
Abstract
Brain edema and swelling is a critical factor in the high mortality and morbidity associated with traumatic brain injury (TBI). Despite this, the mechanisms associated with its development are poorly understood and interventions have not changed in over 30 years. Although neuropeptides and neurogenic inflammation have been implicated in peripheral edema formation, their role in the development of central nervous system edema after brain trauma has not been investigated. This study examines the role of the neuropeptide, substance P (SP), in the development of edema and functional deficits after brain trauma in rats. After severe diffuse TBI in adult male rats, neuronal and perivascular SP immunoreactivity were increased markedly. Perivascular SP colocalized with exogenously administered Evans blue, supporting a role for SP in vascular permeability. Inhibition of SP action by administration of the neurokinin-1 (NIC,) antagonist, N-acetyl-
Introduction
Traumatic brain injury (TBI) is the leading cause of death and disability in people under 40 years of age with the global cost for rehabilitation and care of these individuals exceeding US$50 billion per year (Jacobs et al, 2000). Despite the enormity of this public health problem, no effective treatment currently exists.
It is now accepted that TBI results in the development of neurologic deficits through two main mechanisms (McIntosh et al, 1996). The first is primary injury, which occurs at the time of the trauma and includes mechanical processes, such as shearing, laceration, and stretching of nerve fibers. Secondary injury occurs at later time points, and is composed of biochemical and physiologic factors that are initiated by the primary event and manifest over time. It has been shown earlier that much of the morbidity after brain injury is associated with the development of this secondary injury cascade (McIntosh et al, 1996). A number of secondary injury factors have been identified, including blood-brain barrier (BBB) opening, edema formation, release of excitatory amino acids, ion changes, oxidative stress, and bioenergetic failure, among others. Of the secondary injury factors, the formation of edema has long been known to be critical to outcome after injury (Lobato et al, 1988; Sarabia et al, 1988). Although the mechanisms associated with the formation of edema are still unclear, studies of peripheral tissue edema (Woie et al, 1993) have shown an association between neuropeptides and the development of increased vascular permeability and edema; this event has been termed ‘neurogenic inflammation.’
Neurogenic inflammation is a neurally elicited reaction that has typical characteristics of an inflammatory reaction, including vasodilation, protein extravasation, and tissue swelling. Studies of peripheral nerves have shown that neurogenic inflammation is the result of stimulation of C-fibers, which causes the release of neuropeptides, including substance P (SP), calcitonin gene-related peptide, and neurokinin A (NKA). Although all three neuropeptides are involved in the process, it is SP that has been generally accepted to be associated with increased microvascular permeability and edema formation (Newbold and Brain, 1995). Edema formation in the brain, when left uncontrolled, results in an increase in intracranial pressure that may lead to a decrease in brain tissue perfusion, localized hypoxia and ischemia, and even tissue herniation and death. Therefore, it is of particular interest to attenuate or inhibit such inflammation after TBI. Although a number of studies have investigated the role of classic inflammation in edema formation after TBI (Lenzlinger et al, 1997; Stahel et al, 1998), only one study has examined the role of neurogenic inflammation in this process. Nimmo et al (2004) showed that neuropeptide depletion with preinjury capsaicin administration significantly attenuated the BBB opening, edema formation, and the development of both motor and cognitive deficits. Although this study convincingly showed a role for neurogenic inflammation in TBI, the identification of the neuropeptide primarily involved in the formation of increased BBB permeability and edema formation was not established.
Earlier studies of peripheral edema have shown that SP is the neuropeptide that is most closely associated with capsaicin sensitivity (Saria, 1984; Yonehara et al, 1987). It is also well known that SP is the neuropeptide responsible for increased vascular permeability, whereas calcitonin gene-related peptide is primarily associated with vasodilation. Finally, Kramer et al (1997) have shown in studies of cardiac ischemia that SP release is increased with magnesium depletion; decreases in magnesium concentration have been described widely after TBI (Heath and Vink, 1997; Vink et al, 2003; Cernak et al, 2004). Accordingly, these series of experiments examined the role of SP in TBI, specifically using immunohistochemistry to identify increased SP protein, and an antagonist of the SP neurokinin-1 (NK1) receptor to investigate the role of SP in increased BBB permeability, edema formation, and the occurrence of neurologic deficits after TBI.
Materials and methods
All experimental protocols were approved by the Experimental Ethics committees of the University of Adelaide and the Institute of Medical and Veterinary Science and were conducted according to guidelines established for the use of animals in experimental research as outlined by the Australian National Health and Medical Research Council.
Induction of Traumatic Brain Injury
Injury was induced in halothane-anesthetized male Sprague-Dawley rats (400 ± 20 g) using the acceleration-induced impact TBI model (Foda and Marmarou, 1994; Cernak et al, 2004). This model involves impacting a 10-mm diameter × 3-mm-thick stainless-steel disc fixed centrally to the exposed skull between the lambda and bregma with an accelerating impactor. The impactor can either be a 450 g brass weight dropped from a height of 2 m, or a PC (personal computer)-controlled, high-velocity impactor. Earlier studies have shown that both methods of inducing impact acceleration injury produce similar changes in axonal injury, edema, BBB opening, Mg2+ decline, and moderate-to-severe neurologic deficits (Cernak et al, 2004; Foda and Marmarou, 1994). The weight-drop model was used in all current experiments with the exception of magnetic resonance imaging (MRI) experiments, in which the PC-controlled, high-velocity impactor was used (velocity, 3.25m/sec; distance, 18 mm).
All animals were fed and watered ad libitum before the induction of injury. During surgery, induction of injury, and during the immediate recovery phase, rat rectal temperature was maintained at 37°C using a thermostatically heated warming blanket. Immediately after injury, animals were manually ventilated until stable respiration was restored, usually in < 5 mins. After injury, all wounds were sutured, animals were withdrawn from anesthesia, and returned to their cages after recovery.
Drug Treatment
Animals were treated after injury with either the SP NK1 receptor antagonist, N-acetyl-
Substance P Immunohistochemistry
Animals were injured and perfusion fixed at preselected time points (n = 5 per group). A further five animals were used as sham controls. All brains were perfusion fixed using 4% paraformaldehyde and removed after decapitation. Coronal sections were then cut using a Kopf rodent brain blocker (Kopf; Tujunga, CA, USA) and the resultant 2-mm sections were embedded in paraffin wax. Serial 5-μm sections were cut using a microtome (Microm, Walldorf, Germany) and immunolabeled with SP primary antibody (1:2,000 in normal horse serum (NHS); polyclonal cat. no. SC-9758, Santa Cruz Biotechnology, Santa Cruz, CA, USA) by overnight incubation at room temperature. After washing in PBS (phosphate-buffered saline), slices were then incubated with an anti-goat immunoglobulin G-horseradish peroxidase-conjugated secondary antibody (1:250 in NHS; Sigma-Aldrich, St Louis, MO, USA) for a minimum of 30 mins at room temperature. After this, slides were incubated in the tertiary streptavin peroxidase conjugate (1:1,000 in NHS; Pierce, Rockford, IL, USA) for at least 1 h at room temperature, and the subsequent immunocomplex was visualized using diaminobenzidine tetrahydrochloride as a chromogen in a peroxidase reaction (Sigma-Aldrich, St Louis, MO, USA) and examined using a Leica light microscope (Leica Microsystems, Sydney, NSW, Australia).
Substance P Immune-electron Microscopy
Animals (n = 3) were injured and decapitated at 5 h after injury. Floating vibratome sections (100 mm) were then generated and placed in the PBS buffer (pH 7.4) containing 0.4% Triton X-100 and 1% glycine for 1 h. Blocking of endogenous peroxidase was achieved by incubation in methanol containing 0.6% hydrogen peroxide for 30 mins. The sections underwent antigen retrieval in citrate buffer (pH6.0) for 2.5 h at 65°C, followed by overnight incubation at room temperature in the primary anti-SP (goat anti-SP polyclonal antibody, Santa Cruz Biotechnology, N-18, 1:1,000). The sections were then incubated sequentially with biotinylated mouse anti-goat secondary antibody (Vector, Burlingame, CA, USA) at a dilution of 1:250 for 60 mins, followed by peroxidase-conjugated streptavidin (Pierce) at a dilution of 1:1,000 for 60 mins, and developed with 3,3'-diaminobenzidine tetrahydrochloride for 5 mins. Approximately 4 × 6 mm pieces containing the cerebral cortex were cut from the sections, further fixed in 2.5% glutaraldehyde for 2 h and 1% aqueous osmium for 1 h, dehydrated with ascending ethanol and propylene oxide, and embedded in TAAB (TAAB, Aldermaston, Berks, England) resin. Ultrathin (80 nm) sections were examined under a transmission electron microscope without any further staining with either uranyl acetate or lead citrate. Immunopositive labeled SP appeared as an electron-dense deposit. Negative control vibratome sections were treated similarly but with the omission of the primary antibody.
Confocal Microscopy
Paraffin sections of 10-μm thickness from 5-h injured brains and sham controls (n = 3 per group) were examined using double immunofluorescent staining with the SP antibody (Santa Cruz Biotechnology; 1:200 dilution) and the NK1 receptor antibody (Advanced Targeting Systems, San Diego, CA, USA; 1:150 dilution). Sections were incubated overnight in the primary antibodies, then in the secondary antibodies for 1 h. Secondary antibodies used were Alexa fluor 488 donkey anti-goat (Molecular Probes, Eugene, OR, USA; 1:200 dilution) for SP and Alexa fluor 660 goat antirabbit (Molecular Probes; 1:200 dilution) for NK1. After washing in PBS, slides were mounted using an aqueous mounting solution with anti-fade and sealed using varnish. Positive and negative controls were routinely used for all antibodies. Sections were examined using a confocal fluorescence microscope (BioRad Radiance 2100, Hemel Hempstead, Hertfordshire, UK). A subset of animals (n = 3) that were administered EB dye for the assessment of BBB permeability was also used for the assessment of SP colocalization with EB.
Enzyme-Linked Immunosorbent Assay
Animals were injured (n = 6 per group) using the impact acceleration model of TBI as described above. At 30 mins and 5 h later, injury blood was collected from the tail vein. The plasma was isolated by centrifugation at 10,000 r.p.m. (revolutions per minute) for 10 mins and stored at −80°C until use. A further six animals served as uninjured sham controls. Substance P levels in the plasma were measured using ELISA (enzyme-linked immunosorbent assay) (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions. Briefly, plasma was collected in ethylenediamine tetraacetic acid-lined tubes and diluted in a ratio of 1:2 in an assay buffer. Samples were then plated into individual wells on a 96-well plate, and 50 μL of the SP antibody solution was added. The plates were then incubated for 2 h at room temperature on a horizontal orbital microplate shaker (0.12 inch orbit) set at 500 ± 50 r.p.m. The sample wells were than aspirated and washed thrice with wash buffer before the plate was inverted and blotted against a clean paper towel. A volume of 5 μL of SP conjugate was then added, followed by 200 μL of p-nitrophenyl phosphate substrate, before being incubated for 1 h at room temperature. Addition of stop solution (R80-0247) after 60 mins permitted the optical density of each well to be determined using a microplate reader set at 405 nm with the wavelength correction set between 570 and 590 nm.
N-acetyl-l-tryptophan Dose Optimization
N-acetyl-
Edema Determination
The amount of edema development was calculated using the wet weight—dry weight method. Animals were divided into three groups (n = 6 per group) and either left uninjured (sham controls) or treated after injury with NAT, or with an equal volume of saline vehicle. They were then reanesthetized with 4% halothane at preselected time points and decapitated. The brain was removed rapidly from the skull, the olfactory bulbs and the cerebellum were discarded, and the cortex and subcortex were separated. The cortex and subcortex of each rat were placed separately into pre-weighed and labeled glass vials with quick fit lids (to prevent evaporation) and weighed immediately for wet water content. The vials (glass lids removed) were then placed in an oven at 100°C for 72 h. Vials and brain segments were then reweighed to obtain dry weight content. Edema in each brain sample was calculated using the wet—dry method formula:
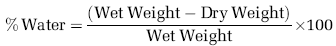
Magnetic Resonance Imaging
At 5 and 24 h after TBI, anesthetized animals (n = 6 per group) were subjected to MRI examination as described earlier (Faden et al, 2003; Cernak et al, 2004). Briefly, animals were placed in the heated plexiglas holder and a respiratory motion detector was positioned over the thorax to facilitate respiratory gating. The plexiglas holder was then positioned in the center of the magnet bore, in which a 72-mm proton-tuned birdcage coil had been positioned. Field homogeneity across the brain was then optimized and sagittal and coronal high-resolution images were acquired with a multislice—multiecho pulse sequence or a rapid acquisition relaxation enhancement pulse sequence; thus, TR (repetition time) = 2 secs, TE (echo time) = 30/60 msecs, FOV (field of view) = 4 cm, matrix = 512 × 512, and slice thickness = 1 mm. Five to seven coronal slices were obtained per animal, which were positioned precisely using a sagittal pilot scan and by measuring a known distance from the anterior pole of the frontal lobe to an index slice. Multislice, multiecho T2-weighted images were acquired to obtain eight contiguous slices from the dorsal end of the olfactory bulb to the rostral margin of the cerebellum; thus, FOV = 4 × 4 cm, slice thickness = 2 mm, resolution 256 × 256, TR/TE = 2,000/20 msecs, 4 echo images, and 2 averages. Diffusion-weighted images were then acquired with a spin echo pulse sequence that had diffusion gradients added before and after the refocusing pulse. Gradient strength was varied in six steps using sensitization values ranging from 20 to 1,000 secs/mm2. A 256 × 256 matrix was used with a 3-cm FOV, TR = 2.0 secs, TE = 502 msecs, slice thickness = 2 mm, and 4 echoes. Diffusion maps were generated by applying the Stejskal—Tanner equation in association with a Marquart algorithm using the commercially available Paravision software (Bruker, Billerica, MA, USA). Apparent diffusion coefficients (ADCs) were calculated for four regions, namely the left cortex, right cortex, left subcortex, and right subcortex. Apparent diffusion coefficients were expressed as 10−5 mm2/sec ± s.e.m.
Functional Outcome
Motor and cognitive outcomes in animals (n = 6 per group) were assessed using the rotarod and Barnes maze, respectively, as described in detail elsewhere (O'Connor et al, 2003). The rotarod device is considered the most sensitive measure of motor outcome after rodent TBI. It consists of a motorized rotating assembly of 18 rods (1 mm in diameter) upon which the animals were placed. Rotational speed of the device is increased from 0 to 30 r.p.m. and the duration (in seconds) when the animals either completed the task and fell from the rods or gripped the rods and spun for two consecutive revolutions rather than walking actively, was recorded as the rotarod score. In the Barnes maze, animals were placed under a cover in the center of an elevated 1.2-m diameter board containing 19 holes around the periphery. One of the holes contained an entrance to a darkened escape tunnel that is not visible from the surface of the board. After activating a series of bright lights aimed at the board and switching on an aversive auditory stimulus, the cover is lifted and the latency (in seconds) for the animal to locate and enter the escape tunnel was recorded. All animals were pretrained on both functional outcome tests twice per day over 5 days before injury to establish a normal, uninjured baseline.
Statistical Analysis
All data are expressed as mean and s.e.m. and, with the exception of the functional outcome data, were analyzed for statistical significance using oneway ANOVA (analysis of variance) followed by Student—Neuman—Keuls tests (GraphPad Prism Software, La Jolla, CA, USA). Functional outcome data were analyzed by repeated measures 2-way ANOVA followed by Student—Neuman—Keuls tests.
Results
Immunohistochemical analysis of tissue after TBI is shown in Figure 1. The cortex of sham animals (Figure 1A) shows light SP immunoreactivity along blood vessels and also within the parenchyma. At 5 h after injury (Figure 1B), there is an unequivocal upregulation of SP immunoreactivity along blood vessels and within the parenchyma. Electron microscopy (Figure 1C) shows that vascular SP immunoreactivity is in the adjacent parenchyma (perivascular) and not within the vessel lumen. There was little evidence of SP immunoreactivity in the vascular endothelial cells. This 5-h time point has been shown earlier to correlate with the maximal formation of vasogenic edema and opening of the BBB (O'Connor et al, 2006). At the level of the trigeminal tract in sham animals, clear SP immunoreactivity could be observed in the tract fibers and to a lesser extent in the trigeminal nuclei (Figure 1D). At 5h after injury (Figure 1E), there was a marked upregulation of SP activity in the trigeminal tract, nuclei, and in the surrounding tissue.
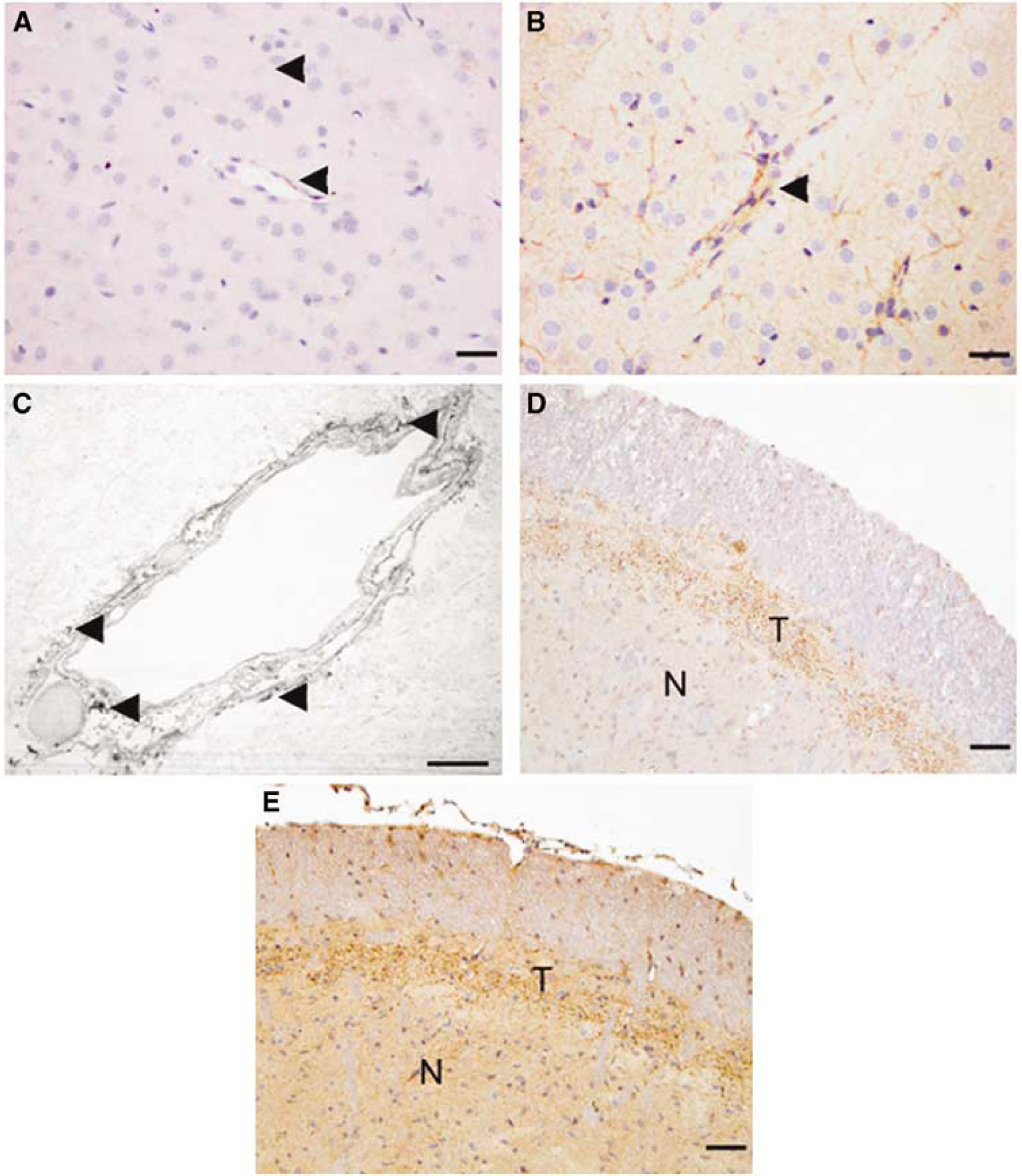
Substance P (SP) immunoreactivity before and 5 h after diffuse TBI in rats. (
Given the high level of perivascular SP at 5 h after TBI, it was of interest to establish whether SP was detected in the blood after TBI. Accordingly, we used a competitive ELISA assay to determine SP concentration within plasma samples of injured animals at 30 mins and 5 h after injury. At 30 mins after injury, there was a significant increase (P < 0.01) in SP concentration to almost double the basal level observed in sham animals (Figure 2). These increased levels of SP in the blood declined to sham levels by 5 h after injury, presumably because of rapid metabolism by vascular proteases. These results suggest that SP is released early after TBI, it is released in the vicinity of the vasculature and enters the bloodstream, and that this release may be associated with the early, but transient, opening of the BBB in this model of TBI.
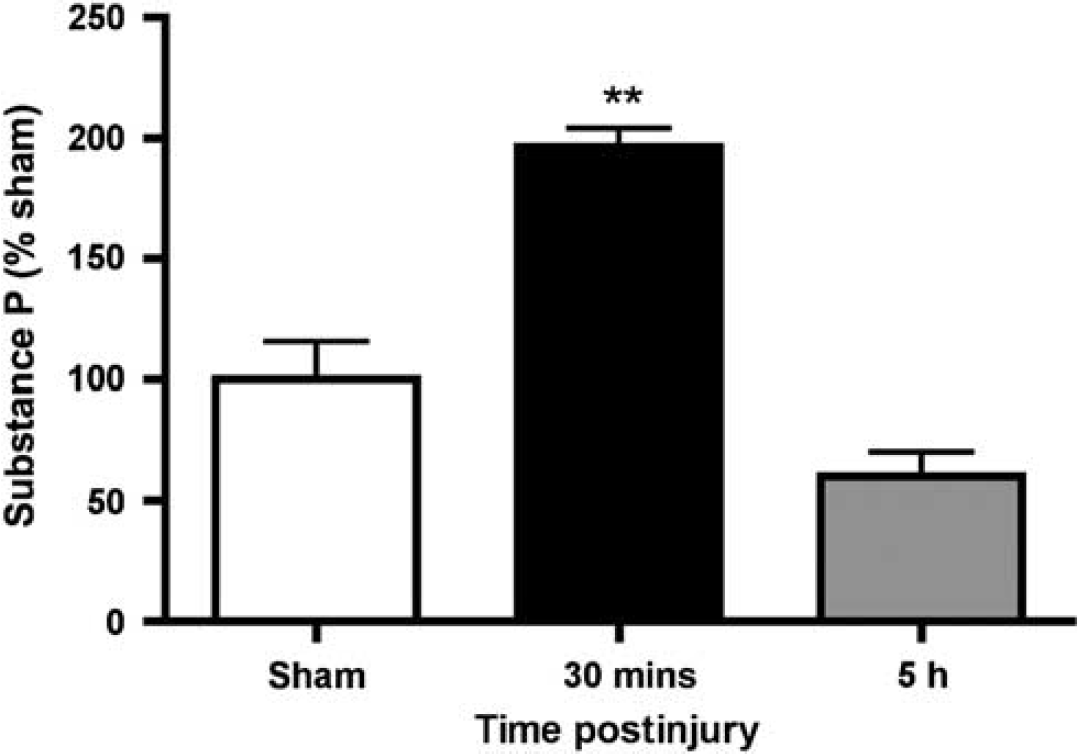
Plasma concentration of SP after diffuse TBI as assessed by ELISA assay (n = 6 per group). **P < 0.001 relative to sham animals.
Having established that SP is increased perivascularly after TBI, we used confocal microscopy to establish whether there was any association between SP and a permeability marker, EB, at the level of the cortical vasculature after TBI. In sham animals, little SP immunoreactivity can be observed throughout the section (Figure 3A). However, at 5 h after TBI, there is clearly a marked increase in perivascular SP immunoreactivity such that it delineates the vessels (Figure 3C). This increase in perivascular SP by confocal microscopy is consistent with the light microscopic and electron microscopy results shown earlier. Colocalization of the SP immunofluorescence with that of an antibody directed toward the neurokinin receptor (results not shown) supports the fact that SP was bound to its NK1 receptor. The cortical blood vessels with increased SP immunoreactivity are clearly permeable at this time point after TBI as confirmed by the extravasation of the vascular permeability marker, EB, which was administered intravenously at 30 mins before tissue removal (Figure 3D). The short circulation time for EB is critical so as to ensure that the dye has not diffused far from the blood vessels. No significant EB was visible in the sham animals (Figure 3B). Super-imposition of the images (Figure 3E) shows a remarkable colocalization of SP with the increased permeability of the blood vessels as shown by the EB dye, supporting an association between SP and the development of increased vascular permeability.
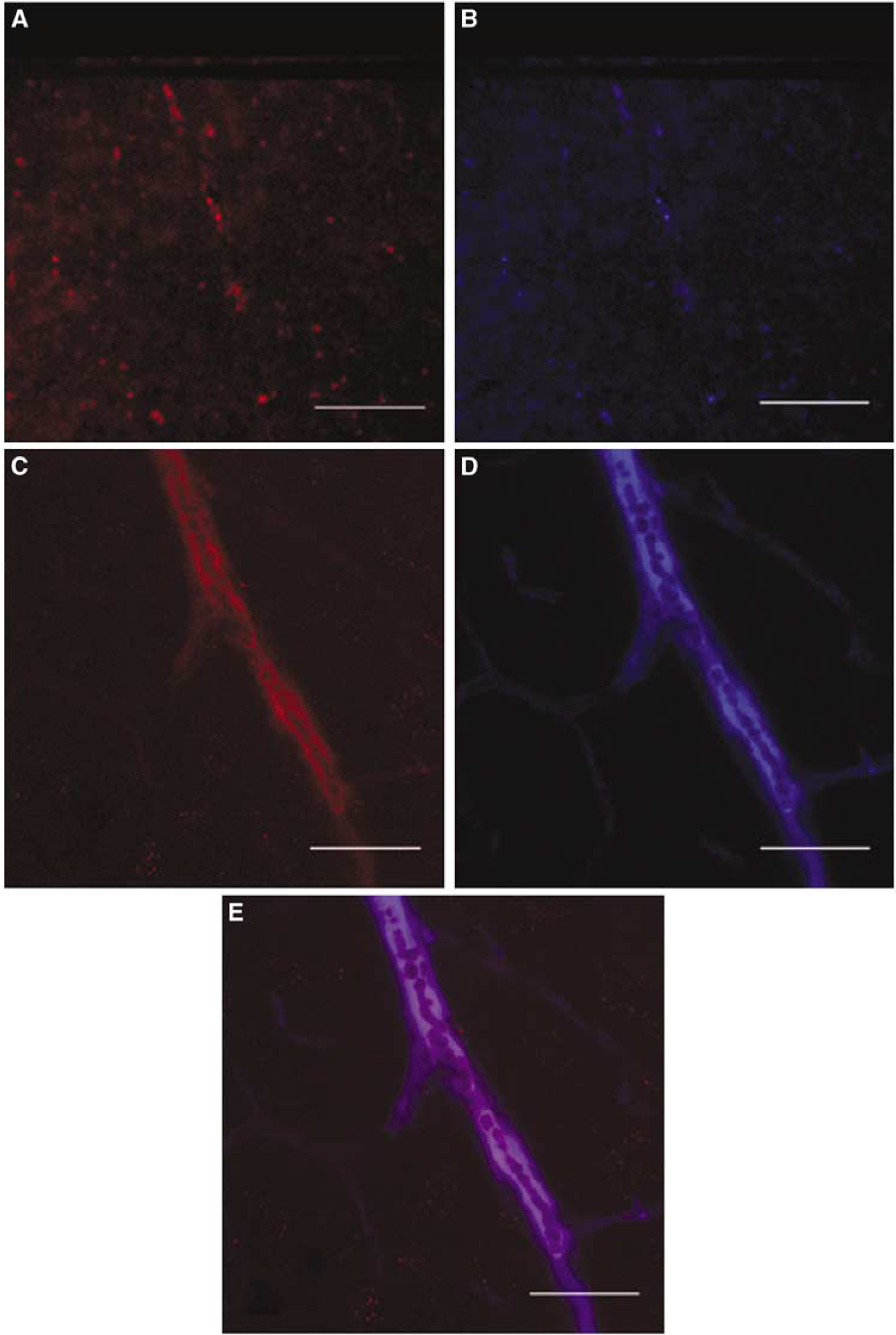
Confocal images of SP and Evans blue (EB) in the rat cortex of sham (uninjured) animals and injured animals at 5 h after diffuse TBI. (
Earlier studies have established that impact acceleration injury results in an increased permeability of the BBB (O'Connor et al, 2006). Having characterized the vascular colocalization of SP with the NK1 receptors, and given their potential role in increasing vascular permeability (Nimmo et al, 2004), we examined the effects of different doses of the NK1 receptor antagonist, NAT, on BBB permeability after injury (Figure 4). In sham animals, the negligible penetration of the EB dye confirmed that the BBB was intact despite induction of anesthesia and surgery. At 5h after injury, a significant increase (P < 0.001) in EB dye was detected indicating increased BBB permeability. In contrast, NAT treatment resulted in a profound reduction in EB penetration (P < 0.001), with the optimal dose being 2.5 mg/kg intravenously.
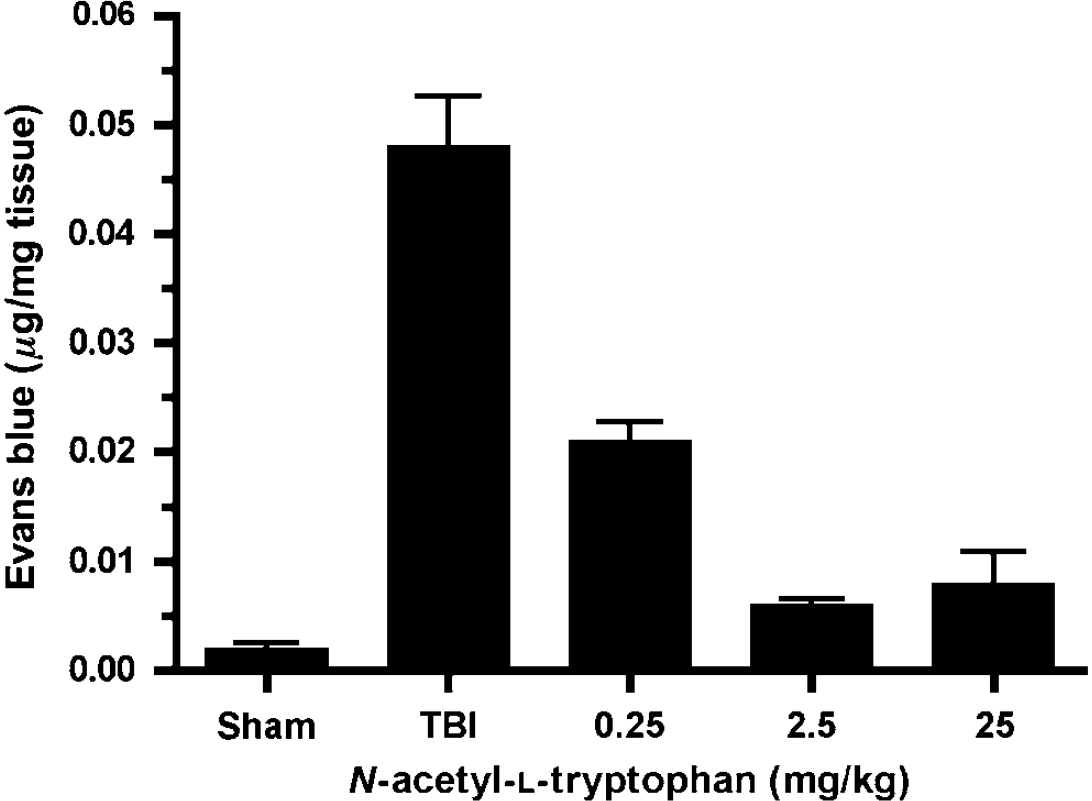
Effects of NAT on brain tissue concentration of Evans blue at 5 h after diffuse TBI (n = 6/group). A concentration of 2.5 mg/kg was the optimum dosage to reduce EB extravasation.
Having established the dose optimum for NAT, its effects on any edema formation after TBI was assessed using MRI. Qualitatively, ADC maps derived from diffusion-weighted MRI show relatively brighter areas of hyperintensity throughout the brain of vehicle (saline)-treated injured animals (Figure 5B) compared with that of sham animals (Figure 5A). This hyperintensity reflects less restriction for water diffusion, consistent with increased extracellular water, and is unlikely to be because of the loss of diffusion barriers (membrane breakdown) given that the images were acquired between 4 and 5h after injury. Thus, the hyperintensity is thought to reflect vasogenic edema formation, consistent with earlier diffusion-weighted image results (Hanstock et al, 1994; Nimmo et al, 2004). In contrast, animals treated with the NK1 receptor antagonist at 30 mins after TBI did not show any hyperintensity throughout the brain, suggesting an attenuation of edema formation after injury (Figure 5C). Quantitatively, the ADC differences are shown in Figure 5D. Sham and naive animals showed similar mean ADC values in all regions of the brain (74.8 ± 2.68 and 72.63 ± 1.09 × 10−5 mm2/sec, respectively), which is similar to earlier published results (Albensi et al, 2000; Hanstock et al, 1994). After injury, there was a significant increase (P < 0.001) in ADC values in vehicle-treated animals (88.5 ± 2.67 × 10−5 mm2/sec) compared with that in sham-treated animals, confirming an increased ADC for water, and consistent with vasogenic edema formation. This increase was significantly inhibited (P < 0.001) in the NAT-treated animals (69 ± 6.7 × 10−5 mm2/sec), which were not significantly different from sham animals.
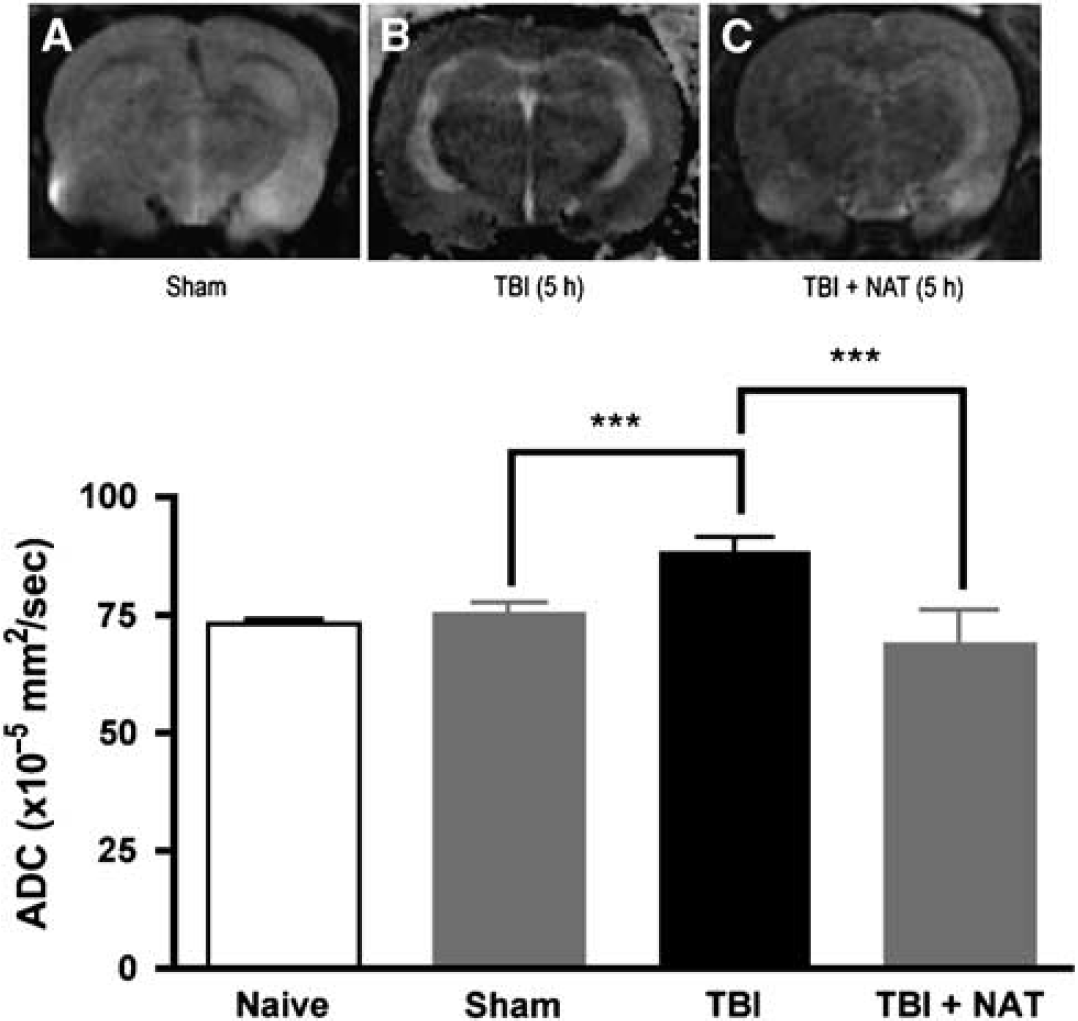
Effects of NAT treatment on apparent diffusion coefficients (ADC) after diffuse TBI. Representative ADC maps are shown for (
The changes in edema described using MRI were confirmed using wet weight—dry weight determinations of brain water content (Figure 6). The percentage of water content in sham animals (77.8 ± 0.2%) was consistent with those published earlier (Bareyre et al, 1997; O'Connor et al, 2006). At 5h after injury, vehicle-treated animals had a significantly increased level of brain water content (P < 0.001), indicating the development of edema after diffuse TBI. Treatment with NAT at 30 mins after TBI led to a significant reduction of edema at 5h after injury (P < 0.001) compared with vehicle controls. This water content after NAT treatment was not significantly different from sham values. These results were consistent with the MRI data shown above.
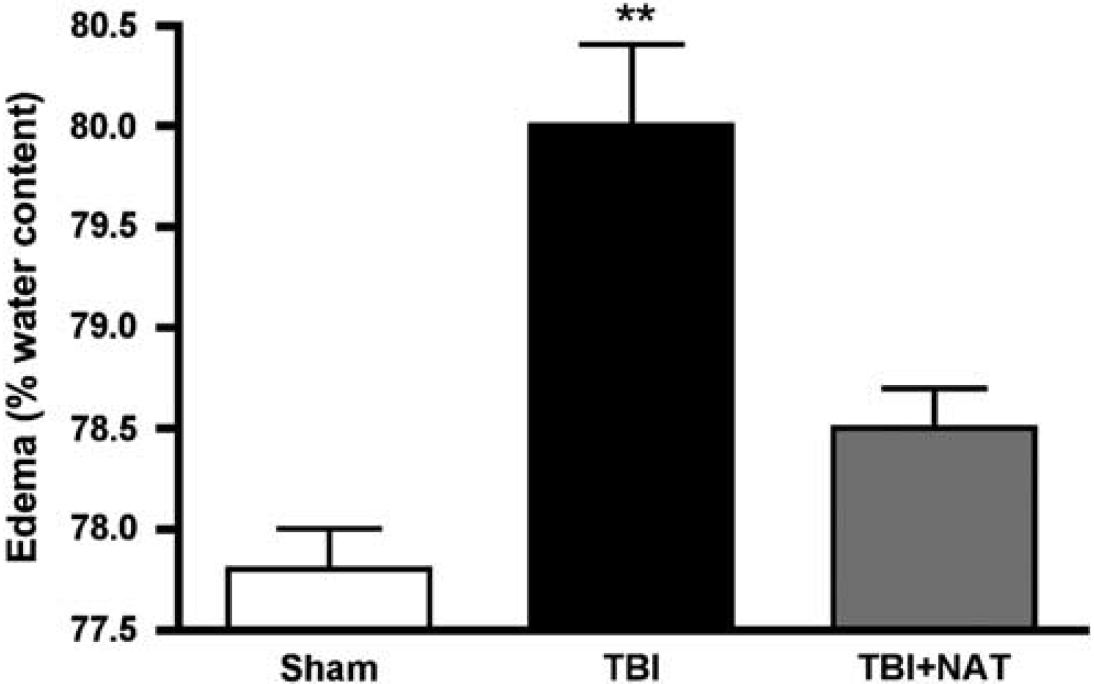
Percentage cortical water content in male rats 5 h after diffuse TBI and after treatment with NAT (n = 6 per group). **P < 0.01 compared with sham levels.
Finally, the effects of the NK1 antagonist on functional outcome were assessed using the rotarod test for motor function and the Barnes maze for cognitive outcome (Figure 7). Before the induction of injury, all animals achieved a mean rotarod score of 111 ± 4 secs. After injury, there was a highly significant decrease (P < 0.001) in the rotarod score in vehicle-treated animals to 23 ± 8 secs at day 1 after injury (Figure 7A). Thereafter, animals improved their performance with repeated exposure to the task. In contrast, animals treated with the NK1 antagonist at 30 mins after TBI performed significantly better (P < 0.001) than vehicle-treated controls, recording a minimum value of 82 ± 11 secs at 1 day after injury and improving thereafter. At no time point were the NK1-treated animals significantly different from sham controls. Similarly, in the Barnes cognitive maze (Figure 7B), animals treated with the NK1 antagonist had significantly reduced cognitive deficits (P < 0.001) compared with vehicle-treated animals. Again, at no time point was the cognitive performance of the NK1-treated animals significantly different from that of sham controls.
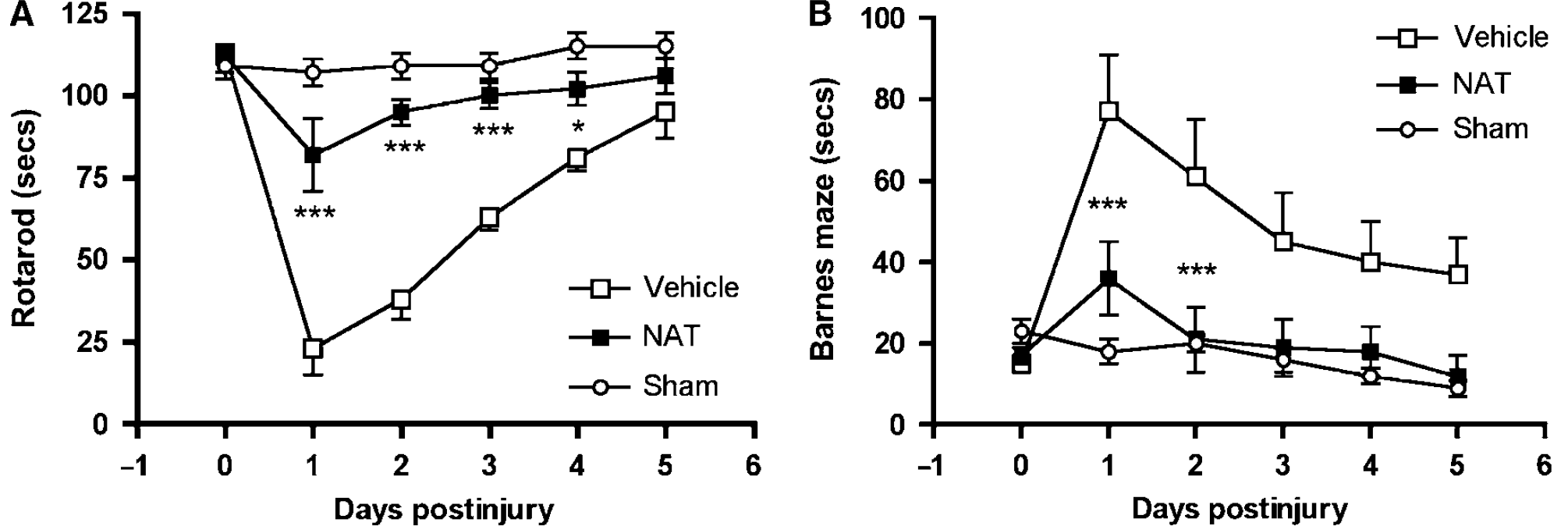
Effects of NAT on neurologic outcome after diffuse TBI in rats as assessed using (
Discussion
In this study, we have shown that the neuropeptide SP is upregulated after TBI, and that administration of the NK1 receptor antagonist NAT results in the attenuation of posttraumatic BBB permeability, edema formation, and neurologic deficits. Before these results, only one study had earlier implicated sensory neuropeptides, and in particular SP, in neurogenic inflammation after TBI. Nimmo et al (2004) showed that an earlier depletion of neuropeptides from sensory nerves led to an inhibition of the BBB opening, edema formation, free Mg2+ decline, and to the development of motor and cognitive deficits after TBI. Before these findings, very few reports describing the role of neuropeptides in nervous system injury had been published. Indeed, only isolated reports in peripheral nerve injury (Malcangio et al, 2000), traumatic spinal cord injury (Sharma et al, 1993), and brain ischemia (Stumm et al, 2001) had described the inflammatory release of sensory neuropeptides. Of these, only the studies of brain ischemia showed an activation of neuropeptide receptors in the endothelium related to edema formation (Stumm et al, 2001), similar to what has been well characterized in peripheral tissue injury (Woie et al, 1993). The results of our study, in which TBI-induced development of edema is associated with increased extravasation of EB across the BBB, are consistent with these earlier observations that activation of neuropeptide receptors leads to edema formation.
The early increase in BBB permeability supports published findings showing that TBI induces a maximal increase in permeability within the first 3 to 6 h after injury, along with early edema formation (O'Connor et al, 2006). This same study described that the edema formation after injury occurred in two phases with an early vasogenic episode directly related to BBB permeability, and a second prolonged cytotoxic episode occurring over the subsequent days independent of increased BBB permeability. In this study, administration of the NK1 receptor antagonist 30 mins after TBI attenuated both the BBB opening and the early vasogenic edema formation, and supports a direct role for SP in these events.
The role of SP in vasogenic edema was confirmed using MRI diffusion-weighted imaging, in which bright areas of hyperintensity throughout the ADC maps of the injured brain occurred 4 to 5h after injury, and a marked reduction occurred after treatment with the NK1 receptor antagonist. Quantitation of the brain water content confirmed the MRI results. This early formation of vasogenic edema was consistent with earlier published results, including those of Hanstock et al (1994) who used the lateral fluid percussion device to show that TBI results in an increased water diffusion distance with the directionality indicating bulk flow of extracellular fluid toward the lateral ventricles (vasogenic edema). Albensi et al (2000) also showed that the fluid percussion model resulted in regions of increased ADC early after TBI that may be associated with vasogenic edema formation. Beaumont et al (2000) showed that diffuse impact acceleration induced-injury resulted in vasogenic edema despite only a very brief BBB opening (Beaumont et al, 2000). Moreover, these authors also showed that the early vasogenic edema is permissive for the subsequent cytotoxic edema, suggesting that the inhibition of the vasogenic episode may also attenuate subsequent cytotoxic edema.
The results of our study also show that SP release and binding to the NK1 receptor may influence functional outcome after TBI. The attenuation of motor deficits with the NK1 receptor antagonist was shown using the rotarod test, whereas the Barnes maze was used to show the efficacy on cognitive outcome. The rotarod test has been described earlier as the most sensitive test for the detection of motor deficits after rodent TBI (Hamm, 2001) and both the rotarod and Barnes maze has been used successfully in earlier diffuse TBI studies to assess the effects of pharmacological agents on motor and cognitive deficits (Heath and Vink, 1997; O'Connor et al, 2003; Vink et al, 2003). The cognitive effects of the NK1 antagonist may be attributed to the high numbers of sensory neuropeptide receptors in the hippocampus and striatum, those parts of the brain that are known to be associated with learning and memory. Supporting this role are studies that have confirmed that alterations in the neuropeptides calcitonin gene-related peptide, neuropeptide Y, and SP in the hippocampus are associated with cognitive deficits as well as with motor function (Gaumann et al, 1990). A number of other studies have also shown that the use of a NK1 receptor antagonist can be beneficial to outcome. Specifically, Pothoulakis et al (1994) showed that pretreatment with a NK1 antagonist significantly inhibited the effects of an inflammatory toxin within the ilium of rats, whereas Rupniak and Kramer (1999) have described the efficacy of NK1 antagonists in the treatment of experimental depression and emesis. A similar beneficial role for NK1 antagonists has been suggested by Nimmo et al (2004), who showed that neuropeptide depletion before induction of TBI attenuated the development of neurogenic inflammation.
Although the mechanisms by which the NK1 receptor antagonists are neuroprotective are unknown, there are a number of possibilities whereby administration could attenuate the actions of SP. A limited number of studies have identified a potential role for SP in the glial response to TBI. Receptor binding sites for SP have been shown to increase on the glia after neuronal injury (Lin, 1995). As SP is known to regulate inflammatory and immune responses in the peripheral tissue, it therefore may regulate the glial response to injury. Subsequent studies have confirmed that in response to SP after injury, astrocytes become ‘reactive’ and mitogenesis and production of several soluble mediators, such as cytokines, prostaglandins, and thromboxane derivatives are induced (Marriott et al, 1991; Palma et al, 1997). This increase was not observed in undamaged areas (Lin, 1995). Moreover, levels of SP have been shown to increase after traumatic spinal cord injury; these increases have been shown to be inhibited by serotonin antagonists (Sharma et al, 1990). There are a number of roles SP may play in the pathophysiology of TBI. Substance P induces endothelial cells to produce nitric oxide (Persson et al, 1991), which has been implicated as a secondary injury factor. Substance P also primes polymorphonuclear cells for oxidative metabolism (superoxide production) (Hafstrom et al, 1998), thus providing a source of reactive oxygen species known to exacerbate the injury process. Weglicki and Phillips (1992) have in fact postulated that SP release may be one of the earliest pathophysiological events associated with injury-induced declines in tissue Mg2+ concentration, leading to stimulation of the inflammatory cytokines, which may then stimulate free radical mechanisms of injury.
Further insight into the possible mechanisms for SP may be gained from earlier studies using NK1 receptor antagonists in a variety of conditions. Neurokinin-1 receptor blockade reduces prenecrotic perivascular inflammatory infiltration, and reducing circulating histamine, prostaglandin E2 and lipid peroxidation products (Kramer et al, 1997). Weglicki and Phillips (1992) have in fact postulated that SP release may be one of the earliest pathophysiological events associated with injury-induced declines in tissue Mg2+ concentration, leading to stimulation of the inflammatory cytokines, which may then stimulate free radical mechanisms of injury. Neurokinin-1 receptor antagonists have also been shown to rapidly improve mood by antagonizing SP-induced anxiety (Kramer et al, 1998; Saria, 1999). Thus, they may be effective in treating posttraumatic depression. The antagonists also have been shown to inhibit pain (Saria, 1999), along with being a beneficial treatment for cerebral ischemia (Yu et al, 1997).
In conclusion, we have shown that inhibition of neurogenic inflammation by posttraumatic administration of the NK1 receptor antagonist NAT inhibits BBB permeability, vasogenic edema formation, and improves functional outcome. These results show sensory neuropeptides, and in particular, SP may play a significant role in the posttraumatic, secondary injury processes and may offer a novel target for the development of interventional pharmacological strategies.