
Abstract
Prostacyclin is the major arachidonic acid metabolite of the vascular endothelium and is produced mainly via the cyclooxygenase-2 pathway. By acting on the prostacyclin (IP) receptor on platelets and vascular smooth muscle cells, prostacyclin exerts vasodilatory and antiaggregative/antiadhesive effects. Previous studies have shown that prostacyclin production increases after brain trauma, but the importance of prostacyclin for posttraumatic hemodynamic alterations and neuron survival has not been investigated. This study evaluated if endogenous prostacyclin plays a role in the pathophysiologic process in the brain after brain trauma. This was performed by comparing prostacyclin (IP) receptor-deficient (IP−/−) mice and mice with functional IP receptor (IP+/+) after a controlled cortical injury regarding contusion volume, cerebral blood flow ([14C]iodoantipyrine autoradiography), number of perfused capillaries (fluorescein isothiocyanate-dextran fluorescence technique), the transfer constant (Ki) for [51Cr]EDTA, and brain water content (wet vs dry weight) in the injured and contralateral cortex. Contusion volume was increased in IP−/− mice compared with IP+/+ mice. Three hours after trauma, cortical blood flow was decreased in the injured cortex of both groups and the reduction in blood flow in the cortex of the IP−/− mice persisted from 3 to 24 h, whereas blood flow approached normal values in the IP+/+ mice after 24 h. No differences could be detected between the two genotypes regarding other hemodynamic parameters. We conclude that the prostacyclin IP receptor is beneficial for neuron survival after brain trauma in mice, an effect that may be mediated by improved cortical perfusion.
Introduction
Prostacyclin is the main product of the arachidonic acid metabolism in endothelial and smooth muscle cells. The synthesis of this labile (t1/2 = 2 to 3 mins) compound includes a cyclooxygenase (COX)-mediated conversion of arachidonic acid into cyclic endoperoxides from which prostacyclin is formed by the action of the enzyme prostacyclin synthase. Although both the COX-1 and COX-2 isoforms contribute to the synthesis of prostacyclin, recent evidence suggests that COX-2 is the main source of substrate for prostacyclin synthase in vascular tissue (McAdam et al, 1999). The main physiologic action of prostacyclin is suggested to be inhibition of aggregation of platelets and adhesion of leukocytes (Kobayashi et al, 2004; Moncada et al, 1976; Murata et al, 1997). Prostacyclin is a vasodilator, but its role in regulation of cerebral vascular resistance during normal conditions is unclear (McCalden et al, 1984). Prostacyclin also has permeability-reducing properties (Bentzer and Grände 2004; Erlansson et al, 1991). Prostacyclin exerts its vascular effects by activation of the IP receptor, which is located on the cell surface of platelets, endothelial cells, and vascular smooth muscle cells (Oida et al, 1995). The IP receptor is a G-protein-coupled receptor, which increases intracellular cAMP via adenylate cyclase, and is suggested to mediate the antithrombotic and vasodilatory actions of prostacyclin (Murata et al, 1997).
It is well known that brain trauma is associated with cerebral ischemia, most probably contributing to cell death several days after the primary insult (Graham et al, 1989; Schröder et al, 1995). The pathophysiology of posttraumatic hemodynamic alterations is complex and poorly understood. Most probably, it involves interplay of different inflammatory vasoactive agents, resulting in vasoconstriction, increased leukocyte adherence, opening of the blood—brain barrier, and increased aggregation of platelets, with ensuing occlusion of microvessels (Hekmatpanah and Hekmatpanah, 1985; Lu et al, 2004; Lundblad et al, 2004). It is suggested from experimental and clinical studies that prostacyclin production may increase after brain trauma (Shohami et al, 1987; Westcott et al, 1987), possibly because of increased expression of COX-2 (Dash et al, 2000). The importance of such an increase in prostacyclin synthesis for posttraumatic hemodynamics and cell death has not been investigated. It has been suggested that, even if synthesis of prostacyclin is increased, imbalance between prostacyclin and vasoconstricting and proaggregatory agents such as thromboxane A2 may contribute to decreases in blood flow during the first hours after brain trauma (Shohami et al, 1987). A beneficial role for prostacyclin in brain trauma is suggested by studies showing reduction in contusion volume and increased cortical blood flow after prostacyclin infusion in rat (Bentzer et al, 2001, 2003). A microdialysis study in man has also indicated that prostacyclin infusion in doses close to the endogenous production improves oxygenation and reduces cell damage in the traumatized brain (Grände et al, 2000). Even though the above-mentioned studies have indicated that prostacyclin is beneficial after brain trauma, there are also studies suggesting that prostacyclin might have adverse effects by showing improved cognitive outcome after brain trauma by COX-2 inhibition (Cernak et al, 2001; Gopez et al, 2005). It has also been suggested that prostacyclin by its vasodilating properties may play a role in edema formation following inflammation in peripheral tissue, effects that may be detrimental after brain trauma (Murata et al, 1997).
The objective of this study was to investigate the significance of endogenous prostacyclin for cell death and hemodynamic alterations following a controlled cortical impact injury in mice. For this purpose, changes in cortical lesion volume, cortical blood flow, number of perfused capillaries, blood to brain transfer constant (Ki) for [51Cr]EDTA, and brain water content (BWC) were analyzed. The study was performed in mice lacking the IP receptor for prostacyclin and their littermates with intact IP receptor.
Materials and methods
Animals
The experimental protocol was approved by the local Ethical Committee for Animal Research (M268-03). Adult male mice (weight 27 to 35 g) lacking the gene for the prostacyclin receptor IP (IP−/−), and their littermates (weight 27 to 35 g) with the intact gene for the IP receptor (IP+/+) were used. In IP−/− mice, the gene encoding the prostacyclin receptor is disrupted by targeted disruption as described previously, and chimeric animals are then back bred into the C57BL/6 background (Murata et al, 1997). IP−/− mice have been reported to have increased prenatal mortality (Murata et al, 1997). After birth, IP−/−mice grow normally, are fertile, and live longer than 1 year. No histologic or morphologic abnormalities have been reported. Both IP−/− and IP+/+ mice have normal production of prostacyclin, but IP−/− mice cannot respond to prostacyclin (Murata et al, 1997). To classify the breeding pairs genetically (gift from Ono Pharmaceutical Co. Ltd., Osaka, Japan), a genotype protocol was used. Briefly, PCR primers CY-37 (5′-GTATCTTTCAGTACCTGAGGACTG-3′), CY-41 (5′-GAGCAGAAAAATTCCCAGAGGCTT-3′), and Neo17 (5′-TGACCGCTTCCTCGTTTAC-3′) (Cybergene AB, Huddinge, Sweden) together with extracted DNA were used in the PCR reaction. The PCR product was then applied to an agarose gel (0.8%). Bands visualized at 1.3 kb correspond to primer pair CY-37/CY-41 and were classified as the wild allele, IP+/+. Bands visualized at 0.9 kb correspond to primer pair CY-37/Neo17 and were classified as the disrupted allele, IP−/−. Two animals from each genotype were also challenged by prostacyclin infusion to ensure lack of blood pressure response in IP−/− mice (Figure 1). As an additional control of the impact on the contusion development of the IP receptor, a group of wild-type C57BL/6 mice (Taconic Europe, Lille Skensved, Denmark) was evaluated histologically. The animals were treated in accordance with the Guide for the Care and Use of Laboratory Animals (publications DFS 2004,4: 15, Swedish Animal Welfare Agency).
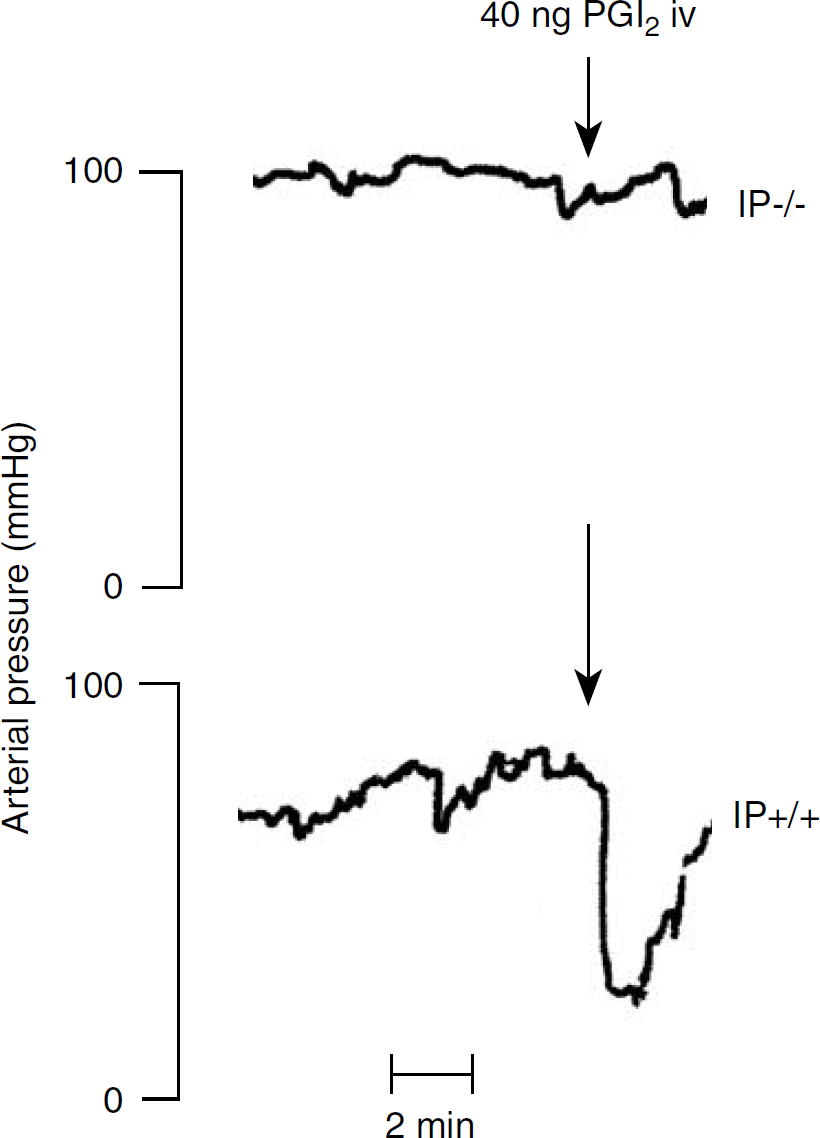
Original recordings showing effect of an intravenous bolus dose of prostacyclin (40 ng) in IP receptor-deficient (IP−/−) mice and in littermates with functional IP receptor (IP+/+).
Controlled Cortical Impact Injury
Sodium pentobarbital (50 mg/kg) and ketamine (50 mg/kg) both administered intraperitoneally were used for anesthesia. The anesthetized animals were placed in a head holder on a heating pad, keeping body core temperature at 37.0°C via a feedback circuit controlled by continuous measurement of rectal temperature. A parasagittal craniotomy of 5 mm in diameter was performed in the left parietal bone between the lambdoid and coronal sutures. Animals with intact dura were placed in a pneumatic injury device and subjected to a controlled cortical impact injury, as described previously (Smith et al, 1995). The discharging gas pressure applied to the piston cylinder was adjusted to 410 kPa giving an impact velocity of approximately 4.3 m/sec. A deformation depth was set at 0.8 mm and the duration was 200 ms. The animals recovered and woke up in an incubator at 33°C to maintain a body coretemperature of 37°C. The controlled cortical impact model has been shown to produce standardized and reproducible brain trauma in mice with loss of both cortical and hippocampal neurons, breakdown of the blood—brain barrier, and cognitive deficiencies (Smith et al, 1995).
Surgical Preparation
Anesthesia was reinduced 2 or 23 h after trauma with isoflurane with an induction concentration of 4% in air and maintained at 1.8% throughout the experiment. The animals were tracheostomized and mechanically ventilated with air by a Mouse ventilator 28025 (Ugo Basile Biological Research apparatus, Comerio-Varese, Italy) using a tidal volume of 0.30 to 0.35 mL and a respiratory rate of 95 to 100/min−1. A positive-end expiratory pressure of 5 mm Hg was applied to prevent pulmonary atelectasis. A bolus dose of pancuronium bromide (0.8 mg/kg intravenously) was administered after cannulation of the external jugular vein to obtain pliability with the ventilator during preparation. All animals received an infusion of isotonic saline (5 μL/min) during the preparation to avoid hypovolemia. The femoral artery was cannulated (PE-10, 30 mm in length) for measurement of PaCO2, PaO2, pH, hematocrit (Hct) (i-STAT, Abbot Scandinavia AB, Stockholm, Sweden) and for blood pressure measurement. Sham animals underwent the same surgical procedures described above, but were not subjected to the craniotomy and trauma.
Measurement of Cortical Blood Flow
[14C]Iodoantipyrine was used as tracer for measurement of cerebral blood flow after controlled cortical impact with autoradiography. Briefly, the animals received an infusion of 0.35 MBq of [14C]iodoantipyrine (Amersham Pharmacia Biotech, Uppsala, Sweden) dissolved in 100 μL isotonic saline with fluorescein isothiocyanate-dextran. Six arterial 10 μL blood samples were collected from the femoral artery during the 45-sec period of tracer infusion. The animals were decapitated simultaneously with the last blood sample, and the brain was removed and immediately frozen in 2-methyl butane (–50°C). Coronal sections from the brain, 20 μm in thickness, were produced in a cryostat at −20°C and dried for 2 h at 50°C. After drying, sections from −2 and −1 mm relative to the bregma (Figure 1) were exposed to an X-ray film (Hyperfilm β-Max, Amersham Pharmacia Biotech) for 2 to 3 days together with a set of calibrated standards. The film was digitized after development and analyzed using a public domain NIH image-processing program. The blood samples collected during the tracer infusion were dissolved in Solvable (Packard, Groningen, the Netherlands) and incubated at 60°C for 1 h. The samples were then bleached with 30% hydrogen peroxide for an additional 1 h. Scintillation fluid was added and the [14C] activity was measured in a liquid scintillation counter. Blood flow was then calculated according to the following equation (Sakurada et al, 1978):
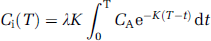
where Ci (T) equals the tissue concentration of a chemically inert diffusible tracer at a given time T after start of the tracer infusion, λ is the tissue/blood partition coefficient, CA the concentration of the tracer in arterial blood, t is time, and K a constant, which incorporates the rate of blood flow in the tissue. The constant K is defined as K = (m/λ)(F/W), where m is a constant that represents the extent to which a diffusion equilibrium between blood and tissue during passage from the arterial to the venous end of a capillary, and F/W is blood flow per unit mass. For iodoantipyrine, λ = 0.8 and m = 1. Blood flow was measured at the two levels −1 and −2 mm relative to the bregma in both the injured cortex and the corresponding area in the contralateral cortex (Figure 2) and is expressed as the mean flow of the two levels.
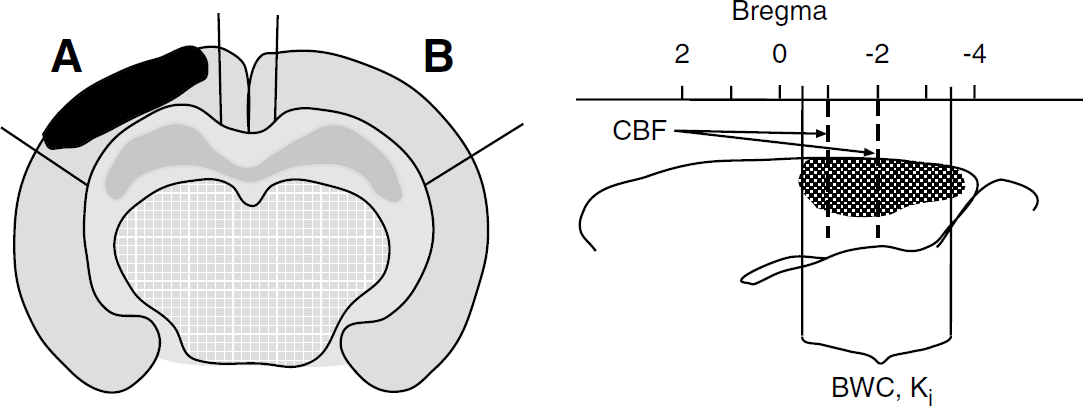
(Left) Schematic drawing of a coronal section of the brain after a brain trauma showing areas for measurement of cortical blood flow and number of perfused capillaries; (
Number of Perfused Capillaries
To investigate the number of perfused capillaries, fluorescein isothiocyanate-dextran (100 mg/kg, 2×106 Da molecular weight; Sigma-Aldrich, Stockholm, Sweden) was administered simultaneously with the iodoantipyrine infusion in some animals. The protocol described by Weiss (1988) was used with minor modifications. In brief, sections adjacent to those chosen for cortical blood flow measurements were collected and the glasses were stored at −20°C until analyzed. The capillary network was observed with an argon laser (488 nm) in a scanning confocal microscope (×10 objective, Zeiss LSM 510;Zeiss, Göttingen, Germany). The capillaries observed to contain fluorescein isothiocyanate-dextran were considered as perfused. Three images from each of the injured and contralateral areas, at both levels −1 and −2 mm relative to the bregma, were collected. The number of perfused capillaries in each image was counted by an image-processing program (SigmaScan Pro 5, Systat Software, Germany). To exclude effects of extravasation, only clearly visible vessels more than 5 μm and less than 12 μm in diameter were counted. Number of perfused capillaries is expressed as the mean of the two levels.
Brain Water Content and the Transfer Constant (Ki) for [51Cr]EDTA
Brain water content and the transfer constant (Ki) for [51Cr]EDTA in the ipsilateral and contralateral cortices were analyzed in a separate series of animals. During preparation, the animals received an intravenous infusion of dextran 70 at a rate of 10 μL/min (Macrodex® 60 mg/mL; Pharmalink, Stockholm, Sweden) to counteract hypovolemia and blood pressure reduction during the subsequent blood sampling (Lundblad et al, 2004). The animals received a bolus infusion of approximately 200 kBq of the tracer [51Cr]EDTA (0.05 mL, 3.7 MBq/mL) (Nycomed Amersham, Stockholm, Sweden), followed by a continuous infusion at a rate of 0.33 mL/h (3.7 MBq/mL). Blood samples (10 μL) were collected at 2.5, 5, 10, 15, 25, 35, and 40 mins after start of the injection. At 37 mins after start of the [51Cr]EDTA infusion, a bolus dose of 25 kBq [125I]albumin, purified from free iodine and dissolved in 0.05 mL of isotonic saline, was administered in the external jugular vein. Albumin was assumed to fully remain within the circulation, and was used for calculation of the plasma volume in the brain. A correction factor of 1.06 was used to convert whole plasma concentration of [51Cr]EDTA into plasma water concentration (Waniewski et al, 1992). Arterial Hct was measured at the start of the tracer infusion and immediately before decapitation 40 mins later, and the mean Hct value was used to convert blood concentrations into whole plasma concentrations. The brain was removed and put on a chilled support, a 3-mm-thick coronal slice (approximately −3.5 to −0.5 mm relative to the bregma, Figure 2) was carefully removed from the brain, and the ipsi- and contralateral cortices were immediately weighed. Tissue and blood tracer activities were counted in a γ-counter. The tissue samples were then dried for 48 h at 80°C, and water content was calculated using the following formula: [(wet weight—dry weight)/(wet weight) × 100].
Ki for [51Cr]EDTA was calculated according to the following equation (Fenstermacher et al, 1981):
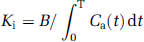
where Ca is concentration of the tracer in arterial plasma as a function of time, T the duration of the experiment, and B the amount of tracer moved from blood to brain, that is tissue uptake of tracer minus regional tracer concentration in plasma. Ki is a function of capillary plasma flow per unit mass of tissue (FV) and the permeability surface area product (PS), the latter reflecting microvascular permeability and surface area available for diffusion exchange. The mathematical expression for this relationship is Ki = FV[1–exp(–PS/FV)] (Fenstermacher et al, 1981). From this expression, it can be concluded that the Ki value approximates PS with an error of less than 6%, when the Ki/FV ratio is less than 0.1.
Lesion Volume
Seven days after trauma, animals were anesthetized with isoflurane and transcardially perfused with isotonic saline followed by phosphate-buffered 4% paraformaldehyde (3 mL/min). The brain was removed and postfixated for 24 h at 8°C and was then dehydrated in stepwise ethanol solutions and embedded in paraffin. Serial coronal sections of 6 μm were taken at 3.8, 3.2, 2.5, and 1.8 mm posterior to the bregma. The sections were mounted (superfrost glasses; Menzel-Gläser, Braunschweig, Germany) and stained with hematoxylin and eosin for evaluation of lesion volume. To evaluate the lesion volume, the brain sections were analyzed in stereologic microscope equipment with an image-analyzing software CAST 2 (Olympus Danmark A/S, Ballerup, Denmark). The demarcation line between the lesioned and the healthy tissue was drawn so that the lesion area contained few or no viable neurons, as judged by cell and nuclear morphology and cytoplasm staining. Lesion volume for each section was calculated as lesion area × thickness of the section (half the distance between the adjacent anterior and posterior sections, for most posterior and anterior sections 2 × half the distance to the adjacent section). Lesion volume was expressed as percentage of total cortical volume and was calculated using the following equation [(contralateral cortex—non-damaged ipsilateral cortex)/(contralateral cortex + non-damaged ipsilateral cortex) × 100]. By comparing adjacent sections stained with hematoxylin and eosin and fluoro-jade, which selectively stains degenerating neurons, we have previously confirmed that this method is a reliable tool to estimate lesion volume (Bentzer et al, 2001).
Experimental Protocol
The animals were traumatized and analyzed blindly regarding genotype. The study aimed at analyzing the following parameters:
Histologic evaluation of the contusion volume 7 days after trauma in the IP−/−, IP+/+ (littermates), and in C57BL/6 mice (wild type). Cortical blood flow and number of perfused capillaries in the injured and contralateral cortex 3 and 24 h after trauma in IP−/− and IP+/+ mice. Brain water content and Ki for [51Cr]EDTA in the injured and contralateral cortex 3 and 24 h after trauma in IP−/− and IP+/+ mice. On a post hoc basis, cortical blood flow was determined in sham animals to determine whether any baseline differences between IP−/− and IP+/+ mice were present.
Statistical Analysis
After tests for equal variance and normal distribution, physiologic data were analyzed with Student's t-test, and histologic data were analyzed with one-way analysis of variance (ANOVA) followed by the Fisher's least significant difference post hoc test. Hemodynamic data in the injured and uninjured cortex, respectively, were analyzed with two-way ANOVA, with genotype and time as explanatory variables. Hemodynamic data at the different time points were analyzed by two-way ANOVA, with location and genotype as explanatory variables. Sigma Stat 3.0 software was used for the statistical analysis. Values are expressed as mean ± s.d.
Results
Physiologic Data
Data for mean arterial pressure, Hct, pH, PaO2, and PaCO2 are given in Tables 1 and 2. Table 1 shows the physiologic data from blood samples in the cerebral blood flow and number of perfused capillary groups taken before the start of the experiments. Table 2 shows physiologic data in the Ki and edema groups presented as mean values of data from the two samples taken before the start of the experiment and at the end of the experiment. There was a higher Hct in the IP−/− compared with the IP+/+ animals in the Ki group at 24 h (P < 0.05). Physiologic data for the sham groups were similar to the other groups and did not differ between groups (data not shown).
Physiologic data for CBF animals
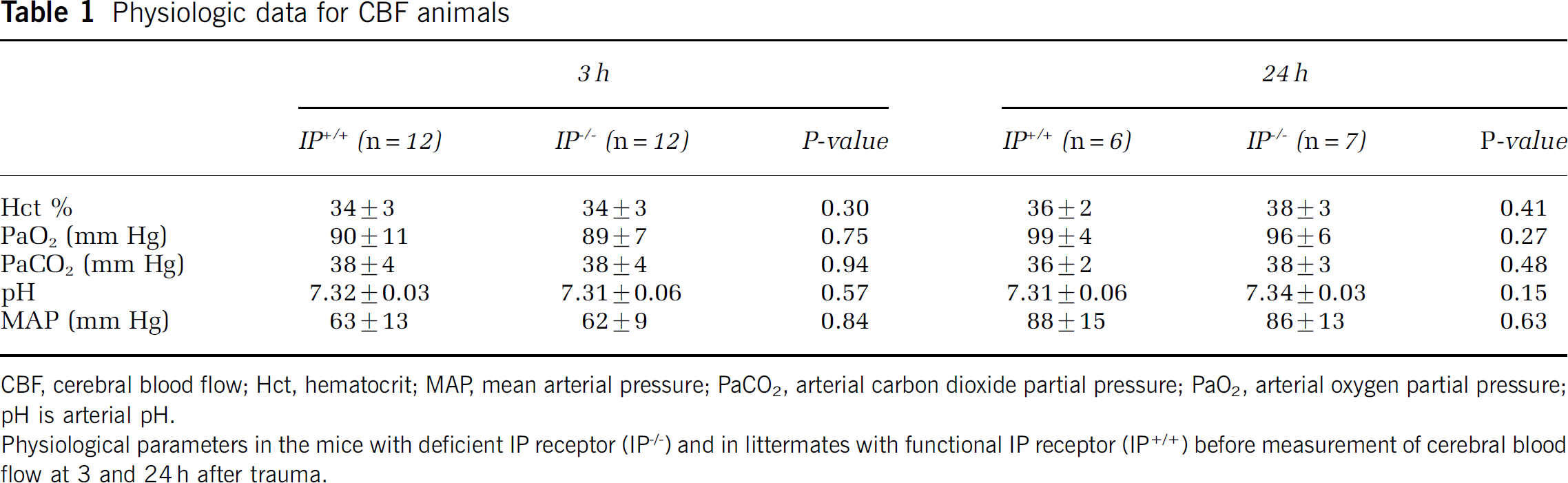
CBF, cerebral blood flow; Hct, hematocrit; MAP, mean arterial pressure; PaCO2, arterial carbon dioxide partial pressure; PaO2, arterial oxygen partial pressure;
pH is arterial pH.
Physiological parameters in the mice with deficient IP receptor (IP−/−) and in littermates with functional IP receptor (IP+/+) before measurement of cerebral blood
flow at 3 and 24 h after trauma.
Physiologic data for the Ki and brain water content animals
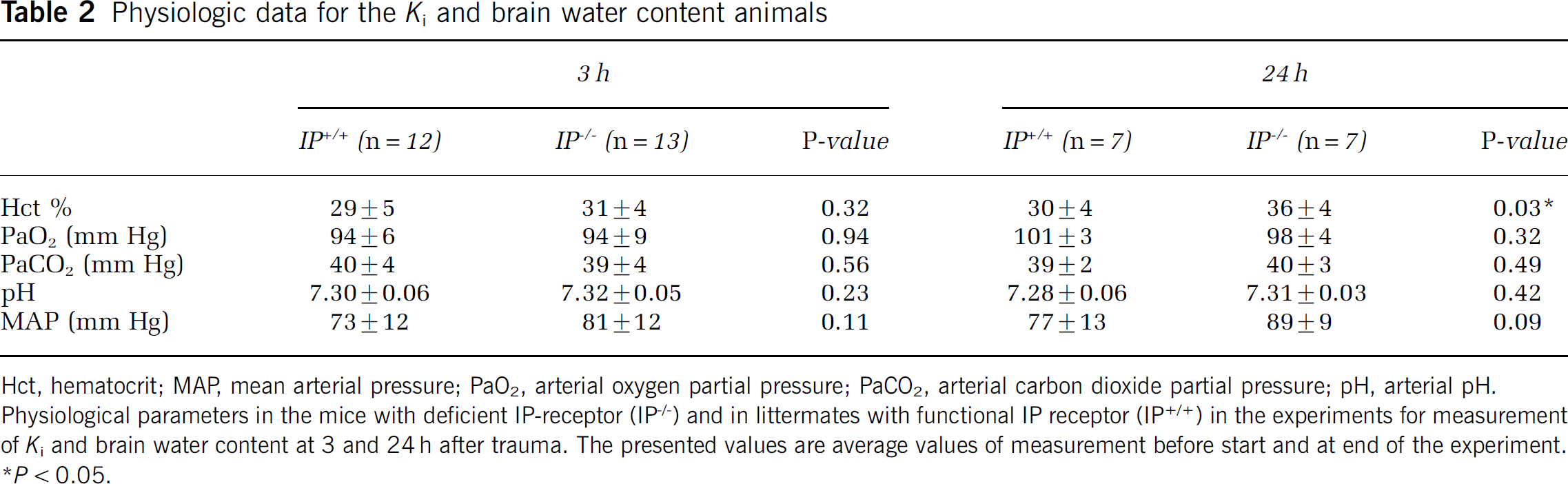
Hct, hematocrit; MAP, mean arterial pressure; PaO2, arterial oxygen partial pressure; PaCO2, arterial carbon dioxide partial pressure; pH, arterial pH.
Physiological parameters in the mice with deficient IP-receptor (IP−/−) and in littermates with functional IP receptor (IP+/+) in the experiments for measurement of Ki and brain water content at 3 and 24 h after trauma. The presented values are average values of measurement before start and at end of the experiment.
P < 0.05.
Cerebral Blood Flow
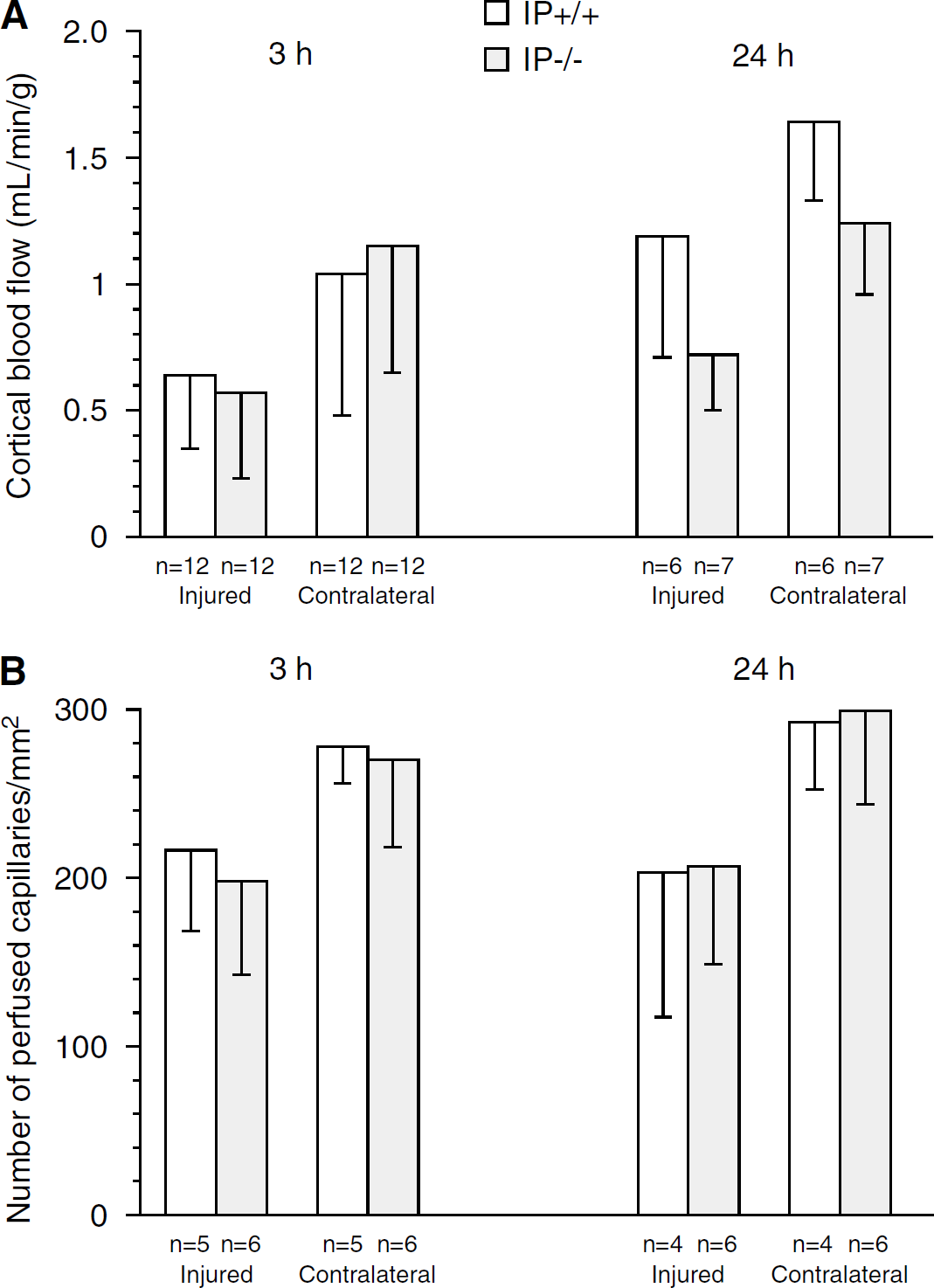
Three hours after trauma, blood flow in the injured cortex was 0.64 ± 0.29 mL/min per g in the IP+/+ animals (n = 12) and 0.57 ± 0.34 mL/min per g in the IP−/− animals (n = 12). Blood flow in the contralateral cortex was 1.04 ± 0.56 mL/min per g in the IP+/+ animals (n = 12) and 1.15 ± 0.50 mL/min g in the IP−/− animals (n = 12, not significant (NS), Figure 3A). There was a significant effect of location on blood flow (location effect, P = 0.000; genotype effect, P = 0.86; genotype × location interaction, P = 0.47).
(
Twenty-four hours after trauma, blood flow in the injured cortex was 0.72 ± 0.23mL/min per g in the IP−/− animals (n = 7) and 1.19 ± 0.48 mL/min per g in the IP+/+ animals (n = 6, P < 0.05). Blood flow in the contralateral cortex was 1.24 ± 0.28 mL/min per g in the IP−/− animals (n = 7) and 1.64 ± 0.30 mL/min per g in the IP+/+ animals (n = 6). One animal in the IP+/+ group was excluded from the study before analysis due to inability to maintain a blood pressure above 50 mm Hg. Twenty-four hours after trauma, a significant effect of both genotype and location on blood flow could be demonstrated (genotype, P = 0.003; location, P = 0.000; genotype × location interaction, P = 0.8). The lack of interaction between genotype and location suggest that the effect of genotype on blood flow is independent of location. Two-way ANOVA with time and genotype as explanatory variables showed a significant effect of both genotype and time on blood flow in the injured cortex (genotype, P = 0.028; time, P = 0.005; genotype × time interaction, P = 0.099). In the uninjured cortex, only time significantly influenced blood flow (genotype, P = 0.392; time P = 0.039; genotype × time interaction, P = 0.12).
Cortical blood flow was determined in both hemispheres of sham animals to determine if the observed difference in blood flow could be attributed to differences in baseline blood flow. Cortical blood flow in PP−/− animals was 1.97 ± 0.48 and 1.91 ± 0.54 mL/min per g in the ipsilateral and contralateral cortices, respectively (n = 7). Cortical blood flow in IP+/+ animals was 1.94 ± 0.74 mL/min per g and 1.92 ± 0.72 mL/min per g in the ipsilateral and contralateral cortices, respectively (n = 6). No genotype or location effects could be demonstrated.
Number of Perfused Capillaries
Three hours after trauma, number of perfused capillaries in the injured cortex was 217 ± 48/mm−2 in the IP+/+ animals (n = 5) and 198 ± 56/mm−2 in the PP−/− animals (n = 6, NS). Number of perfused capillaries in the contralateral cortex in the injured cortex was 278 ± 22/mm−2 in the IP+/+ animals and 270 ± 52/mm−2 in the IP−/− animals (Figure 3B). A significant effect of location on number of perfused capillaries could be demonstrated (genotype effect, P = 0.52; location effect, P = 0.004; genotype × location interaction, P = 0.80).
Twenty-four hours after trauma, number of perfused capillaries in the injured cortex was 203 ± 86/mm−2 in the IP+/+ animals (n = 4) and 207 ± 58/mm−2 in the IP−/− animals (n = 6). Number of perfused capillaries in the contralateral cortex was 293 ± 40/mm−2 in the IP+/+ animals and 299 ± 55/mm2 in the IP−/− animals (Figure 3B). A significant effect of location could be demonstrated also at this time point (genotype effect, P = 0.85; location effect, P = 0.005; genotype × location effect, P = 0.96). No effects of time or genotype were found when performing two-way ANOVA with genotype and time as the explanatory variables in each of the two locations.
Ki and Brain Water Content
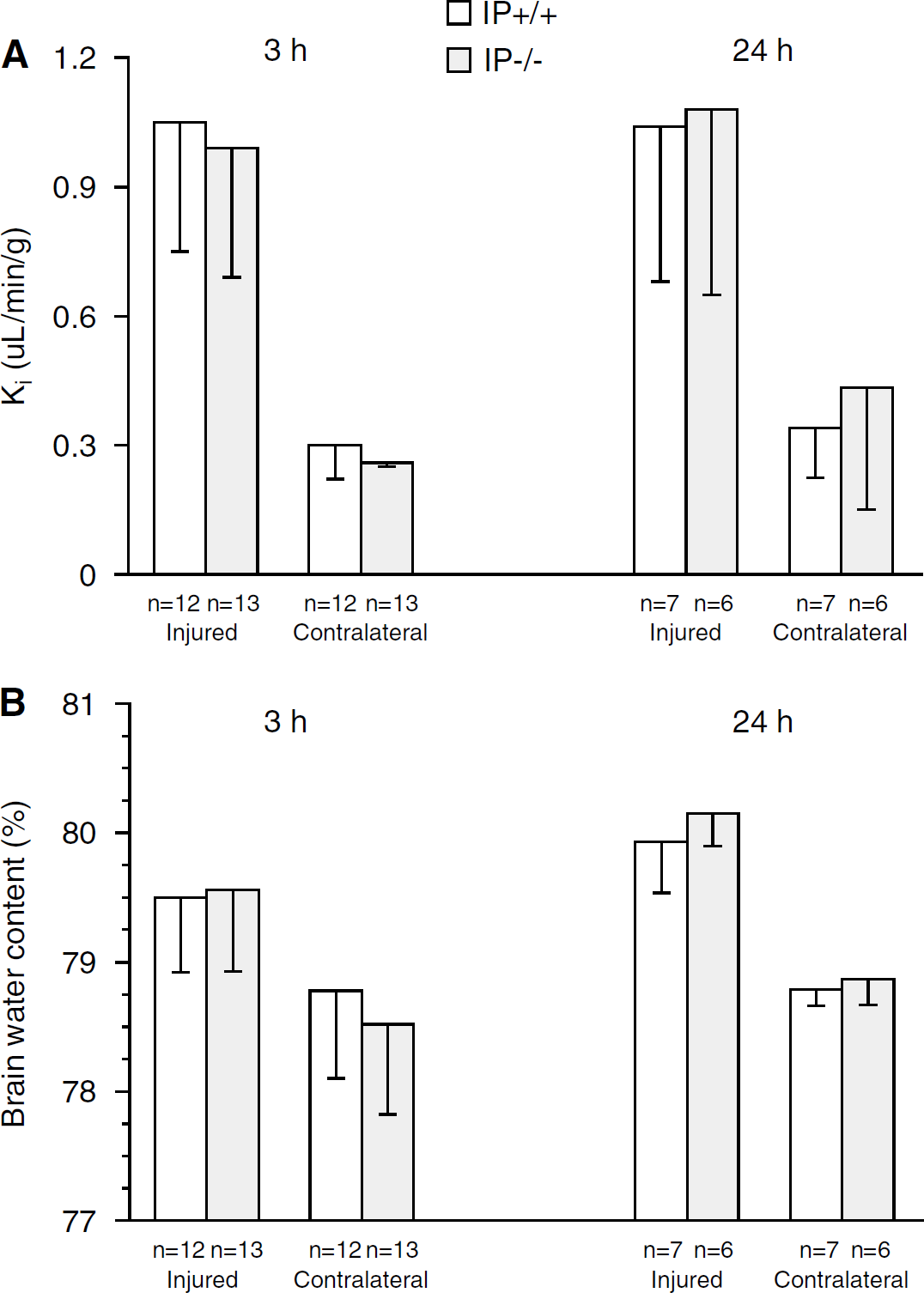
Three hours after trauma, Ki was 1.05 ± 0. 32 μL/min per g in the injured cortex of the IP+/+ animals (n = 12) and 0.99 ± 0.31 μL/min per g in the IP−/− animals (n = 13). In the contralateral cortex, Ki was lower than in the injured cortex and was 0.30 ± 0.1 μL/min per g in the IP+/+ animals (n = 12) and 0.26 ± 0.1 μL/min per g in the IP−/− animals (n = 13) (genotype effect, P = 0.22; location effect, P = 0.000; genotype × location interaction, P = 0.61). Twenty-four hours after trauma, Ki was 1.04 ± 0.36 μL/min per g in the injured cortex of the IP+/+ animals (n = 7) and 1.08 ± 0.43 μL/min per g in the IP−/−animals (n = 6). In the contralateral cortex, Ki was lower than in the injured cortex and was 0.34 ± 0.12 μL/min per g in the IP+/+ animals and 0.43 ± 0.28 μL/min per g in the IP−/− animals (Figure 4A) (genotype effect, P = 0.524; location effect, P = 0.000; genotype × location interaction, P = 0.69). No effects of time or genotype were found when performing two-way ANOVA with genotype and time as the explanatory variables in each of the two locations.
(
Three hours after trauma, BWC was 79.5 ± 0.6% in the injured cortex in the IP+/+ animals (n = 12) and 79.6 ± 0.6% in the IP−/− animals (n = 13, NS). Brain water content in the contralateral cortex was lower than in the injured cortex and was 78.8 ± 0.5% in the IP+/+ animals and 78.6 ± 0.7% in the IP−/−animals (genotype effect, P = 0.58; location effect, P = 0.000; genotype × location interaction, P = 0.41). Twenty-four hours after trauma, BWC in the injured cortex was 79.9 ± 0.4% in the IP+/+ (n = 7) and 80.2 ± 0.6% in the IP−/− animals (n = 6, NS). Brain water content in the contralateral cortex was lower than in the injured cortex and was 78.8 ± 0.3% in the IP+/+ animals and 78.9 ± 0.5% in the IP−/− animals (Figure 4B) (genotype effect, P = 0.38; location effect, P = 0.000; genotype × location interaction, P = 0.66). In the injured cortex, BWC increased from 3 to 24 h (genotype effect, P = 0.47; time effect, P = 0.016; genotype × time interaction, P = 0.64). No effects of time or genotype were found when performing two-way ANOVA with genotype and time as the explanatory variables in each of the two locations.
Lesion Volume
At 7 days after trauma, a lesion could be observed in the posterior-lateral ipsilateral cortex. The lesion was well demarcated, contained no or very few viable neurons and was densely infiltrated with leukocytes. No histologic abnormalities were noted in the IP−/− animals compared with IP+/+ (littermates) and wild-type C57BL/6 animals. Lesion volume was 11.2 ± 4.2% in the IP−/− animals (n = 11) and 6.8 ± 3.2% in the IP+/+ animals (n = 6, P < 0.05; Figure 5). Lesion volume in C57BL/6 animals was very similar to the IP+/+ animals at 7.6 ± 3.4% (n = 10).
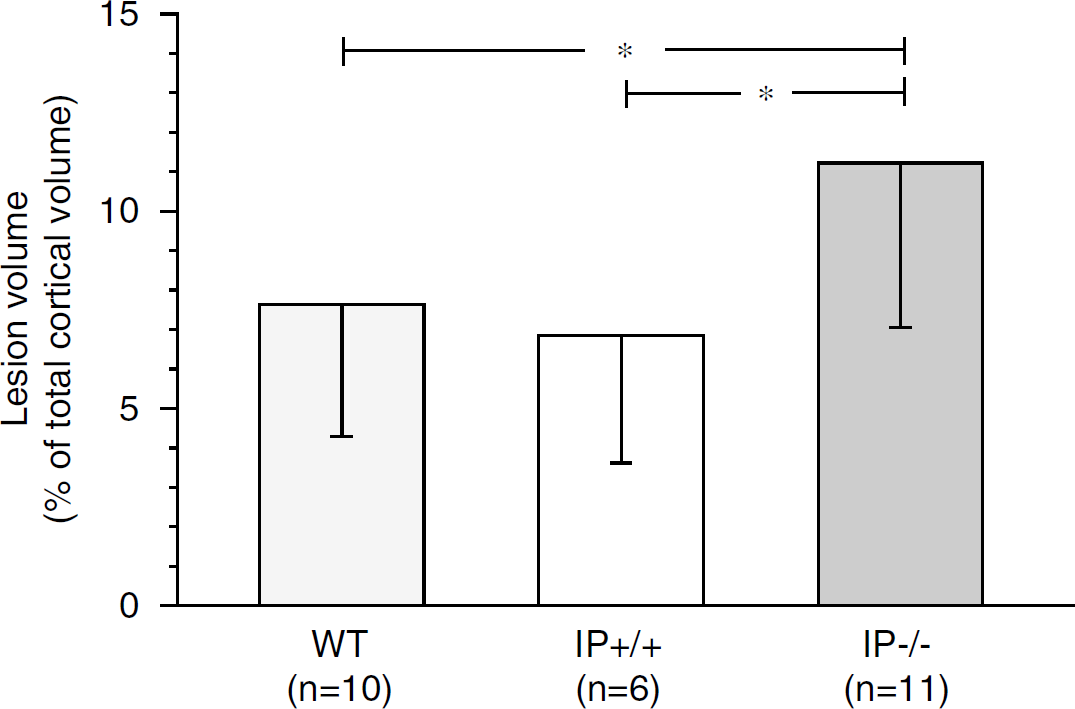
Lesion volume, expressed as % of total cortical volume 7 days after trauma in IP receptor-deficient (IP−/−) mice, in littermates with functional IP receptor (IP+/+) and in C57BL/6 mice (WT)(*P < 0.05).
Discussion
The present results showed that IP−/− mice develop a larger cortical lesion after brain trauma than littermates and wild-type mice. This finding was associated with the observation that posttraumatic reduction in blood flow in the injured and contralateral cortex of the IP−/− mice persisted from 3 to 24 h, whereas blood flow in the IP+/+ mice increased from 3 to 24 h and approached normal values 24 h after trauma. No genotype effects could be detected regarding number of perfused capillaries, brain edema, or Ki.
Previous studies have shown that IP−/− mice have normal blood pressure and bleeding time, and no histologic or morphologic abnormalities have been described (Murata et al, 1997). However, IP−/− mice have been shown to display an increased response to a variety of vascular injuries and are also prone to development of atherosclerosis in genetically predisposed animals, such as apoE-deficient mice (Egan et al, 2004; Kobayashi et al, 2004). The results in this study confirm that IP−/− mice do not differ from littermates with regard to blood pressure and blood gases. In addition, our results suggest that baseline cortical blood flow in IP−/− mice do not differ from that of littermates.
Our result of an increased contusion volume in the IP−/− mice compared with littermates and wild-type mice suggests that the endogenous prostacyclin production in the injured cortex is of importance for neuronal cell survival after traumatic brain injury. This result is compatible with the results in a previous study from our group showing reduced cortical cell loss after infusion of a low dose of prostacyclin in brain-injured rats (Bentzer et al, 2001). Our observation that the posttraumatic reduction in blood flow both in the injured and in the contralateral cortex of the IP−/− mice persisted from 3 to 24 h, whereas blood flow in the IP+/+ mice increased from 3 to 24 h and approached normal values at 24 h after trauma suggest that prostacyclin is important for recovery of the reduced cortical blood flow occurring within the first 24 h after brain trauma. Also previous studies have showed that, early after trauma, mild transient reductions in blood flow can be detected in areas remote from the impact (Marklund et al, 2002, Yamakami and McIntosh, 1991). Our result of a persistent reduction in blood flow in the contralateral cortex of the IP−/−mice suggests that prostacyclin is important for posttraumatic normalization of blood flow also in regions where no cell death occurs. The blood flow alterations observed in IP−/− mice are in line with a previous study showing that the posttraumatic increase in prostacyclin production is limited early after trauma, but subsequently increases and peaks approximately 24 h after trauma both in the injured and contralateral cortex (Shohami et al, 1987).
It has been suggested that in rat, short-term reductions in cerebral blood flow to levels below 0.18 mL/min per g cause energy failure and cell death, and blood flow below 0.55 mL/min per g inhibit protein synthesis (Mies et al, 1991). If blood flow reductions persist for a longer period of time, the threshold for energy failure approaches that of protein synthesis. Assuming that these thresholds apply also to mice, it could be argued that the blood flow in injured cortex is above the threshold for ischemic cell death in both groups of animals, both 3 and 24 h after trauma. However, blood flow within the injured cortex is heterogenous, and in subregions below the threshold for protein synthesis (Lundblad et al, 2004), suggesting that ischemia may play a part in posttraumatic cell death in this model. In IP−/− mice, the posttraumatic decrease in cortical blood flow persisted at a level close to the threshold for protein synthesis from 3 to 24 h after trauma, whereas in the IP+/+ mice blood flow approaches normal levels at 24 h. It is therefore possible that blood flow in parts of the injured cortex of the IP−/− animals could have been below the threshold for ischemic injury, which might have contributed to the observed difference in cortical lesion volume between the genotypes.
As mentioned, progressive microvascular thrombosis has been discussed as a possible mechanism behind blood flow reductions after trauma (Lu et al, 2004) and may explain the observed reductions in the number of perfused capillaries in the injured cortex after brain trauma (Lundblad et al, 2004). On the basis of previous observation that IP−/− mice have an increased susceptibility to thrombosis after vascular injury (Murata et al, 1997), we hypothesized that IP−/− mice would display a decreased number of perfused capillaries. Our finding that IP−/−mice and their littermates have a similar number of perfused capillaries indicate that IP receptor activation by endogenous prostacyclin does not influence the number of occlusive microvascular thrombi or the size of the microvascular network available for exchange of oxygen and other nutrients after brain trauma. A tentative explanation to this result is the recent observation that the ability of prostacyclin to counteract platelet aggregation is dependent on the type of stimulus to which the platelet is exposed, with a preference for thromboxane A2 (Manganello et al, 1999). If so, it could be speculated that the major stimulus for platelet aggregation after trauma could be relatively insensitive to IP receptor stimulation. However, it should be noted that our method does not detect the effect of non-occlusive thrombi and cell aggregates and we cannot exclude that prostacyclin, to a minor extent, yet may have improved microcirculation via effects on coagulation and cell aggregation.
Our result of a persistent reduction in cortical blood flow in IP−/− mice, which is not accompanied by a decrease in number of perfused capillaries, indicate that prostacyclin may increase blood flow by mechanisms unrelated to its effects on intravascular coagulation. Instead, it is likely that the vasodilatory action of IP receptor activation by prostacyclin is involved in the recovery of blood flow after a brain trauma. The observed reduction in blood flow in IP−/− mice may, however, have an additional explanation. It was recently shown that IP−/− mice display an increased proliferative response and decreased luminal area weeks after vascular injury (Cheng et al, 2002; Rudic et al, 2005), and we cannot exclude that the decreased blood flow 24 h after trauma may, in part, be explained by such an effect.
Changes in Ki reflect the net effect of changes in permeability surface area product (PS), provided that plasma flow is highly relative to Ki (see Materials and methods). It can be calculated from the present results with a lowest blood flow of approximately 0.14 mL/min per g, hematocrit of approximately 30%, and a highest Ki value of 1.6 μL/min per g in individual experiments that Ki approximates PS with an error of less than 6% in all situations analyzed. Ki thus reflects the net effect of changes in microvascular permeability and/or changes in surface area available for diffusional exchange, the latter being due to alterations in the number of perfused capillaries. As number of perfused capillaries is reduced after brain trauma (Lundblad et al, 2004), the increase in Ki in the injured cortex compared with the contralateral cortex after trauma in both groups must reflect an increase in blood—brain barrier permeability rather than an increase in surface area. The present observation of no difference in edema, Ki, or number of perfused capillaries between the IP−/− mice and littermates suggests that the effect of IP receptor activation by endogenous prostacyclin on the blood—brain barrier is slight, and its contribution to edema formation after brain trauma, if present, must be small.
The observed neuroprotective effect of prostacyclin may not only be due to hemodynamic effects. Recently, it was shown that IP−/− mice were more susceptible to ischemia—reperfusion injury in the heart (Xiao et al, 2001). Interestingly, this effect was suggested to be independent of vascular tone and was also present during cell-free perfusion, indicating a cytoprotective effect of prostacyclin unrelated to cell components in plasma. It was suggested that the cytoprotective effect was mediated via activation of ATP-dependent potassium channels. Such a mechanism could be operational also in the brain, as activation of ATP-dependent potassium channels may be neuroprotective after brain ischemia (Liu et al, 2002).
As mentioned in the introduction, the main source of substrate for prostacyclin synthase is the COX-2 enzyme in vascular tissue. There are conflicting reports in the literature with regard to the effect of selective COX-2 inhibition on outcome after traumatic brain injury. Some studies suggest a beneficial effect of COX-2 inhibition on functional outcome (Cernak et al, 2001; Gopez et al, 2005), whereas others suggest no or even adverse effects (Dash et al, 2000; Kunz et al, 2006). The results of this study indicate that, if COX-2 inhibition is neuroprotective, such an effect is mediated via mechanisms other than inhibition of prostacyclin synthesis, and that COX-2 inhibition is a potentially harmful intervention following brain trauma.
We conclude that IP receptor activation by prostacyclin may be beneficial for outcome after brain injury in mice by reducing the neuronal cell loss. This effect may, at least in part, be mediated by an improved cortical perfusion.
Conflict of interest
The authors report no conflicts of interest.