
Abstract
Aging is associated with cerebrovascular dysregulation, which may underlie the increased susceptibility to ischemic stroke and vascular cognitive impairment occurring in the elder individuals. Although it has long been known that oxidative stress is responsible for the cerebrovascular dysfunction, the enzymatic system(s) generating the reactive oxygen species (ROS) have not been identified. In this study, we investigated whether the superoxide-producing enzyme NADPH oxidase is involved in alterations of neurovascular regulation induced by aging. Cerebral blood flow (CBF) was recorded by laser-Doppler flowmetry in anesthetized C57BL/6 mice equipped with a cranial window (age = 3, 12, and 24 months). In 12-month-old mice, the CBF increases evoked by whisker stimulation or by the endothelium-dependent vasodilators acetylcholine and bradykinin were attenuated by 42, 36, and 53%, respectively (P < 0.05). In contrast, responses to the nitric oxide donor S-nitroso-D-penicillamine or adenosine were not attenuated (P > 0.05). These cerebrovascular effects were associated with increased production of ROS in neurons and cerebral blood vessels, assessed by hydroethidine microfluorography. The cerebrovascular impairment present in 12-month-old mice was reversed by the ROS scavenger Mn (III) tetrakis (4-benzoic acid) porphyrin chloride or by the NADPH oxidase peptide inhibitor gp91ds-tat, and was not observed in mice lacking the Nox2 subunit of NADPH oxidase. These findings establish Nox2 as a critical source of the neurovascular oxidative stress mediating the deleterious cerebrovascular effects associated with increasing age.
Keywords
Introduction
Aging has profound cerebrovascular effects that are thought to underlie the increased incidence of ischemic stroke and vascular cognitive impairment affecting older individuals (Farkas and Luiten, 2001; Rothwell et al, 2005). Aging is associated with a reduction in resting cerebral blood flow (CBF) and a dysfunction of the mechanisms regulating the cerebral circulation (Faraci and Heistad, 1998; Farkas and Luiten, 2001). For example, the cerebrovascular relaxation induced by endothelium-dependent vasodilators, such as acetylcholine, is impaired in large and small cerebral arteries of older animals (Mayhan et al, 1990; Paterno et al, 1994) (for a review, see Faraci and Heistad, 1998). Furthermore, the increases in CBF induced by activation of central cholinergic pathways, hypercapnia, hypoxia, or hypotension are attenuated (Hoffman et al, 1981, 1984, 1982; Sato et al, 2002). These cerebrovascular alterations reduce cerebrovascular reserves and increase the susceptibility of the brain to vascular insufficiency and ischemic injury (Farkas and Luiten, 2001). However, less is known about the effects of aging on the increase in CBF induced by synaptic activity, a critical homeostatic response that matches the energetic needs of the active brain with the delivery of nutrients through blood flow (Iadecola, 2004).
There is increasing evidence that the impairment in cerebrovascular function induced by aging is mediated by oxidative stress, but the cellular sources of reactive oxygen species (ROS) and the enzymatic systems generating them have not been defined (Faraci and Heistad, 1998; Farkas and Luiten, 2001). There are several potential sources of ROS in brain, including xanthine oxidase, mitochondrial enzymes, and enzymes involved in nitric oxide synthesis or arachidonic acid metabolism (Faraci, 2006). However, NADPH oxidase has recently emerged as a major source of ROS in neurons, glia, and cerebral blood vessels (Abramov et al, 2005; Infanger et al, 2006; Kazama et al, 2004; Miller et al, 2006). NADPH oxidase is a multiunit enzyme initially discovered in neutrophils (Bedard and Krause 2007). It is composed of membrane-bound (p22phox and gp91phox) and cytoplasmic subunits (p40phox, p47phox, and p67phox). The catalytic subunit of the enzyme gp91phox, also termed ‘Nox2,’ is present in several homologs (Nox1 through Nox5) (Bedard and Krause, 2007). Activation of the enzyme depends on phosphorylation of p47phox, which, in concert with the small GTPase Rac1, leads to assembly of the cytoplasmic and membrane-bound subunits and production of superoxide (Bedard and Krause, 2007). Although the role of NADPH oxidase in the cerebrovascular dysfunction induced by hypertension, diabetes, and amyloid-β is well established (Kazama et al, 2004; Mayhan et al, 2006; Miller et al, 2006; Park et al, 2005), it is unknown whether this enzyme is also involved in the cerebrovascular alterations induced by aging.
In this study, we examined the effect of aging on the neurovascular and endothelial mechanisms regulating the cerebral microcirculation. We found that aging, in addition to its deleterious effects on endothelium-dependent vasodilation, also disrupts the increase in CBF induced by synaptic activity in the mouse neocortex. Furthermore, we found that NADPH oxidase-derived ROS, originating from neurons and blood vessels, can account in full for the cerebrovascular disturbances observed in older mice. The findings expand our understanding of the cerebrovascular effects of aging and identify NADPH oxidase as a key source of ROS mediating the alterations in critical homeostatic mechanisms controlling the cerebral microcirculation.
Materials and methods
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College.
Mice
Experiments were performed in male C57BL/6 and Nox2 (gp91phox)-deficient mice aged 3, 12, or 24 months. Nox2-deficient mice (Pollock et al, 1995) were obtained from our own colony and were congenic with the C57BL/6 strain (Park et al, 2005).
General Surgical Procedures
Procedures for surgical preparation of the mice have been described previously in detail (Niwa et al, 2001; Park et al, 2005) and are only summarized here. Mice were anesthetized with isoflurane (induction: 5%; maintenance: 1% to 2%). One of the femoral arteries was cannulated for recording of arterial pressure and collection of blood samples. Mice were incubated and artificially ventilated with oxygen-nitrogen mixture adjusted to provide an arterial pO2 (paO2) of 120 to 140 mm Hg (Supplementary Table 1). Rectal temperature was maintained at 37°C using a thermostatically controlled rectal probe connected to a heating pad. After surgery, isoflurane was gradually discontinued and anesthesia was maintained with urethane (750 mg/kg; intraperitoneally) and α-chloralose (50 mg/kg; intraperitoneally). Throughout the experiment, the level of anesthesia was monitored by testing corneal reflexes and motor responses to tail pinch.
Monitoring of Cerebral Blood Flow
A small craniotomy (2 × 2 mm) was performed to expose the somatosensory cortex. The dura was removed, and the site was superfused with a modified Ringer's solution (37°C; pH 7.3 to 7.4) (see Iadecola, 1992 for composition). Cerebral blood flow was continuously monitored at the site of superfusion with a laser-Doppler probe (Vasamedic, St Paul, MN, USA) positioned stereotaxically on the neocortical surface and connected to a computerized data acquisition system. Cerebral blood flow values were expressed as percent increase relative to the resting level. Zero values for CBF were obtained after the heart was stopped by an overdose of isoflurane at the end of the experiment.
Detection of Reactive Oxygen Species
Reactive oxygen species production was assessed using hydroethidine as a marker, as described previously (Park et al, 2005). Hydroethidine is quickly taken up by cells and reacts with superoxide to form a stable fluorescent product (2-hydroxyethidium), which binds to DNA. Hydroethidine (2 μmol/L; Molecular Probes, Eugene, OR, USA) was topically superfused on the somatosensory cortex for 60 mins (Park et al, 2005). The brain was then removed and frozen. Brain sections (thickness, 20 μm) were cut through cortex underlying the cranial window using a cryostat and collected at 100 μm intervals. Sections were examined under an Eclipse E800 fluorescence microscope (Nikon, Melville, NY, USA) equipped with a custom filter set designed to maximize 2-hydroxyethidium fluorescence. Images were acquired with a computer-controlled digital monochrome camera (Coolsnap, Roper Scientific, Trenton, NJ, USA) using the same fluorescence settings in all cases. The analysis of ROS production in the different conditions studied (see Experimental Protocol) was performed in a masked manner using the IPLab software (Scanalytics, Fairfax, VA, USA). After background subtraction of the camera dark current, pixel intensities of the fluorescent signal were quantified. Fluorescent intensities of all sections (15 to 20 per animal) were added, divided by the total number of pixels analyzed, and expressed as relative fluorescence units (RFU) (Park et al, 2005).
Immunocytochemistry and Confocal Microscopy
At the end of the hydroethidine superfusion, mice were deeply anesthetized with 5% isoflurane and perfused transcardially with heparin sulfate (1000 U/mL) followed by 4% paraformaldehyde. Brains were removed and frozen, and the brain area underlying the cranial window was sectioned in a cryostat (thickness, 14 μm). To identify hydroethidine-positive cells, sections were processed for immunocytochemistry for the neuronal marker NeuN (1:100, Chemicon International, Temecula, CA, USA), the astrocytic marker glial fibrillary acidic protein (GFAP, 1:1000, Sigma, St Louis, MO, USA) or the endothelial cell marker CD31 (1:100, BD Biosciences, San Diego, CA, USA). Sections were washed and incubated with cyanine dye (Cy5)-conjugated goat anti-mouse immunoglobulin G (for NeuN and GFAP; Jackson ImmunoResearch, West Grove, PA) and goat anti-rat immunoglobulin G (for CD31; Jackson ImmunoResearch) secondary antibodies. The specificity of the labeling was established by omitting the primary antibody or by preadsorption with the antigen. Images were sequentially acquired using a confocal laser scanning microscope (Leica, Nussloch, Germany). Ethidium and Cy5 signals were pseudocolored red and green, respectively.
Experimental Protocol
Cerebral blood flow recordings and ROS measurements were started after arterial pressure and blood gases were in a steady state (Supplementary Table 1). All pharmacological agents were dissolved in a modified Ringer's solution, unless otherwise indicated.
Effect of Aging on Cerebrovascular Responses in Wild Type and Nox-2-Null Mice: The cranial window was superfused with Ringer's solution, and the CBF increases evoked by whisker stimulation (functional hyperemia) and endothelium-dependent and -independent vasodilators were tested. Cerebral blood flow responses to whisker stimulation were recorded while gently stroking the whiskers with a cotton-tipped applicator for 60 secs. The endothelium-dependent vasodilators acetylcholine (10 mmol/L; Sigma), bradykinin (50 μmol/L; Sigma), or the calcium ionophore A23187 (3 μmol/L; Sigma) were topically superfused for 3 to 5 mins and the evoked CBF increases recorded. Cerebral blood flow responses to the nitric oxide donor S-nitroso-
Effect of MnTBAP or gp91ds-tat: In some experiments, the effect of the ROS scavenger manganic (I-II)meso-tetrakis (4-benzoic acid) porphyrin (MnTBAP, 100 μmol/L; Porphyrin Products, Logna, UT, USA) was studied. The cranial window was superfused with Ringer's solution and the CBF increases evoked by whisker stimulation, acetylcholine, bradykinin, A23187, SNAP, adenosine, or hypercapnia were tested. Next, the superfusion solution was switched to Ringer containing MnTBAP (100 μmol/L) and CBF responses were tested again 30 mins later. In other experiments, we examined the effect of the NADPH oxidase peptide inhibitor gp91ds-tat (Rey et al, 2001) as described previously (Kazama et al, 2004; Park et al, 2005). Cerebrovascular responses were assessed before and 30 to 40 mins after superfusion with gp91ds-tat (1 mmol/L; YGRKKRRQRRRCSTRIRRQL-NH2) or its scrambled control (1 mmol/L; YGRKKRRQRRRCLRITRQSR-NH2; Bio Synthesis, Lewisville, TX, USA).
Reactive Oxygen Species Production: The experimental protocol for these studies was identical to that in which CBF responses were examined. The cranial window was superfused for 60 mins with Ringer's solution containing hydroethidine with or without MnTBAP, gp91ds-tat, or the scrambled peptide. Nox2-deficient mice were also treated with the SOD inhibitor diethyldithiocarbamate (10 mmmol/L; topical application) to test their ability to generate ROS independently of NADPH oxidase. At the end of superfusion, brains were removed and processed for ROS assessment as described above.
Data Analysis
Data in text and figures are expressed as means ± s.d. Two-group comparisons were analyzed by the two-tailed t-test for dependent or independent samples, as appropriate. Multiple comparisons were evaluated by the analysis of variance and Tukey's test. Statistical significance was considered for P < 0.05.
Results
Aging has Profound Effects on Cerebrovascular Regulation
To examine the effect of aging on cerebrovascular reactivity, we compared selected CBF responses in mice aged 3, 12, and 24 months. The increases in CBF evoked by whisker stimulation and by superfusion with acetylcholine (10 μmol/L) or bradykinin (50 μmol/L) were markedly attenuated starting at 12 months of age (Figures 1A to 1C; P < 0.05; n = 5/ group). The CBF response to hypercapnia was reduced only at 24 months (Figure 1G; P < 0.05), whereas responses to A23187 (3 μmol/L), SNAP (50 μmol/L), and adenosine (400 μmol/L) were not altered (Figures 1D to 1F; P > 0.05). Therefore, the increases in CBF evoked by whisker stimulation and by selected endothelium-dependent vasodilators are attenuated in older mice.
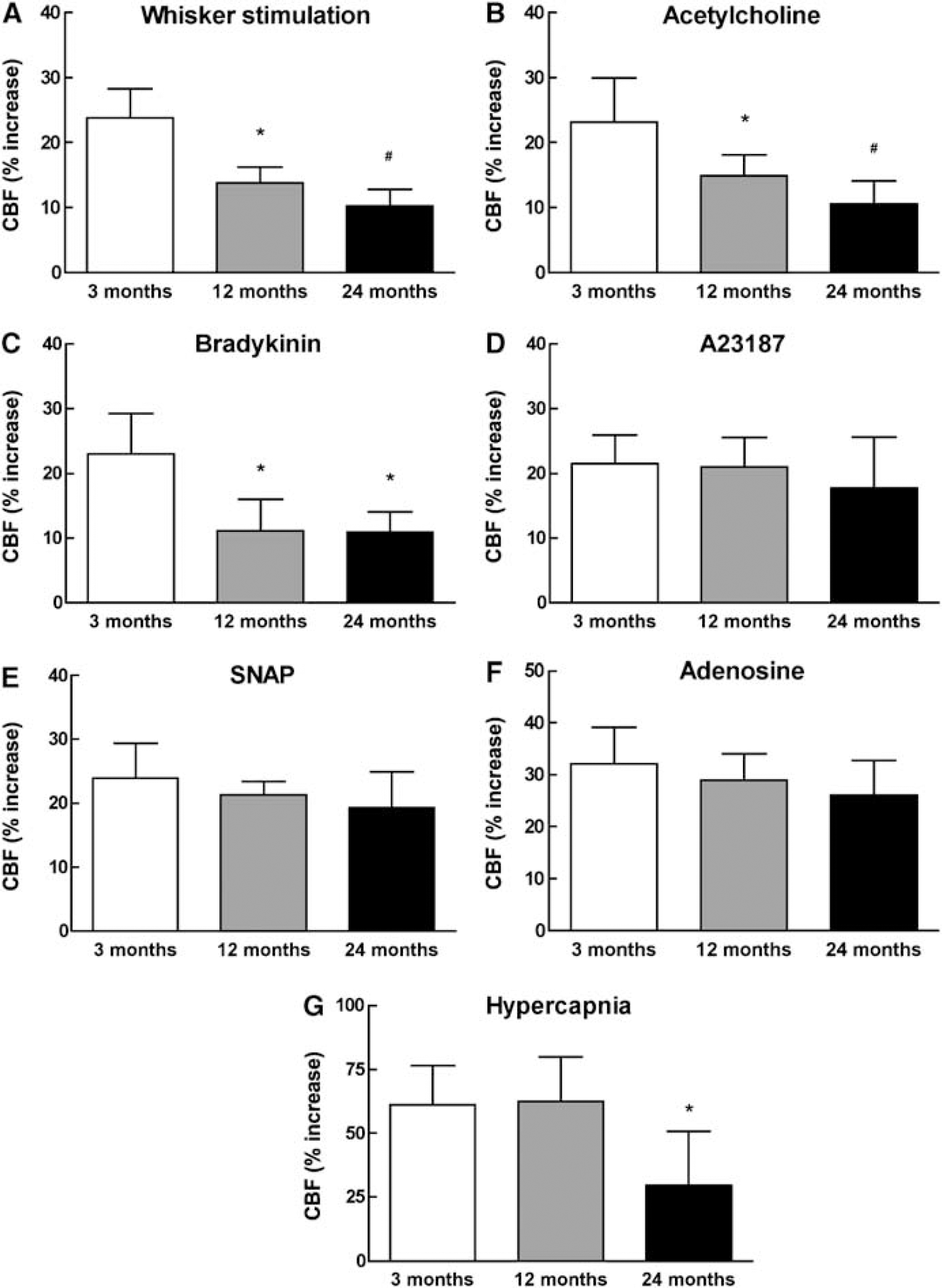
Increase in CBF produced by whisker stimulation (
Advanced Age Leads to Vascular and Neuronal Oxidative Stress
Aging is associated with increased ROS production in brain and other organs (Balaban et al, 2005). To determine whether the cerebrovascular alterations induced by aging were associated with oxidative stress, we used hydroethidine microfluorography. Because the cerebrovascular alterations were well developed at 12 months of age (Figures 1A to 1C), in this and subsequent studies we focused on 12-month-old mice (n = 5/group). First, we compared ROS production in 3- and 12-month-old mice. As illustrated in Figures 2A to 2C, the 12-month-old mice had an increase in fluorescent signal reflecting increased ROS production. To determine the cell type in which ROS production was increased, we immunostained hydroethidine-treated brains with endothelial, astrocytic, and neuronal markers. The increased ROS production was observed in cells positive for the endothelial marker CD31 or the neuronal marker NeuN, but not in GFAP-positive astrocytes (Figures 2A and 2B).
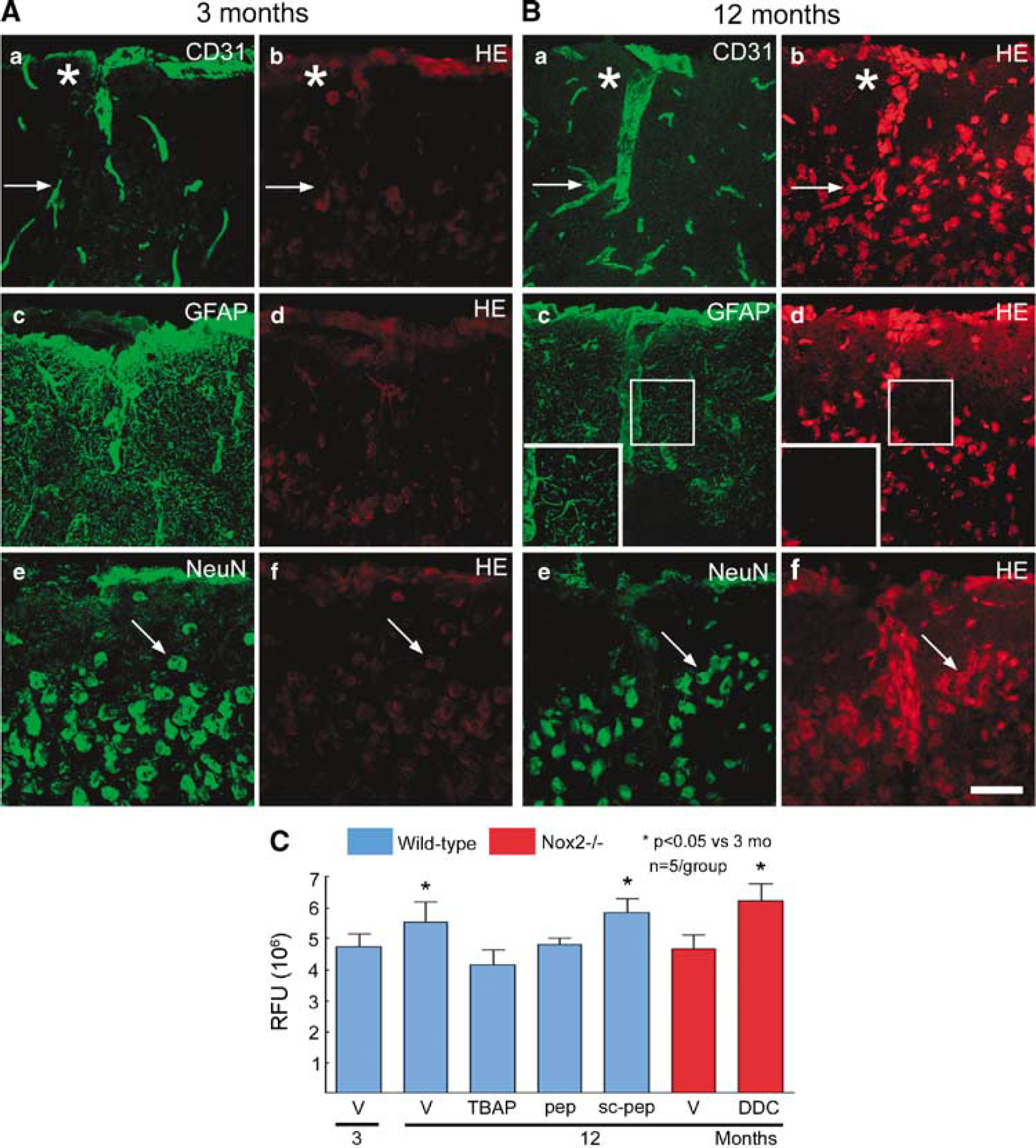
Increase in ROS production in 12-month-old mice. (
The Reactive Oxygen Species Scavenger MnTBAP Rescues the Cerebrovascular Dysfunction in 12-Month-Old Mice
Next, we sought to determine whether the increased ROS production was responsible for the cerebrovascular dysfunction observed in 12-month-old mice. Neocortical superfusion of the ROS scavenger MnTBAP (100 μmol/L) did not alter resting CBF (Figure 3A; n = 5/group; P> 0.05), but it reversed the attenuation of the CBF increases induced by whisker stimulation, acetylcholine, and bradykinin in 12-month-old mice (Figures 3B to 3D; P < 0.05). Responses to A23187, SNAP, adenosine, and hypercapnia were not affected (Figures 3E to 3H; P > 0.05). To determine whether MnTBAP attenuated the ROS increase induced by aging, we assessed ROS production in mice treated with MnTBAP. As illustrated in Figure 2C, MnTBAP blocked the ROS increase observed in 12-month-old mice (n = 5/ group; P > 0.05 from 3-month-old mice). These observations implicate ROS in the impairment of functional hyperemia and endothelium-dependent responses observed in older mice.
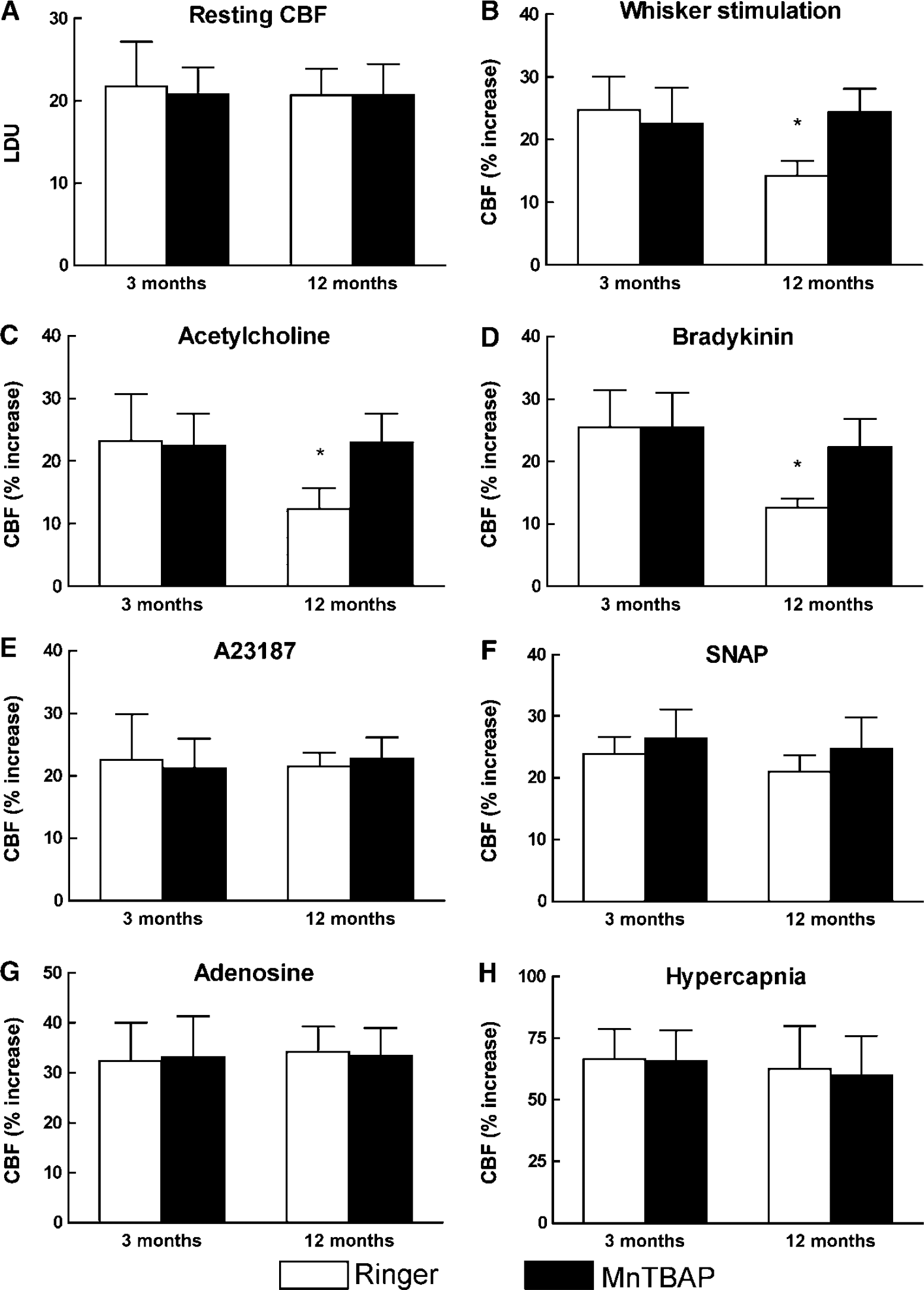
Effect of topical superfusion of MnTBAP (100 μmol/L) on resting CBF (
The NADPH Oxidase Peptide Inhibitor gp91ds-tat Reverses the Cerebrovascular Dysfunction
In these experiments, we used the NADPH oxidase peptide inhibitor gp91ds-tat (Kazama et al, 2004; Park et al, 2005; Rey et al, 2001) to establish whether the age-related alterations in neurovascular function are mediated by NADPH oxidase. In 3-month-old mice, neocortical application of gp91ds-tat (1 μmol/ L; n = 5/group) or its scrambled control (1 μmol/L; n = 5/group) did not alter resting CBF or the cerebrovascular responses studied (Figures 4A to 4H). In 12-month-old mice, gp91ds-tat had no effect on resting CBF and responses to A23187, SNAP, adenosine, and hypercapnia (Figures 4A, 4E to 4H; P > 0.05), but it reversed the age-induced attenuation of the increases in CBF produced by functional hyperemia, acetylcholine, and bradykinin (Figures 4B to 4D; P < 0.05). In contrast, the scrambled peptide failed to reverse these alterations. In parallel studies in which the effect of gp91ds-tat on ROS production was studied, we found that the peptide inhibitor, but not its scrambled control, blocked the increase in ROS observed in 12-month-old mice (Figure 2C).
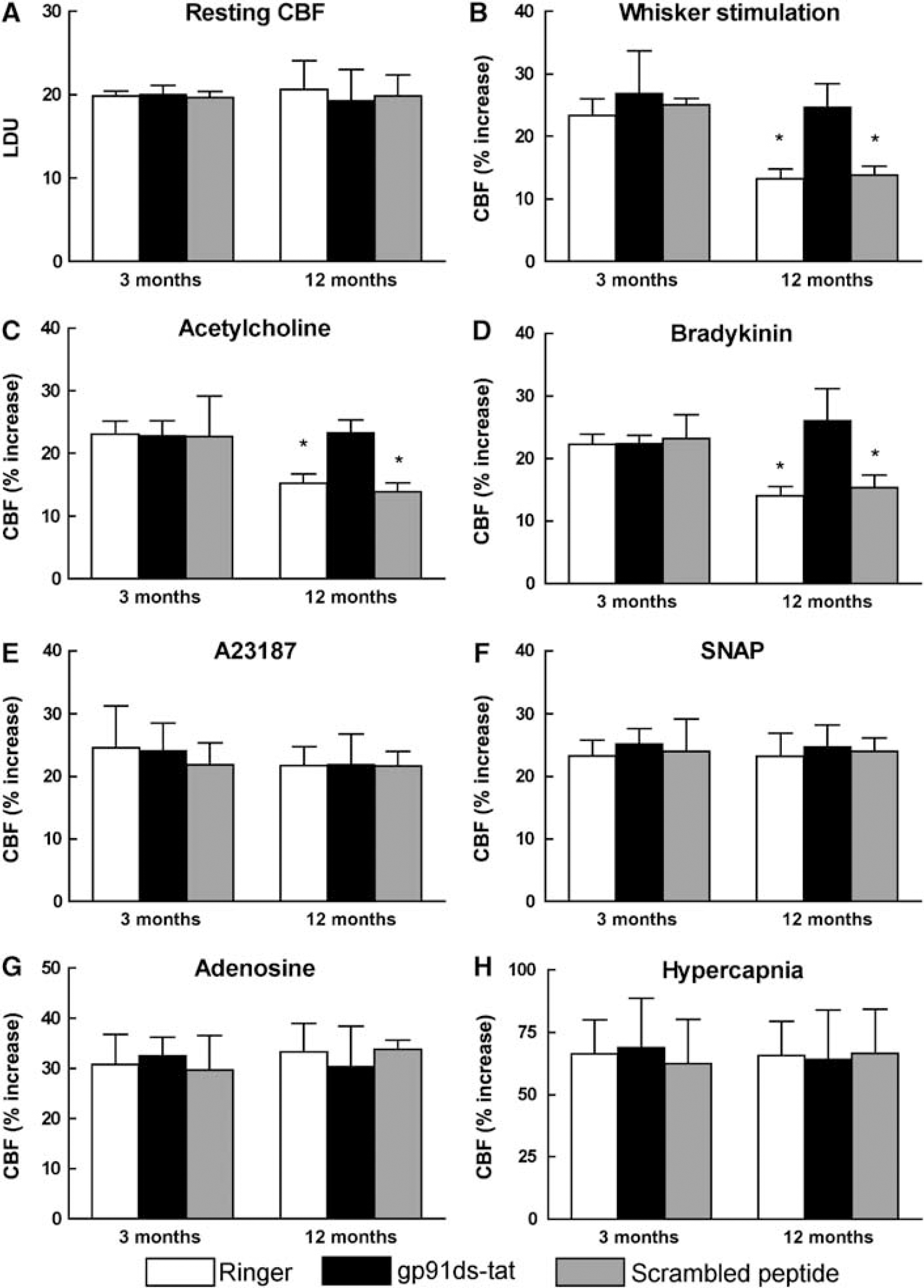
Effect of topical superfusion of gp91ds-tat (1 μmol/L) or its scrambled version (1 μmol/L) on resting CBF (
Nox2-Deficient Mice are Protected from the Cerebrovascular Effects of Advanced Age
To provide further evidence that NADPH oxidase is involved in the cerebrovascular effect of aging, mice lacking the Nox2 (gp91phox) subunit of the enzyme were studied. At 3 months of age, cerebrovascular responses to functional hyperemia and endothelium-dependent or independent relaxing agents did not differ between Nox2 + /+ and Nox2–/– mice (Figures 5A to 5H; P > 0.05; n= 5/group). However, the attenuation in functional hyperemia and in the cerebrovascular responses to acetylcholine and bradykinin observed in 12-month-old Nox2 + /+ mice was not present in Nox2–/– mice (Figures 5B to 5D; P < 0.05). Furthermore, the increase in ROS production was not observed in 12-month-old Nox2–/– mice (Figure 2C). The lack of ROS increase was not a consequence of a global failure in ROS production because superfusion with the SOD inhibitor diethyldithiocarbamate (10 mmol/L) was able to increase ROS in Nox2–/– mice (Figure 2C). Thus, Nox2–/– mice are protected from the oxidative stress and cerebrovascular dysfunction induced by advancing age.
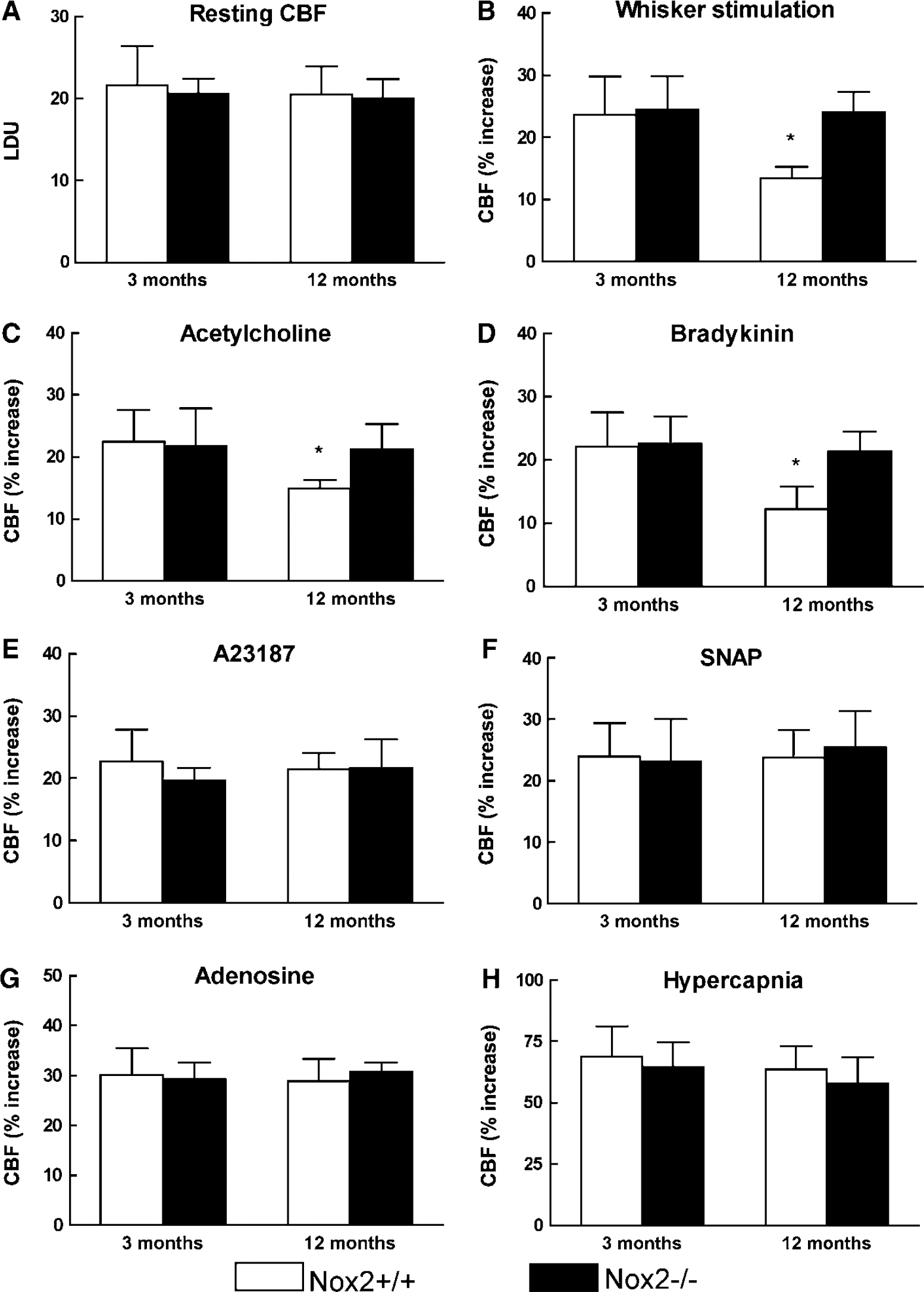
Resting CBF (
Discussion
We investigated the alterations in cerebrovascular regulation induced by aging and we sought to provide an insight into their mechanisms. We found that aging leads to profound alterations in selected aspect of cerebrovascular regulation. Specifically, the increases in CBF induced by topical application of acetylcholine or bradykinin are attenuated, whereas responses to A23187, SNAP, and adenosine are not reduced. In addition, the increase in CBF produced by whisker stimulation is also attenuated by aging. The cerebrovascular effects of aging are well developed at 12 months of age, except for the response to hypercapnia that is reduced at 24 but not at 12 months. These cerebrovascular alterations are associated with vascular and neuronal oxidative stress and are reversed by a ROS scavenger. The age-induced alterations in vascular reactivity are abrogated by a peptide inhibitor of NADPH oxidase, a major source of ROS in vessels and neurons, and are absent in mice lacking the Nox2 subunit of NADPH oxidase. These observations provide the first demonstration that aging impairs not only endothelium-dependent relaxation but also neurovascular coupling, and that these cerebrovascular alterations are mediated by Nox2-derived ROS.
The findings of this study cannot be attributed to differences in arterial pressure or blood gases, factors that have profound effects on CBF (Faraci and Heistad, 1998), because these variables were carefully controlled and did not differ among the groups of mice studied. Furthermore, the observations of this study cannot be attributed to age-related differences in the sensitivity to anesthesia leading to a generalized depression in cerebrovascular reactivity, because only selected CBF responses are attenuated in older mice. Therefore, the reduction in cerebrovascular reactivity observed in older mice is not a result of instability of the preparation or artifacts resulting from anesthesia.
Cerebral blood flow responses to whisker stimulation and acetylcholine were attenuated more in 24-than in 12-month-old mice, whereas the response to bradykinin was attenuated equally at 12 and 24 months. Conversly, the CBF response to hypercapnia was attenuated only in 24-month-old mice and responses to A23187, SNAP, and adenosine were not reduced even at 24 months of age. These differences in the temporal profile of the attenuation suggest that the susceptibility to the effects of aging is not identical for all vascular responses of the cerebrovascular bed. Interestingly, the temporal differences in the attenuation are not related to the mechanism of the vasodilation. For example, the increase in CBF evoked by bradykinin and A23187 is mediated by endothelial COX-1 reaction products (Niwa et al, 2001); yet, the attenuation of the response to bradykinin is maximal at 12 months of age, whereas the response to A23187 is not attenuated even at 24 months. One possible explanation for this discrepancy is that the CBF response to bradykinin depends on activation of bradykinin receptors, whereas the response to A23187 is receptor-independent (Faraci and Heistad, 1998). Therefore, the attenuation of the response to bradykinin could reflect an age-related disruption of bradykinin receptors, as described in the heart (Kintsurashvili et al, 2005). Similarly, a reduction in muscarinic receptors could contribute to the attenuation of the CBF response to acetylcholine. However, in the case of acetylcholine, a reduction in the synthesis and/or bioavailability of nitric oxide could also play a role (van der Loo et al, 2000).
One of the new findings of this study is that aging attenuates the increase in CBF evoked by whisker stimulation. Functional hyperemia is essential for the normal workings of the brain and its impairment leads to brain dysfunction, as observed in models of hypertension and Alzheimer's disease (Kazama et al, 2004; Niwa et al, 2000). Reduced hyperemia during brain activation limits the delivery of energy substrates and oxygen to the active brain and impairs the removal of metabolic byproducts. The impairment in functional hyperemia occurs in concert with alterations of other key regulatory mechanisms of the cerebral circulation, such as responses to endothelium-dependent vasodilators, hypoxia, hypercapnia, and hypotension (Hoffman et al, 1981, 1984, 1982; Sato et al, 2002). These alterations act synergistically to increase the susceptibility of the brain to vascular insufficiency and ischemia, and are likely to play a role in the increased incidence in ischemic stroke and vascular cognitive impairment observed with advancing age (Farkas and Luiten, 2001).
Aging increases the stiffness of cerebral blood vessels, and leads to thinning and loss of endothelial cells as well as tortuosity and narrowing of arterioles and capillaries (Hajdu et al, 1990; Hutchins et al, 1996). Although these morphological changes have been implicated in the cerebrovascular alterations observed with increasing age they are unlikely to play a major role in our model. This is because the cerebrovascular responses tested were not uniformly disrupted. Rather, responses to the endothelium-dependent vasodilator A23187 and responses mediated by agents that act directly on vascular smooth muscles, such as adenosine and SNAP, were not attenuated. Therefore, our data suggest that the altered reactivity is mediated by dysfunction of the vessels rather than mechanical factors deriving from structural alterations.
Reactive oxygen species have long been implicated in the cerebrovascular dysfunction induced by aging (Faraci and Heistad, 1998; Farkas and Luiten, 2001), but their cellular and enzymatic sources have not been defined. We found that 12-month-old mice exhibit an increase in ROS production in the cerebral cortex. Using double-label immunocyto-chemistry with specific cell-type markers, we found that the cellular source of the radicals is primarily neurons and cerebral blood vessels. Surprisingly, increased ROS production was not detected in astrocytes in 12-month-old mice. The reasons for this finding remain unclear, but astrocytes are endowed with greater antioxidant defenses than neurons and, consequently, are more resistant to oxidative stress (Makar et al, 1994; Raps et al, 1989; Savolainen, 1978). However, we cannot rule out the possibility that astrocytic ROS production is increased in mice older than 12 months. Indeed, experiments in cultured astrocytes indicate that with increasing age the astrocytic antioxidant defenses become insufficient, leading to development of oxidative stress (Papadopoulos et al, 1998).
The cerebrovascular alterations observed in 12-month-old mice were completely reversed by the ROS scavenger MnTBAP. This finding is in agreement with reports by others (Brown et al, 2006; Didion et al, 2006) and indicates that oxidative stress is involved in the attenuation of cerebrovascular responses induced by aging. We then investigated the enzymatic source of ROS focusing on NADPH oxidase, a superoxide-generating enzyme that has been implicated in the cerebrovascular dysfunction induced by other conditions associated with oxidative stress (Fang et al, 2006; Kazama et al, 2004; Miller et al, 2006; Park et al, 2005). We found that inhibition of NADPH oxidase attenuates ROS production and normalizes the cerebrovascular alterations observed in 12-month-old mice. Furthermore, mice lacking Nox2 were protected from the oxidative stress and the resulting alterations in cerebrovascular function. These findings provide evidence that Nox2-containing NADPH oxidase is the source of the ROS mediating the cerebrovascular dysfunction observed in 12-month-old mice.
Recent studies have examined the role of NADPH oxidase in the oxidative stress observed in the heart and systemic blood vessels during aging. However, these studies have been largely inconclusive. Some reports have described an increase in NADPH oxidase subunits in systemic blood vessels (Adler et al, 2003; Hamilton et al, 2001; Oudot et al, 2006), whereas others have not (Bachschmid et al, 2004; Csiszar et al, 2002; Newaz et al, 2006). Other studies have used nonspecific pharmacological inhibitors, such as DPI or apocyanin, to assess the contribution of NADPH oxidase to ROS production and/or cardiovascular dysfunction (Adler et al, 2003; Csiszar et al, 2002; Hamilton et al, 2001). In this study, using both a peptide inhibitor and Nox2-null mice we have been able to provide, for the first time, converging evidence that a Nox2-containing NADPH oxidase is a key factor in the cerebrovascular dysfunction produced by aging. However, we cannot rule out that NADPH oxidase-derived ROS serve to ‘prime’ oxidative stress by activating other ROS-generating systems, such as mitochondrial enzymes or xanthine oxidase (Brandes, 2005; Newaz et al, 2006).
In conclusion, we have showed that aging, in addition to its well-established effects on endothelium-dependent vasodilation, induces profound changes in neurovascular coupling. These alterations are mediated by ROS derived from a Nox2-containing NADPH oxidase in neurons and cerebral blood vessels. The data identify, for the first time, Nox2 as a key source of ROS mediating the alterations in vascular reactivity induced by advancing age. Modulation of Nox2-derived ROS may provide a new strategy to counteract the deleterious consequences of the cerebrovascular dysfunction induced by aging.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.