferrocyanide (KFeCN6) for 30 mins, washed in water, stained with 1% aqueous uranyl acetate for 30 mins followed by dehydration in grades of alcohol (50, 70, and 95, 2 × 100%), and then infiltrated and embedded in TAAB Epon (Marivac Canada Inc., St Laurent, Canada). Ultrathin sections (60 to 80 nm) were cut on a Reichert Ultracut-S microtome, placed on copper grids, stained with 0.2% lead citrate, and examined in a Tecnai G2 Spirit BioTWIN Transmission electron microscope. Images were taken with a 2k AMT CCD camera.
Statistical Analyses
Propidium iodide-positive cell counts from time-course experiments using Protocols 1 and 3, and PI-TUNEL cell counts (Protocol 1) were analyzed by analysis of variance on ranks and Dunnett's test for nonparametrically distributed data, or by analysis of variance and Tukey's test for parametric data. Cell count data for PI and H&E staining were analyzed using Pearson's product-moment correlation. For all tests, P < 0.05 was considered significant.
Results
Spatiotemporal Distribution of Cell Death after Controlled Cortical Impact Assessed by Hematoxylin and Eosin Staining
Previous studies using CCI in mice reported selective vulnerability to neuronal death in CA3 and the hilar regions, as well as dentate gyrus (Hall et al, 2005; Saatman et al, 2006; Smith et al, 1995). Using a depth of CCI set at 0.6 mm, we also observed robust cellular injury using H&E staining in ipsilateral cortex, CA3 and hilus, and dentate gyrus. Figure 1 shows the typical progression of cellular injury and loss from ipsilateral dentate gyrus at 1 to 3 days after CCI. Injured cells were only occasionally detectable by H&E staining in contralateral hemispheric brain regions (Figure 1A and data not shown). By 21 days, the 0.6 mm impact setting typically produced significant gross tissue loss in ipsilateral hippocampus (including dentate gyrus) and cortex (Figure 1E).
Propidium Iodide Label Cells in Injured Brain Regions Selectively Vulnerable to Cell Death after Controlled Cortical Impact
We next determined the spatiotemporal distribution of PI-labeled cells after CCI using PI labeling Protocols 1 and 2 (Figure 2). Propidium iodide labeling appeared in distinct populations of traumatically injured brain cells in regions known to be vulnerable to traumatic cell death (Clark et al, 1997; Colicos et al, 1996; Hannay et al, 1999; Smith et al, 1995), including cortex, CA3, the hilar region, and dentate gyrus, with relative sparing of CA1. Propidium iodide-positive cells were detected as early as 5 mins after CCI in ipsilateral dentate gyrus (Figure 2G). At 1 h after CCI, PI-positive cells were robustly detected in ipsilateral cortex directly under and adjacent to the impact site, and in ipsilateral dentate gyrus and CA3 (Figures 2A to 2C, 2F to 2H, and 2J). Propidium iodide-positive cells were also sometimes found in CA1, a brain region that is relatively resistant to cell death in our CCI model (Figures 2A to 2C). Propidium iodide-positive cells could be detected in vulnerable brain regions as late as 72 h after CCI (Figure 2H). In all injured brain regions, PI labeling was heterogeneous, with PI-positive cells with abnormal nuclear morphology juxtaposed with (apparently uninjured) PI-negative cells. In contrast to injured mice, PI-positive cells were not detected in brain regions from naïve or sham-injured mice, or in the contralateral hemisphere of injured mice using either Protocol 1 or 2 (Figure 2D and data not shown).
PI-positive cells exhibited two cellular labeling patterns that may suggest that injured cells die by more than one mechanism (Figure 2K): (1) cells with cytoplasmic, nuclear, or cytoplasmic + nuclear PI labeling and round, smooth nuclei (nuclear morphology consistent with necrosis); and (2) cells with cytoplasmic, nuclear, or cytoplasmic + nuclear PI labeling and condensed fragmented nuclei (nuclear morphology consistent with apoptosis). Cells with apoptotic nuclear morphology (condensed, fragmented) were often, but not always, PI-positive, although many of these cells were not as intensely labeled as surrounding cells with pyknotic nuclei.
Figure 3 shows representative photomicrographs of cell types vulnerable to plasmalemma permeability early (1 h) after CCI. Neurons in cortical and hippocampal brain regions showed increased plasmalemma permeability (Figures 3A and 2B), as were cortical cells with morphological features of microglia and glial fibrillary acidic protein-positive astrocytes (Figures 3C and 3D), and cortical endothelial cells identified by specific lectin staining (Figures 3E and 3F). These and other cell types such as those residing within white matter tracts were also detected at early and later times after CCI.
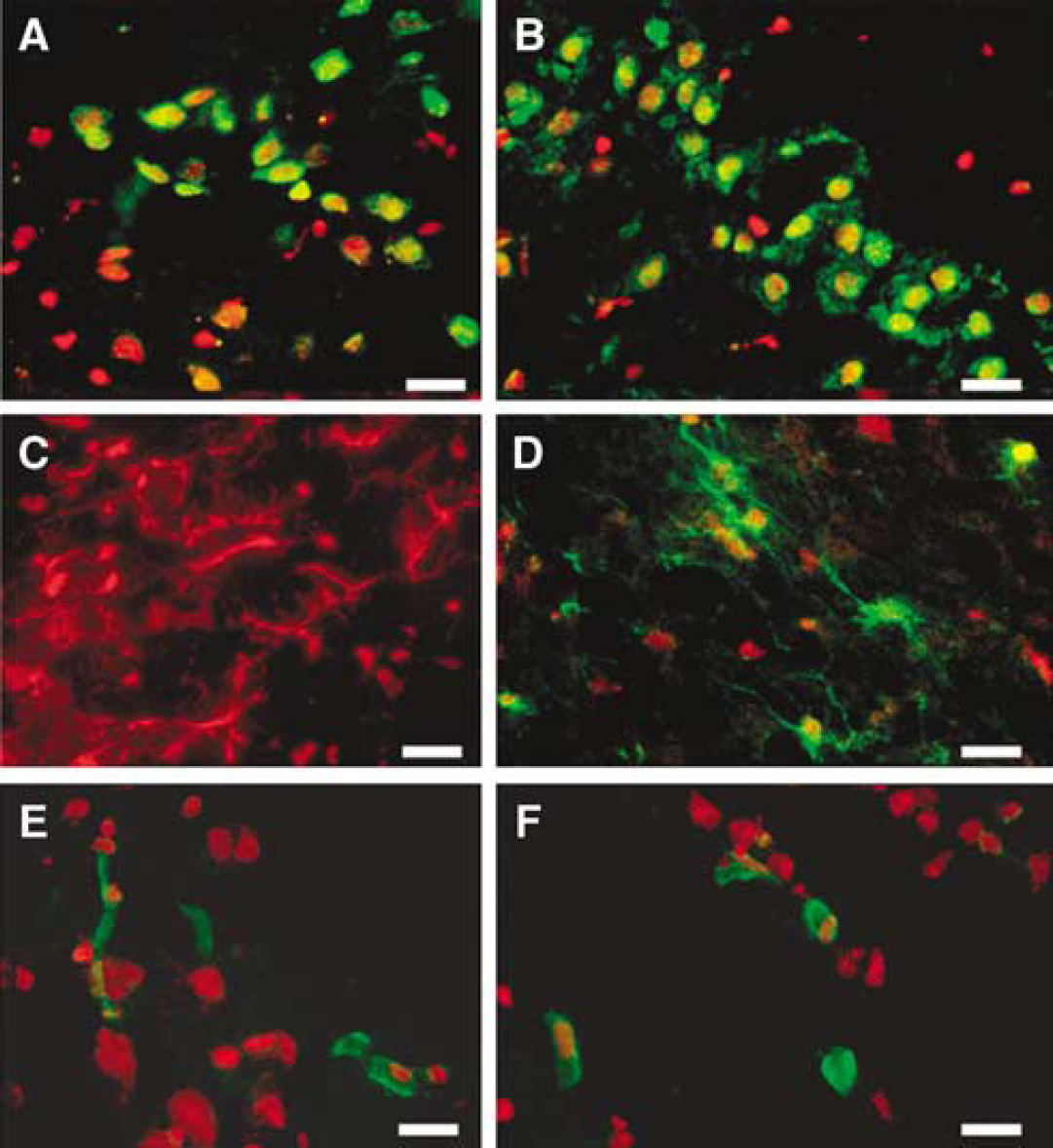
Acute plasmalemma permeability to PI occurs in multiple cell types early after controlled cortical impact (CCI). At 1 h after CCI, PI colocalized with neurons labeled with anti-NeuN (green) in cortex (
Figure 4 shows the temporal distribution and response to injury severity of PI-positive cells after CCI using Protocols 1 and 2 (for the 5-min time point, PI was administered as described in Protocol 2 (Methods); however, owing to differences in the route of PI administration between Protocols 1 and 2, mice in the 5-min time point were not included in statistical analyses). The number of PI-positive cells in injured cortex, dentate gyrus, CA3, and CA1 peaked at 1 to 6 h after CCI (Figure 4A). By 7 days, PI+ cells were nearly undetectable in all injured brain regions examined. Because previous studies have shown a direct relationship between depth of impact and hippocampal cell death after CCI in mice (Saatman et al, 2006), we determined the effect of increasing injury severity on plasmalemma permeability at 1 h, the time of peak PI labeling after CCI. Figure 4B shows that increasing depth of CCI was associated with increased numbers of PI-positive cells in injured hippocampus, but not cortex. Taken together with the finding that PI-positive cells are found in brain regions with overt cell loss, the data suggest that in vivo PI labeling may be a marker of eventual cell death in our CCI model.
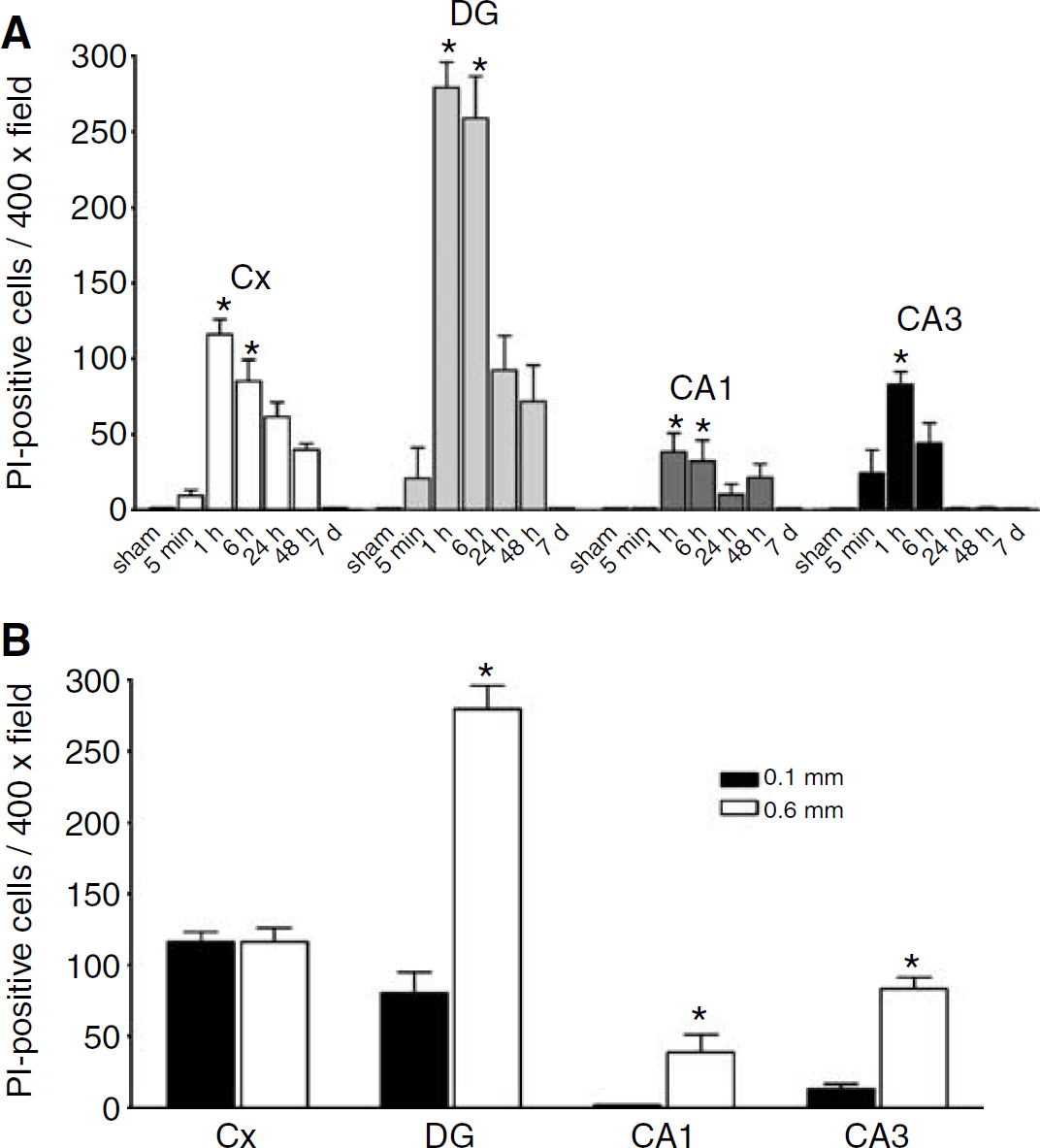
(
Propidium Iodide Labeling Corresponds with Markers of Cellular Degeneration after Controlled Cortical Impact
To test the hypothesis that in vivo PI labeling predicts posttraumatic cell death, we asked whether PI labeling corresponds with other markers of cellular degeneration. Previous studies in experimental TBI have shown that cells labeled with eosinophilic cytoplasm and intense nuclear basophilia by H&E staining degenerate after injury (Newcomb et al, 1999). To examine whether PI permeability is also a marker for cellular degeneration after CCI, we correlated the number of PI-positive cells with degenerating (pyknotic) cells labeled by H&E staining in randomly chosen × 400 cortical and hippocampal fields at 24 and 48 h after CCI. Comparison of the two staining methods shows that degenerating cells correspond to those labeled by PI (Figures 5A and 5B). Of cells labeled by PI, 95% were degenerative by H&E staining (Figure 5C; r=0.99, P < 0.001). The finding that PI positivity and degeneration identified by H&E staining were strongly correlated suggests that PI may be a surrogate marker for eventual cell death in the CCI model, as reported previously in studies of experimental stroke (Unal Cevik and Dalkara, 2003).
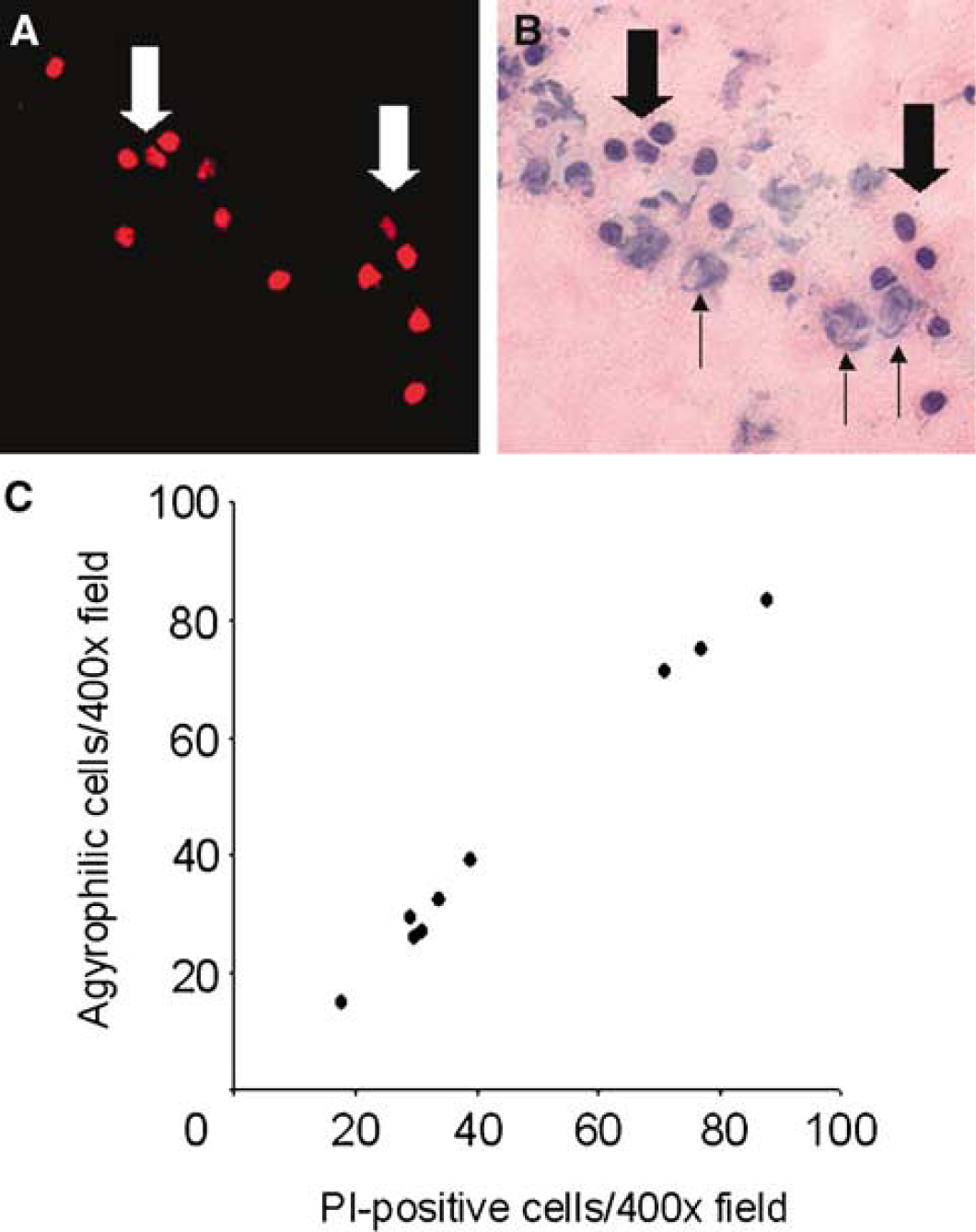
Propidium iodide (PI) identifies degenerating cells after controlled cortical impact (CCI). Representative micrographs showing correspondence between PI labeling (
Plasmalemma Permeability Occurs Independently of DNA Fragmentation after Controlled Cortical Impact
We next sought to determine the relationship between plasmalemma permeability and double-strand DNA fragmentation, a marker of cellular injury, using PI labeling Protocol 1 and ex vivo TUNEL histochemistry. Although PI labeling was maximal at 1 h after CCI, no TUNEL-positive cells were observed at this early time point in cortex or hippocampus (Figures 1 and 2 and TUNEL data not shown). Thus, plasmalemma permeability typically occurs earlier than and can be independent of DNA fragmentation after CCI.
Figure 6 shows representative photomicrographs of TUNEL and PI labeling in hippocampal and cortical brain regions at 6 h after CCI. At least three cell death phenotypes were consistently identified: (1) PI-positive/TUNEL-negative cells (PI+/TUNEL–, consistent with plasmalemma injury without significant DNA fragmentation; red cells in Figures 6Ac and 6Af); (2) PI-negative/TUNEL-positive cells (PI–/ TUNEL+; intact cell membrane integrity with DNA fragmentation; green cells in Figures 6Ac and 6Af); and (3) cells that were both PI-positive/TUNEL-positive (PI+/TUNEL+; loss of cell membrane integrity and significant DNA fragmentation, yellow cells in Figures 6Ac and 6Af). Injured cells with PI staining were differentiated from erythrocytes (that lack a nucleus) or infiltrating leukocytes (with characteristic nuclear shape) and other potential sources of autofluorescence by Hoechst 33342 nuclear staining (Figures 6Ad and 6Ae) and by absence of fluorescence emission using inappropriate excitation spectra. The spatial distribution of each of the three cell death phenotypes was remarkably heterogeneous in cortex and hippocampus. Some microscopic fields contained multiple cellular labeling phenotypes, whereas a single phenotype (usually PI+/TUNEL– or PI+/TUNEL+) predominated in others (Figure 6Af). In quantitative estimates of PI+/TUNEL+ cells in injured cortex, the majority of injured cortical cells detected at 24 h were PI+/TUNEL– or PI+/TUNEL+ (Figure 6B). In contrast, relatively few PI–/TUNEL+ cells were observed in injured cortex.
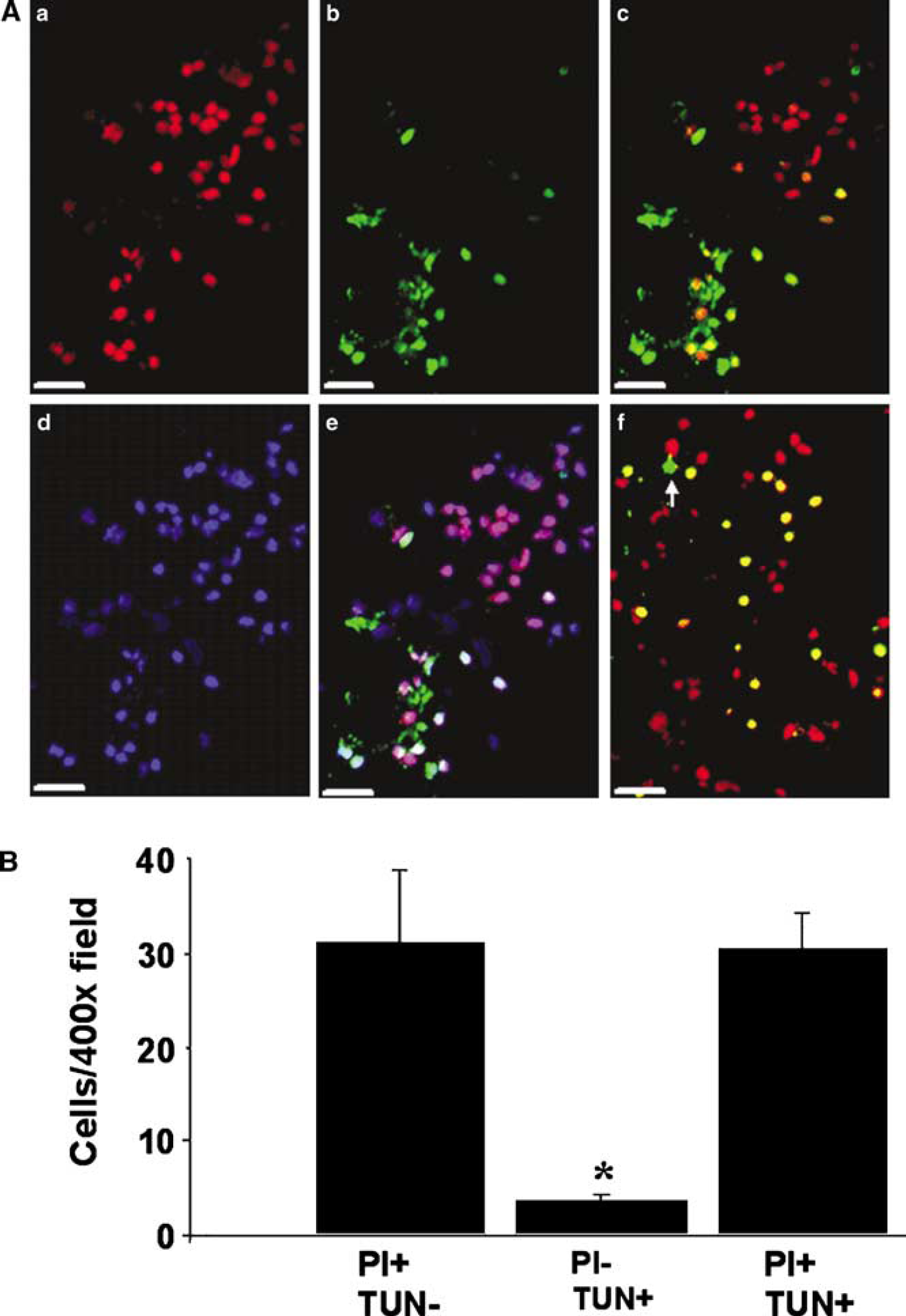
Loss of plasmalemma integrity and DNA fragmentation in cells injured by controlled cortical impact (CCI). (
Protracted Clearance of Propidium Iodide-Positive Cells from Brain after Controlled Cortical Impact
We next sought to estimate the survival time of PI-positive cells in contused brain regions after CCI. To this end, we developed a PI pulse-labeling protocol (Protocol 3) to follow the fate of a distinct population of cells injured within 0 to 2 h after CCI. We based the pulse-labeling protocol on the assumptions that (1) PI injected intraperitoneally at the time of CCI has a short in vivo brain elimination time, and that (2) PI bound to DNA/RNA does not leak out of labeled cells. To address the former, we injected PI into naïve mice, waited for variable amounts of time after injection, performed CCI, and examined brains at 1 h after CCI for the presence or absence of PI-positive cells. The 1 h postinjury time point was chosen to examine for PI-positive cells because PI labeling in injured brain peaks at 1 h after CCI (Figure 4). Of eight mice injected with PI at the time of CCI, all eight had PI+ cells in injured cortex and hippocampus. However, only one of the seven mice injected with PI 1 h before CCI had PI+ cells in contused brain, and no mice injected with PI at 2, 3, or 4 h before CCI (n = 3 to 4/group) had detectable PI+ cells in cortical or hippocampal brain regions. Thus, the time window for PI-labeling in the injured brain at the doses and route of administration used in Protocol 3 was within the first 0 to 2 h after CCI.
We then performed a series of experiments to assess the time course of PI+ cell loss, and to show that cellular uptake of PI is irreversible within the experimental period. Propidium iodide was administered to mice immediately after CCI (Protocol 3, n = 3 to 4/time point), and mice were killed at various time points after injury. Propidium iodide-positive cells were readily detected at 24 and 48 h, and as late as 72 h, in injured cortex and hippocampus (Figures 7A to 7F). At 7 days, only occasionally a PI+ cell was detected in the margins of the (cavitary) cortical lesion and none were detected in injured hippocampus (Figures 7G and 7H). However, choroid plexus cells that were labeled at the time of CCI showed robust PI staining at all time points, including 7 days (Figure 7I), showing that once inside normal cells, PI staining is retained for the duration of the experimental period.
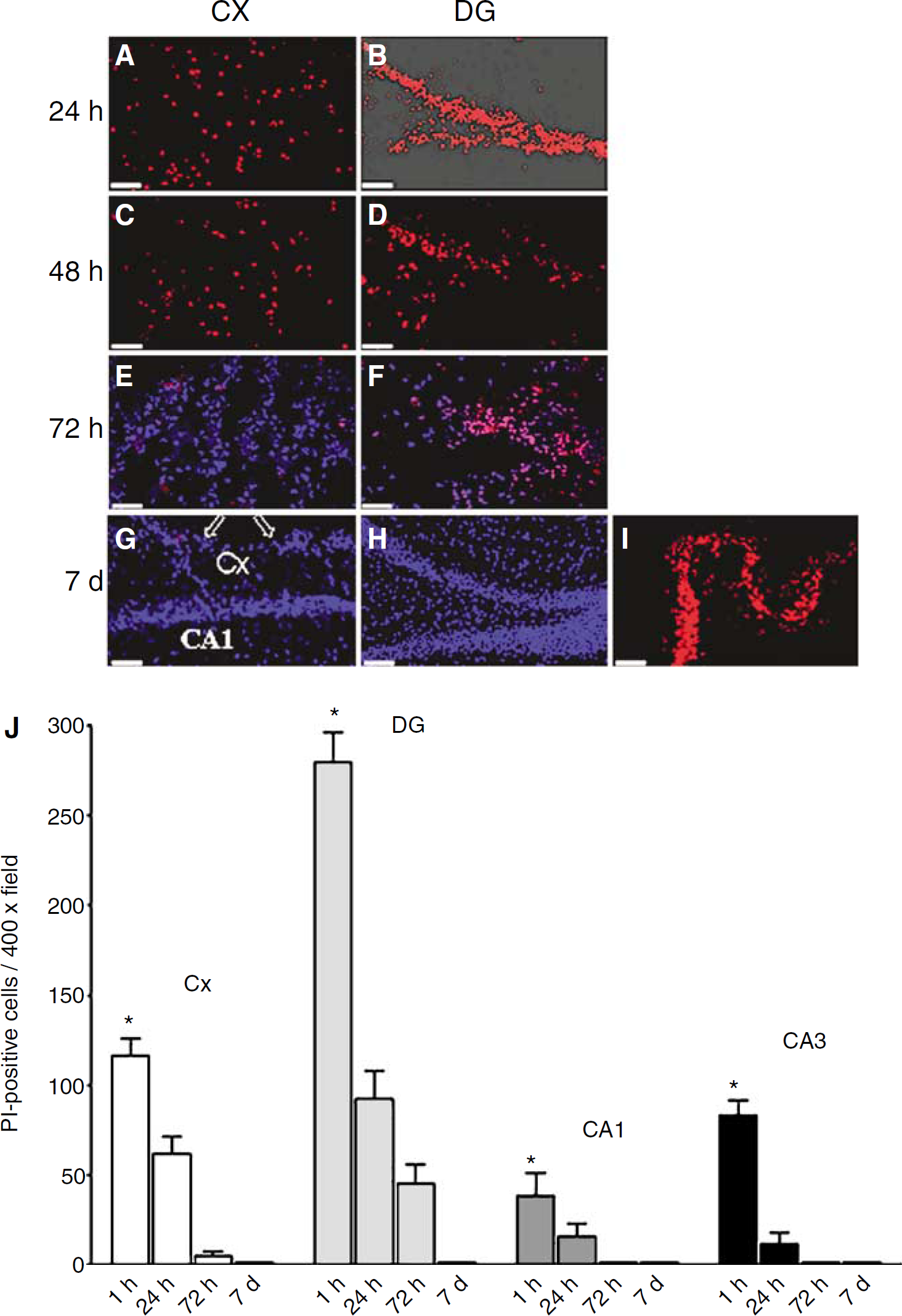
Protracted clearance of propidium iodide (PI)-positive cells in contused brain after controlled cortical impact (CCI). Mice (n = 3 to 7/time point) were administered PI at the time of CCI (Protocol 3) and killed at 24, 48, or 72 h, or 7 days. Propidium iodide-positive cells were photographed in representative cortical (
To determine whether injured parenchymal cells may retain PI, we injected PI into normal mouse cortex and examined for PI-positive cells. At 7 days after intraparenchymal PI administration, numerous PI-positive cells surrounding the needle tract were observed (n = 3 mice, data not shown). Thus, even injured cells rendered PI-permeable retained PI for at least 7 days.
Figure 7J shows a quantitative assessment of PI pulse-labeled cells (Protocol 3) remaining in injured cortical and hippocampal brain regions over the first 7 days after CCI. In cortical and hippocampal regions, the number of pulse-labeled PI-positive cells peaked at 1 h after CCI and then decreased to near zero in a time-dependent manner (see also Figures 7G and 7H). These data strongly suggest that PI-positive cells die or are removed from injured brain regions within several days of injury, and further support the hypothesis that loss of plasmalemma integrity is a marker of eventual cell death in our CCI model.
Development of DNA Fragmentation in Propidium Iodide Pulse-Labeled Cells after Controlled Cortical Impact
To more precisely determine the temporal relationship between loss of plasmalemma integrity and development of DNA fragmentation after CCI, mice (n = 4 to 5/time point) were administered PI at the time of CCI (pulse label; Protocol 3) and killed at 24 or 48 h after injury. Frozen brain sections were subjected to TUNEL histochemistry, and PI+/TUNEL+ cells were quantitated in × 400 cortical fields (Methods). At each time point, PI+/ TUNEL– cells comprised approximately 40 to 60% of the total number of cells counted, suggesting that DNA fragmentation may be quite delayed or may not occur in some PI-positive cells (24 h: PI+/TUNEL–, 34.1+4.9 and PI+/TUNEL+, 23.6+2; 48 h: PI+/ TUNEL–, 20.5+3.6 and PI+/TUNEL+ 24.1+7 cells/ × 400 field), assuming equal labeling efficiency of the TUNEL reagent in all experiments.
Evidence for Membrane Resealing in Propidium Iodide-Permeable Cells Injured Early after Controlled Cortical Impact
Previous studies using a diffuse TBI model lacking overt tissue damage suggested that some permeable neurons may regain plasmalemma integrity at later times after injury (Farkas et al, 2006). To determine whether loss of plasmalemma integrity may be reversible after CCI, we used Protocol 3 and performed double-labeling experiments with YOYO-1, a membrane-impermeable dye with green fluorescence. In a series of pilot studies, we first administered PI and YOYO-1 intracerebroventricularly to mice (n = 4) concomitantly at the time of CCI and showed that at 1 h after injury, YOYO-1 consistently colabeled with the majority of PI+ cells but not with PI– cells (n = 5 cortical fields (450 × 450 μm) examined per mouse; Figures 8A to 8C). Thus, YOYO-1 labeling is specific for PI+ cells early after CCI, but, because YOYO-1 did not colabel 100% of PI-positive cells, is probably a less sensitive marker for loss of cell membrane integrity compared with PI.
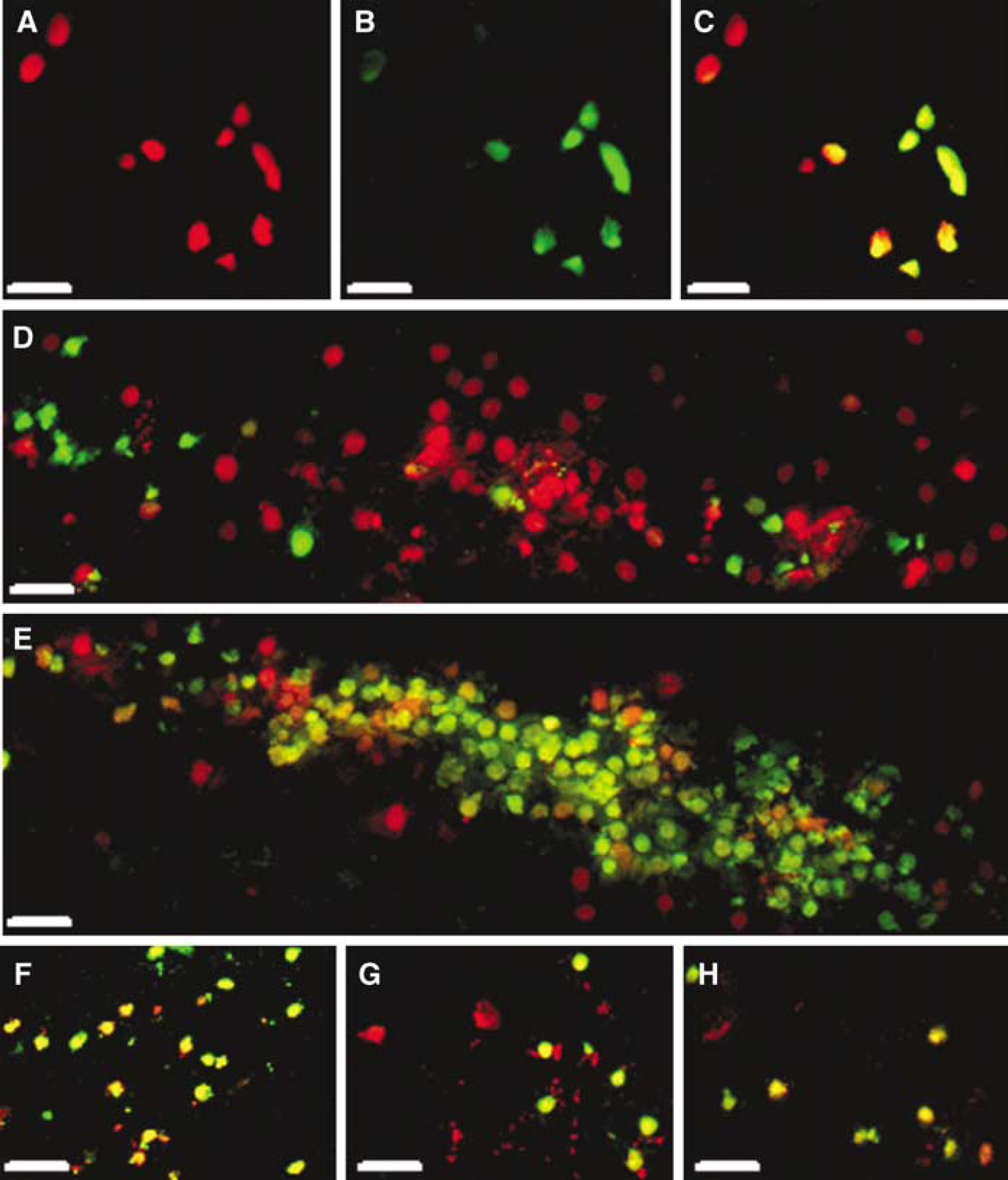
Evidence for plasmalemma resealing after controlled cortical impact (CCI). (
We next administered PI to mice (n = 4) at the time of CCI, and administered YOYO-1 intracerebroventricularly 1 h before killing at 24 h. Frozen brain sections were examined for the presence or absence of PI and YOYO-1 staining. Figures 8D to 8H show cells with three PI/YOYO-1 labeling patterns: (1) PI+/YOYO-1–, (2) PI+/YOYO-1+, and (3) PI–/ YOYO-1+. The distribution of the three cellular phenotypes was remarkably heterogeneous, with persistent membrane permeability predominating in some brain regions (Figure 8E) and membrane resealing predominating in others (Figure 8D). Assuming 100% labeling efficiency for YOYO-1 in each animal, the data suggest that, despite their eventual demise, some PI-positive cells may undergo membrane resealing, whereas loss of membrane integrity (PI+/YOYO-1+) may persist for up to 48 h in other PI-positive cells. In addition, the presence of PI–/YOYO-1+ cells implies that, after CCI, delayed loss of membrane integrity may occur in brain cells that initially escape plasmalemma damage.
Ultrastructural Analysis of Propidium Iodide-Positive Cells
To gain further insight into mechanisms of cellular injury and death in PI-positive cells, we used transmission electron microscopy to assess ultrastructural features of pulse-labeled PI-positive cells microdissected by laser capture microscopy from injured dentate gyrus at 1 or 24 h after CCI (PI Protocol 3). We focused our studies on dentate gyrus because the high density of cells in this region allowed us to isolate groups of PI-positive cells that were devoid of PI-negative cells (documented by Hoechst 33342 counterstain in alternate brain sections).
Figure 9 shows typical nuclear ultrastructure of PI-positive cells isolated from dentate gyrus at 1 h after CCI. Compared with the normal ultrastructure of cells isolated from naïve (uninjured) mouse brain (Figure 9A), PI-positive cells showed marked nuclear chromatin condensation (Figures 9B and 9C), which could be easily identified in groups of PI-positive cells in microdissected brain tissue sections (Figure 9D). As early as 1 h after CCI, PI-positive cells displayed a spectrum of plasmalemma and nuclear membrane abnormalities, from relatively intact membranes (rarely seen; Figures 10D and 10E) to partially fragmented (Figures 10A and 10B) or even complete loss of visible plasma and nuclear membrane structures in most cases (Figures 10C and 10E). By 24h after CCI, most PI-positive cells examined showed evidence of extensive nuclear membrane damage including partial dissolution (Figure 11A), blebbing (Figure 11B), and fragmentation of the double-membrane structure (Figure 11C). Mitochondrial swelling was also observed in many PI-positive cells (Figure 11C), and infrequently apoptotic bodies were also observed. Although some PI-positive cells at 24 h maintained an intact plasmalemma (Figure 11A), most PI-positive cells examined (n = 45 from a total of three mice) had evidence of extensive plasmalemma damage/loss by 24 h after CCI. Despite the frequent finding of mitochondrial swelling in PI-positive cells, mitochondrial membranes were generally intact in PI-positive cells even at 24 h after injury, suggesting that plasmalemma and nuclear membranes may be particularly susceptible to damage after CCI. In contrast to PI-positive cells, PI-negative cells did not exhibit plasma or nuclear membrane gaps or other identifiable pathological ultrastructural changes (Figures 11D and 11E).
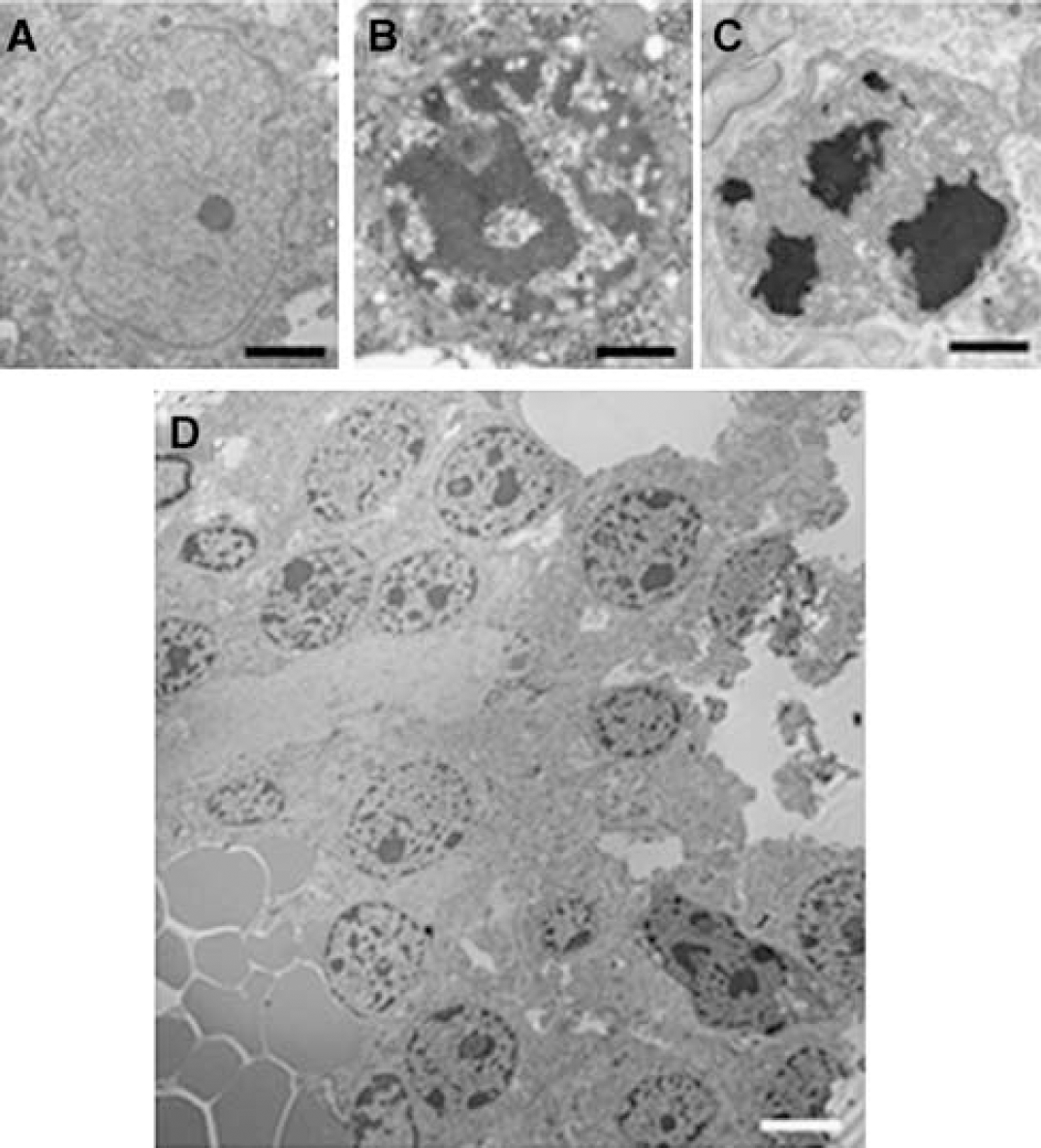
Nuclear ultrastructure in normal brain cells and in brain cells labeled with propidium iodide (PI) after controlled cortical impact (CCI). Mice (n = 5) were administered PI at the time of CCI and killed at 1 or 24 h (Protocol 3). The cell (
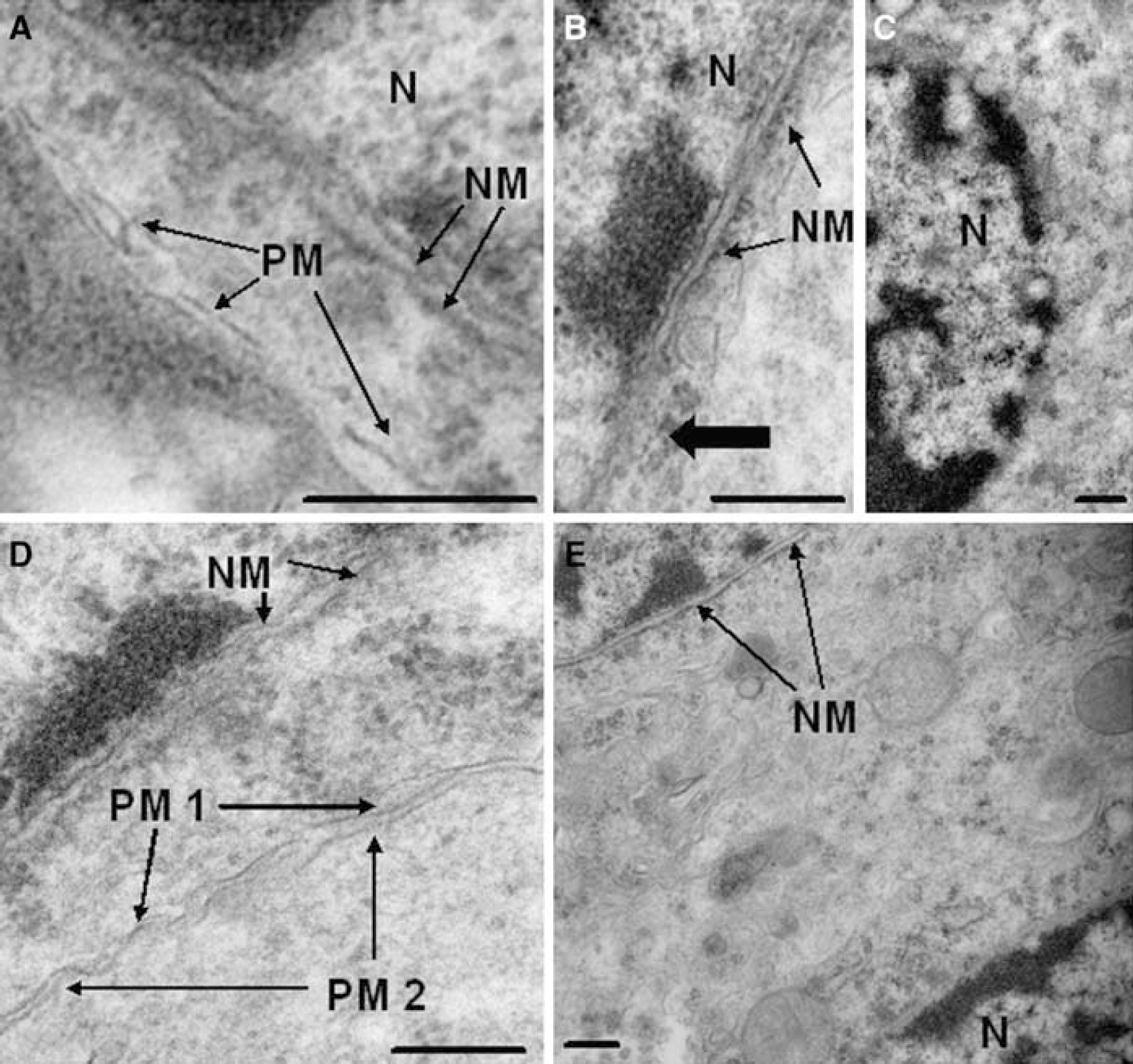
Ultrastructural changes in propidium iodide (PI) pulse-labeled cells at 1 h after controlled cortical impact. (
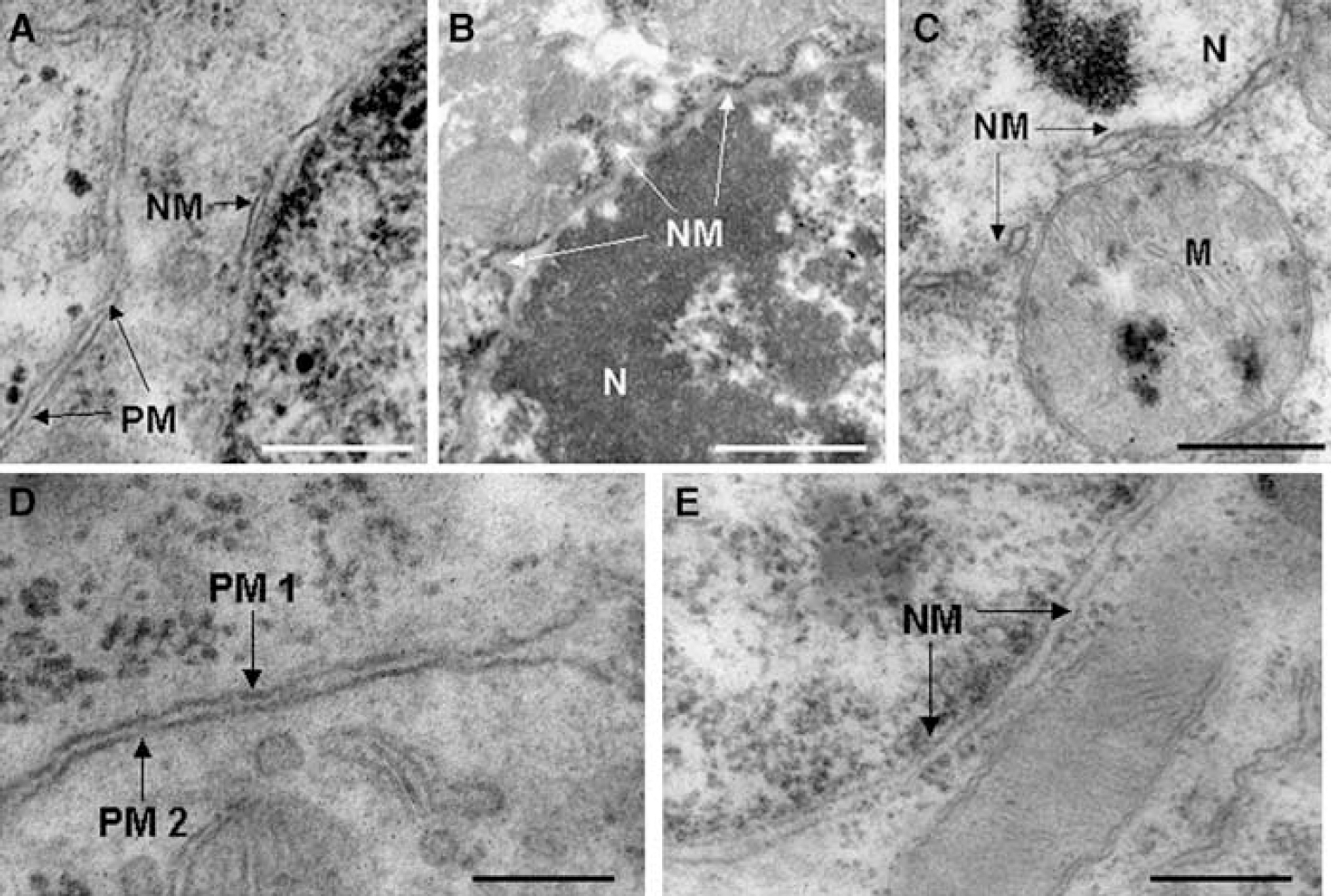
Ultrastructural changes in propidium iodide (PI) pulse-labeled cells at 24 h after controlled cortical impact. (
Discussion
Using in vivo PI labeling to follow the fate of cells injured by CCI, we found that plasmalemma damage occurs early and before the onset of DNA fragmentation in brain regions vulnerable to traumatic cell death. The close correspondence between PI labeling and H&E staining, and the ultrastructural findings in PI+ cells at 1 and 24 h suggest that PI permeability may be a marker for eventual cell death in this model. Nonetheless, the findings that PI-positive cells may regain plasmalemma integrity and remain in brain for up to several days after injury suggest that a prolonged therapeutic window might exist to rescue PI-positive cells using neuroprotective therapies. Although specific mechanisms leading to PI permeability after CCI remain to be defined, our findings help to clarify the nature and time course of cell death and removal after cerebral contusion.
A striking finding of this study is that PI-permeable cells were observed within 1 h after CCI (Figures 2 and 4). In contrast, significant PI permeability was delayed until approximately 6 h after cerebral ischemia/reperfusion in mice (Unal Cevik and Dalkara, 2003). The different kinetics of membrane failure in CCI versus stroke possibly reflects fundamental differences in injury mechanisms between the two models, rather than PI dosing (20 in stroke versus 1 mg/kg in CCI) or PI bioavailability (Unal Cevik and Dalkara, 2003).
Early PI labeling after CCI could be mediated by direct physical mechanoporation (Farkas et al, 2006). However, data from our studies implicate secondary injury mechanisms as well. First, the number of PI-positive cells increased between 5 mins and 1 h after CCI. If physical damage was the sole mechanism of membrane permeability, higher numbers of PI-permeable cells would be expected at earlier times after CCI (i.e., 5 mins; Toth et al, 1997). Second, delayed plasmalemma permeability appeared in some cells as late as 24 to 48 h after CCI, implicating secondary mechanisms at these prolonged times after mechanical insult.
An alternative explanation for early PI labeling in CCI versus experimental stroke may be injury-specific differences in the kinetics of programmed cell death mechanisms. We have reported previously decreased cortical PI-positive cells at 6 h after CCI in mice deficient in bid (BH3 interacting domain death agonist) (Bermpohl et al, 2006) as well as tumor necrosis factor-a and Fas receptor (Bermpohl et al, 2007). Necroptosis, a recently described form of programmed necrosis mediated by tumor necrosis factor-α and Fas, induces loss of plasmalemma integrity in cultured cells and contributes to cell death in experimental stroke (Degterev et al, 2005). Focal stroke is characterized by delayed onset of plasmalemma permeability (Unal Cevik and Dalkara, 2003) and a prolonged therapeutic window for necroptosis inhibitors (Degterev et al, 2005), whereas our preliminary findings suggest a short therapeutic window to inhibit necroptosis after CCI (MJ Whalen, unpublished data). Thus, differences in the kinetics of necroptosis, and perhaps other programmed cell death mechanisms, may contribute to the more rapid onset of plasmalemma permeability in CCI versus stroke.
Several lines of evidence suggest that plasmalemma permeability may be a marker of fatal cellular injury. First, virtually the entire cohort of cells that were pulse labeled within the first 0 to 2 h disappeared from injured brain by 7 days. Second, PI-positive cells were numerous in brain regions undergoing overt cell loss, and were distributed in a pattern similar to that of neurodegeneration reported in other mouse CCI models (Hannay et al, 1999; Saatman et al, 2006; Smith et al, 1995). Third, the number of hippocampal PI-positive cells increased with increasing CCI depth, consistent with a direct effect of CCI injury severity on hippocampal cell death (Saatman et al, 2006). Fourth, PI labeling correlated strongly with cellular injury assessed by H&E staining. Fifth, a significant fraction of PI-positive cells developed DNA fragmentation, consistent with fatal injury. Finally, necrotic-like ultrastructural derangements (e.g., plasma and nuclear membrane damage and nuclear karyorrhexis) were present in virtually all pulse-labeled PI-positive cells analyzed at either 1 or 24 h after CCI. The ultrastructural changes observed in PI-positive cells were reported in pathological samples of contused human brain in patients with severe TBI (Castejon, 2004; Castejon and Arismendi, 2004).
Taken together, the data suggest that PI permeability may portend a fatal outcome in cells injured early after CCI. In contrast, previous studies using other fluorescent tracers in diffuse TBI models showed that some cells with early plasmalemma permeability were detectable in the brain for up to 8 h, and that many of these cells lacked ultrastructural changes and regained their plasmalemma integrity (Farkas et al, 2006; Singleton and Povlishock, 2004). These latter findings were interpreted as evidence that some permeable cells may be reversibly injured and survive TBI in an experimental model that lacks overt tissue damage. The differences between our findings (mainly evidence against reversible injury) and those of the aforementioned studies (reversible or fatal injury) may relate to differences in the pathogenesis of cell death between focal and diffuse TBI, as well as the later time points evaluated in this study.
Our study extends the 8-h time course of cellular injury reported previously in diffuse TBI to several days in CCI (Farkas et al, 2006). Previous investigators have shown progressive cell death and brain tissue loss for as long as 1 year after CCI in rats (Dixon et al, 1999); however, no published studies have determined the kinetics of injury and clearance of labeled cells in a CCI model. The prolonged clearance of PI-positive cells is somewhat surprising given the severity of cortical contusion injury and its associated microenvironment. The notion that necrotic-like cell death in contused brain (indicated by ultrastructural data in PI-positive cells) might take several days to complete runs counter to the widely held view that necrosis is a relatively rapid event in vivo (Festjens et al, 2006). However, the observations that protein synthesis is maintained during necrotic cell death (Saelens et al, 2005) and that necrosis (like apoptosis) is orchestrated by intrinsic cell death programs (Degterev et al, 2005; Festjens et al, 2006) support the possibility that protracted programmed necrosis might contribute to prolonged clearance of PI-positive cells after CCI.
Our data have important implications for neuroprotective strategies designed to rescue injured cells. The findings that many PI-positive cells resealed their plasma membranes and were TUNEL-negative as late as 24 h after CCI implies that an extended time window may exist to rescue this cohort of injured cells. However, protracted clearance of permeable cells does not mean that these cells are rescuable. Indeed, given the ultrastructural features of PI-positive cells, it is not clear that neuroprotective strategies administered after overt plasmalemma damage or loss can permit PI+ cells to survive TBI. Data from this study suggest the feasibility of examining this important question in the future.
Propidium iodide and TUNEL staining have been widely used to characterize modes of death in cultured cells based on early plasmalemma permeability in necrosis (PI+/TUNEL– or PI+/TUNEL+) and intact plasmalemma function early in apoptosis (PI–/TUNEL+). Recently, several investigators have extended this analysis to ischemic kidney and brain (Kelly et al, 2003; Unal Cevik and Dalkara, 2003). Our finding that all PI-positive cells at 1 h after CCI were TUNEL-negative, that few PI–/TUNEL+ cells were observed, and that most PI-positive cells examined had ultrastructural features of necrosis suggests that necrotic-like cell death predominates in CCI, similar to what has been reported in other TBI models (Colicos et al, 1996; Knoblach et al, 2004; Rink et al, 1995; Singleton and Povlishock, 2004). However, we cannot rule out the possibility that with a 6 mins/sec impact velocity, shearing forces may play a unique and important role in direct cellular disruption (Gallyas et al, 1992), making it difficult to extrapolate findings of cell permeability in other injury models to TBI. Our data provide a starting point for future mechanistic studies aimed at clarifying the relationship between loss of plasmalemma integrity and mode of cell death after TBI.
We conclude that, under the stated experimental conditions, PI permeability predicts severe injury and quite possibly death of cells injured early after CCI in mice. Further studies are needed to determine whether PI permeability portends a similar fate for cells that lose plasmalemma integrity at more delayed times after injury, and whether PI permeability is a marker for, or cause of, eventual cell death. If the latter holds true, membrane-resealing agents might be therapeutically useful to rescue injured brain cells in patients with TBI, as has been suggested in experimental models of spinal cord injury and other acute central nervous system injuries (Serbest et al, 2006).