
Abstract
Normobaric hyperoxia (NBO) has been shown to extend the reperfusion window after focal cerebral ischemia. Employing diffusion (DWI)- and perfusion (PWI)-weighted magnetic resonance imaging (MRI), the effect of NBO (100% started at 30 mins after middle cerebral artery occlusion (MCAO)) on the spatiotemporal evolution of ischemia during and after permanent (pMCAO) and transient suture middle cerebral artery occlusion (tMCAO) was investigated (experiment 3). In two additional experiments, time window (experiment 1) and cell death pathways (experiment 2) were investigated in the pMCAO model. In experiment 1, NBO treatment reduced infarct volume at 24 h after pMCAO by 10% when administered for 3 h (P > 0.05) and by 44% when administered for 6 h (P < 0.05). In experiment 2, NBO acutely (390 mins, P < 0.05) reduced in situ end labeling (ISEL) positivity in the ipsilesional penumbra but increased contralesional necrotic as well as caspase-3-mediated apoptotic cell death. In experiment 3, CBF characteristics and CBF-derived lesion volumes did not differ between treated and untreated animals, whereas the apparent diffusion coefficient (ADC)-derived lesion volume essentially stopped progressing during NBO treatment, resulting in a persistent PWI/DWI mismatch that could be salvaged by delayed (3 h) reperfusion. In conclusion, NBO (1) acutely preserved the perfusion/diffusion mismatch without altering CBF, (2) significantly extended the time window for reperfusion, (3) induced lasting neuroprotection in permanent ischemia, and (4) although capable of reducing cell death in hypoperfused tissue it also induced cell death in otherwise unaffected areas. Our data suggest that NBO may represent a promising strategy for acute stroke treatment.
Introduction
Thrombolysis with recombinant tissue plasminogen activator remains the only approved therapy for acute ischemic stroke but its use is substantially limited to less than 5% of stroke patients owing to numerous contraindications to treatment and most importantly a narrow therapeutic time window (Wardlaw et al, 2003). However, it is well recognized that early reperfusion leads to smaller ischemic lesion volumes and good clinical recovery in humans suffering from acute ischemic stroke (Christou et al, 2000). Promising strategies to extend the treatment window include rigorous patient selection based on imaging criteria, intraarterial thrombolysis, combined intravenous and intraarterial thrombolysis, and endovascular approaches to reperfusion (Furlan et al, 2006; Khatri et al, 2005; Smith et al, 2005). However, these approaches have the common disadvantage that they require patient transport to a suitable hospital for advanced neuroimaging before treatment.
Tissue hypoxia plays a critical role in the primary and secondary events leading to cell death after cerebral ischemia (Zauner et al, 2002). Therefore, improving brain tissue oxygenation is a logical and important stroke treatment strategy to delay the transition of ischemia to infarction (‘buying time’). However, the concept of brain injury due to reperfusion (Aronowski et al, 1997) and the theoretical potential for exacerbating damage due to the detrimental tissue effects of oxygen-derived free radicals caused considerable concern and uncertainty for oxygen use (Kent et al, 2001; Sugawara and Chan, 2003). Oxygen therapy can enhance free radical formation that may promote lipid peroxidation, blood–brain barrier integrity breakdown, glutamate-induced excitotoxic cell death, neuroinflammation, and increase brain hemorrhage, particularly when combined with reperfusion (Aronowski et al, 1997; Dubinsky et al, 1995; Elayan et al, 2000; Kent et al, 2001; Kim et al, 2005; Mink and Dutka, 1995). Nevertheless, several experimental studies showed impressive reductions in stroke lesion volumes with normobaric hyperoxia (NBO) (Flynn and Auer, 2002; Kim et al, 2005; Miyamoto and Auer, 2000; Singhal et al, 2002a, b). Most importantly, hyperoxia salvaged ischemic brain tissue without increasing the risk of oxygen free-radical injury (Kim et al, 2005; Singhal et al, 2002b). Although these studies indicated that normobaric hyperoxia may be a safe and effective stroke therapy, the mechanisms of neuroprotection remain largely unclear.
In this study, we sought to evaluate the neuroprotective potential of NBO when administered for different durations after permanent middle cerebral artery occlusion (pMCAO) in the rat. Concomitantly, we assessed cell death pathways that are affected by oxygen treatment, with in situ end labeling (ISEL) and cleaved caspase-3. Lastly, repetitive in vivo diffusion (DWI)- and perfusion (PWI)-weighted magnetic resonance imaging (MRI) was employed to monitor the effect of NBO on the spatiotemporal lesion evolution and treatment effectiveness during and after pMCAO as well as 3 h of temporary suture MCAO (tMCAO).
Materials and methods
Animal Preparation
All procedures used in this study were performed in accordance with our institutional guidelines and all experiments were performed in a masked, randomized manner. Spontaneously breathing male Sprague–Dawley rats (n = 72, Taconic Farms, Hudson, NY, USA) weighing 270 to 330 g were anesthetized with isoflurane (5% for induction, 2% for surgery, 1.2% for maintenance) in room air. PE-50 polyethylene tubing was inserted into the femoral artery for monitoring of mean arterial blood pressure and for obtaining blood samples to measure blood gases (pH, PaO2, PaCO2), electrolytes (Na+, K+, Ca2+), and plasma glucose before as well as 30, 60, 120, 180, 240, 300, and 360 mins after MCAO. Note that the last two time points were examined exclusively in animals subjected to 6 h NBO or air treatment. Body temperature was monitored continuously with a rectal probe and maintained at 37.0°C ± 0.5°C with a thermostatically controlled heating lamp.
Focal cerebral ischemia was produced by intraluminal suture occlusion of the right middle cerebral artery using a 4–0 silicone-coated nylon filament for pMCAO (Meng et al, 2004) or a 3–0 uncoated nylon filament for in-bore reperfusion at 3 h (Li et al, 1998a). Neurologic evaluation was performed at 24 h, as previously described (Menzies et al, 1992).
Study Design
The Study Consisted of Three Different Experiments: Experiment 1 aimed to investigate the neuroprotective potential of 100% NBO when administered for different durations after pMCAO. Infarct volume was defined at 24 h after MCAO using 2,3,5-triphenyltetrazolium chloride (TTC) staining with edema correction (Meng et al, 2004). To correct for the effects of brain edema, a corrected infarct volume was calculated by the following formula: corrected infarct volume — left hemisphere volume — (right hemisphere volume — infarct volume).
Animals remained anesthetized during the first 390 mins of the experiment and received 100% oxygen or room air via a face mask at a flow rate of 2 L/min. Rats were randomized to one of the following groups: start of 6 h NBO at 30 mins after MCAO (control, n = 8), start of 3 h NBO at 30 mins after MCAO followed by 3 h of room air (3 h NBO, n = 8), and 6 h of room air after MCAO (6 h NBO, n = 8).
In experiment 2, potential differences in the mechanism and severity of cell death between treatment (3 h NBO) and control (room air) animals were assessed. Control and NBO rats were subjected to permanent ischemia, and brains were removed at 210 mins, 390 mins, and 24 h (n = 3 per group and time point) as well as at 30 mins (n = 3). The brains of three nonstroke animals served as negative controls.
In experiment 3, the in vivo effects of NBO on apparent diffusion coefficient (ADC) and cerebral blood flow (CBF) characteristics as well as the spatiotemporal evolution of the ischemic lesion in light of permanent (experiment 3a, n = 12) or delayed (3 h after MCAO) reperfusion (experiment 3b, n = 12) were evaluated. Immediately after occlusion, the animals were placed into the magnet. Normobaric hyperoxia was started at 30 mins after MCAO and maintained for 3 h. Control animals received room air. After the last MR-scan at 24 h after MCAO, animals were electively euthanized under pentobarbital anesthesia. The brains were removed and sectioned coronally into seven 1.5-mm-thick slices corresponding to the MR slices and stained with TTC.
All histological analyses were conducted by authors who were masked to the experimental protocol (TTC by JB, ISEL and caspase-3 by JN).
Magnetic Resonance Imaging Measurements
Magnetic resonance imaging experiments were performed on a 4.7 T/40 cm horizontal magnet equipped with a Biospec Bruker console (Billerica, MA, USA), and a 20 G/cm gradient insert (inner diameter = 12 cm, 120 μs rise time). A surface coil (inner diameter = 2.3 cm) was used for brain imaging and an actively decoupled neck coil for perfusion labeling (Meng et al, 2004). Animals were imaged at 20 (baseline), 45, 60, 90, 120, 150, 180, 210, 240, and 270 mins, as well as 1 day after MCAO. Three ADC maps were separately acquired with diffusion-sensitive gradients applied along the x, y, or z direction (Meng et al, 2004). Single shot, echo-planar images (EPI) were acquired over 2.5 mins with matrix = 64 × 64, spectral width = 200 kHz, TR = 2 secs (90° flip-angle), TE = 37.5 msecs, b = 8 and 1300 secs/mm2, Δ = 24 msecs, δ = 4.75 msecs, field of view (FOV) = 2.56 × 2.56 cm, seven 1.5 mm slices, and 16 averages. Quantitative CBF measurements were made using the continuous arterial spin-labeling technique with single-shot, gradient-echo, echo-planar images acquisition (Henninger et al, 2006b). Sixty paired images (for signal averaging) were acquired over 4 mins, alternately, one with arterial spin labeling and the other (control) without spin-labeling preparation. The MRI parameters were similar to ADC measurements except TE = 13.5 msecs. Arterial spin labeling utilized a 1.78-second, square radiofrequency pulse in the presence of 1.0 Gauss/cm gradient along the flow direction. The sign of the frequency offset was switched for nonlabeled images. Lastly, time of flight (TOF)-magnetic resonance angiography (MRA) was performed, using a three-dimensional gradient echo sequence with matrix = 256 × 256 × 64, FOV = 3.03 × 2.68 × 2.00 cm, TR = 16 msecs, TE = 5.6 msecs, pulse angle = 25°, and 1 average (imaging time ∼4.3 mins).
Calculation of In Vivo Lesion Size
Images were analyzed using MATLAB (MathWorks Inc., Natick, MA, USA), STIMULATE (University of Minnesota) and QuickVol II (http://www.quickvol.com/) (Schmidt et al, 2004). Quantitative CBF and ADC maps and their corresponding threshold-derived lesion volumes were calculated, as described previously (Meng et al, 2004). The viability thresholds were 0.53 × 10−3 mm2/sec for ADC and 0.3 mL/g per min for CBF (Meng et al, 2004). Mean intensity projection analysis was used to qualitatively assess arterial occlusion.
Region of Interest Analysis
Two ROIs (each 4 × 4 pixels) were manually defined at all imaging time points in the ipsilesional hemisphere on the initial ADC and CBF maps that were coregistered with the TTC results, and compared with corresponding contralesional areas. The position of these ROIs remained constant over time and described two possible tissue compartments (Henninger et al, 2006b) (1) core defined by initially (20 mins) reduced ADC values below the critical threshold, a corresponding TTC abnormality, and CBF values below the critical threshold, and (2) mismatch defined by initial (20 mins) ADC values above the critical threshold and reduced CBF values below the critical threshold. Because NBO-treated rats subjected to transient MCAO showed persistent preservation of mismatch tissue up to 24 h, the definition of the mismatch ROIs was solely based on the ADC and CBF maps.
Histology
Animals were euthanized by an overdose of pentobarbital (200 mg/kg, intraperitoneal) followed by perfusion with phosphate-buffered saline (PBS) and 4% paraformaldehyde (hematoxylin and eosin stain (H&E) and immunohistochemistry). The cerebrum was removed, placed in 4% paraformaldehyde for 4 h and stored in 30% sucrose in PBS at 4°C until further processing. The cerebrum was then sliced into 16 μm coronal brain sections. DNA fragmentation as a marker of overall cell death was ascertained by ISEL staining according to the protocol developed by Wijsman et al (1993). Sections were treated with Pronase E (1 g/mL; VWR International, Bridgeport, NJ, USA) and incubated with DNA polymerase I (50 μg/mL; Fisher Scientific, Pittsburgh, PA, USA), biotin-21-2-deoxyuridine 5-triphosphate (10 μmol/L; BD Biosciences, San Jose, CA, USA), and deoxyadenosine triphosphate, deoxycytidine triphosphate, and deoxyguanosine triphosphate (10 μmol/L; Fisher Scientific). Vectastain Elite ABC Kit was used for amplification (Vector Laboratories, Burlingame, CA, USA) and DAB peroxidase substrate kit (Vector Laboratories) was used for detection of DNA fragmentation. Caspase-mediated apoptotic cell death was evaluated by staining for cleaved caspase-3 (Asp 175). Sections were first treated with blocking solution, 10% normal horse serum in PBS with 0.1% triton to block nonspecific staining and to permeabilize the tissue. Then, sections were treated with caspase-3 antibody (Cell Signaling Technology, Inc., Danvers, MA, USA; polyclonal immunoglobulin G, 1:100 in blocking solution) for 1 h and were rinsed three times for 20 mins with PBS. Next, sections were treated with fluorescent conjugated anti-rabbit immunoglobulin G secondary (Molecular Probes, Carlsbad, CA, USA; 1:1000 in blocking solution) for 1 h and rinsed three times for 20 mins with PBS. Nuclei were labeled with 4′-6-diamidino-2-phenylindole (Sigma, St Louis, MO, USA; 1:1000 in blocking solution). Ipsilesional ROI delineating ischemic core and penumbral tissues as well as spatially corresponding contralesional ROIs (0.28 × 0.22 mm) were analyzed at × 40. To facilitate coregistration of the MR-defined ROIs on the histological slices, ROIs were first positioned on H&E-stained coronal sections (Bregma −1 mm) with core ROIs located in the caudate putamen, and penumbral ROIs in the sensorimotor cortex. Care was taken that ROIs were then positioned in the same location on H&E, ISEL as well as caspase-3-stained sections across animals. Lastly, since the ISEL procedure stains predominantly apoptotic (and to a lesser degree necrotic) cells (Wijsman et al, 1993), H&E staining was used to exemplarily confirm cell death. Briefly, neurons were classified as necrotic when they exhibited pyknosis, karyorrhexis, karyolysis, cytoplasmic eosinophilia (‘red neuron’), or loss of affinity for hematoxylin (‘ghost neuron’) (Li et al, 2000). Apoptotic neurons were identified by the criteria of Li et al (1998b), that is, via the presence of membrane-bound apoptotic bodies of roughly round or ovoid shape. All histological analyses were performed by an investigator masked to the animal groups (JN).
Statistical Analysis
Data are presented as mean ± s.d. unless otherwise stated. Statistical comparisons (Sigma-Stat 3.1, SPSS, Chicago, IL, USA) were performed using analysis of variance with post hoc Holm–Sidak or Dunn's test for multiple comparisons and two-tailed paired or unpaired Student's t-test, where appropriate. P < 0.05 was considered significant.
Results
Physiologic Parameters and Neurological Score
There were no differences in physiological parameters between corresponding groups of the three experiments (data not shown). To document the changes in physiological variables, data are provided exclusively for experiment 3 (Table 1). Blood gases, electrolytes, and pH did not differ between time points and groups. In accordance with previously reported data, 100% NBO increased PaCO2 and PaO2 levels compared with baseline values as well as compared with controls (Table 1), the former presumably via induction of mild hypoventilation (Sicard and Duong, 2005). Interestingly, mean arterial blood pressure of NBO-treated animals was significantly elevated at 120 and 180 mins compared with controls. Lastly, in both NBO and control animals, glucose levels were significantly higher at baseline compared with subsequently obtained values. Compared with controls, the 24 h neurological scores were improved in all NBO groups (Table 2).
Physiological parameters obtained from experiment 3 (MRI)
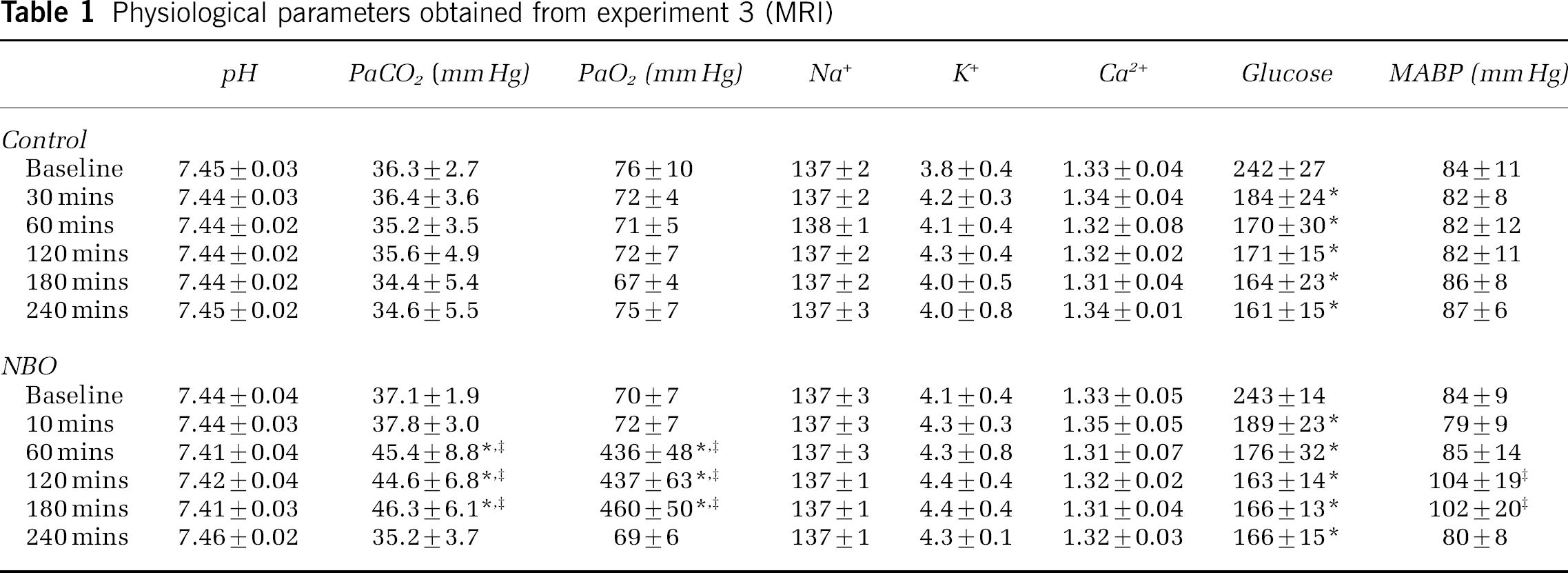
There was no difference between transient and permanent middle cerebral artery occlusion experiments, and thus, data were pooled (n = 12 per time point). Electrolytes and pH did not differ between time points and groups throughout the study. Mean arterial blood pressure (MABP) of normobaric hyperoxia (NBO)-treated animals was significantly elevated at 120 and 180 mins compared with room air-treated controls (control). PaCO2 and PaO2 levels were significantly higher in the NBO group compared with baseline values as well as compared with controls. Lastly, in both NBO and control animals, glucose levels were significantly higher at baseline compared to subsequently obtained values.
P < 0.05 versus baseline
P < 0.05 versus control.
Neuroscores at 24 h
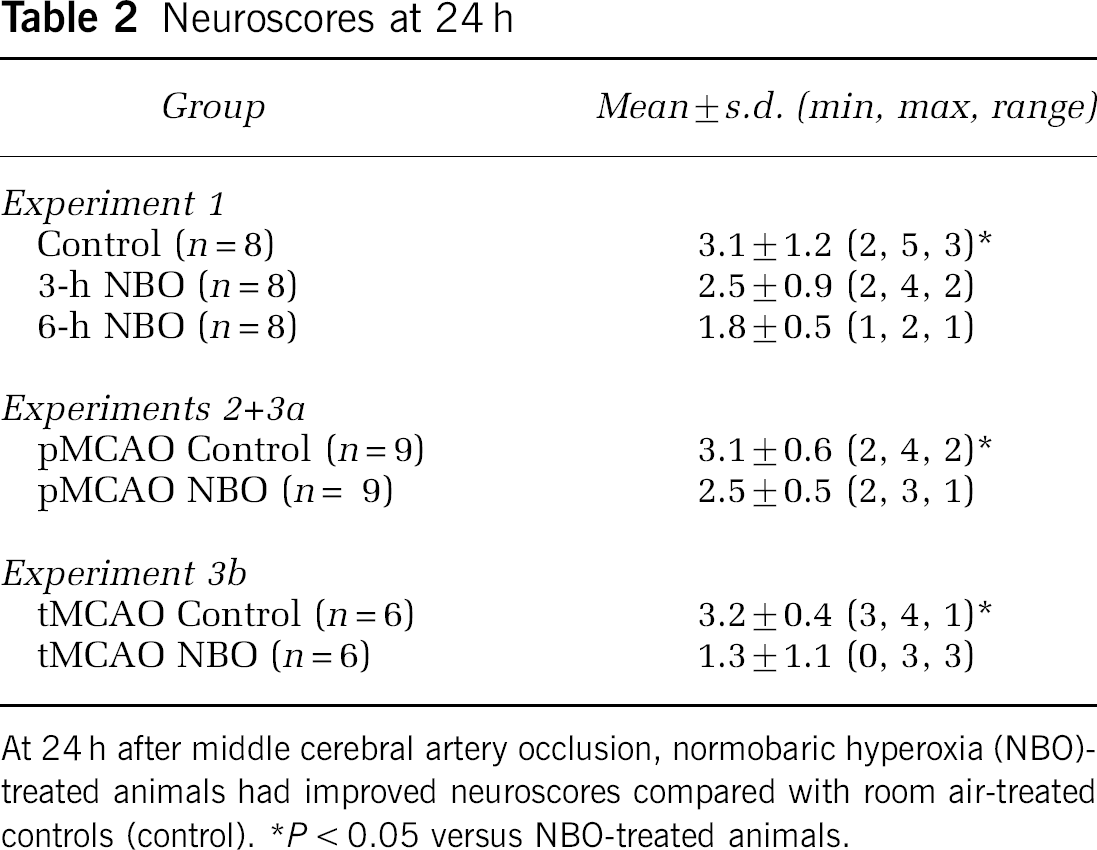
At 24 h after middle cerebral artery occlusion, normobaric hyperoxia (NBO)-treated animals had improved neuroscores compared with room air-treated controls (control).
P < 0.05 versus NBO-treated animals.
Infarct Volume and Survivability
No animal died prematurely. In experiment 1 (Figure 1), infarct volumes derived by post-mortem TTC staining at 24 h were 214 ± 50 mm3 (room air-treated controls) versus 193 ± 50 mm3 (3-h NBO) and 120 ± 81 mm3 (6-h NBO, P < 0.05). In the experiment 3a (Figure 3A), infarct volumes were 258 ± 55 mm3 (control) versus 232 ± 96 mm3 (NBO, P = 0.578), and in experiment 3b (Figure 3B) infarct volumes were 235 ± 42 mm3 (control) versus 64 ± 45 mm3 (NBO, P < 0.001). With the exception of four control animals (67%) subjected to 3-h reperfusion that showed intracerebral microhemorrhages, no cerebral hemorrhages were observed at 24 h after MCAO (data not shown).
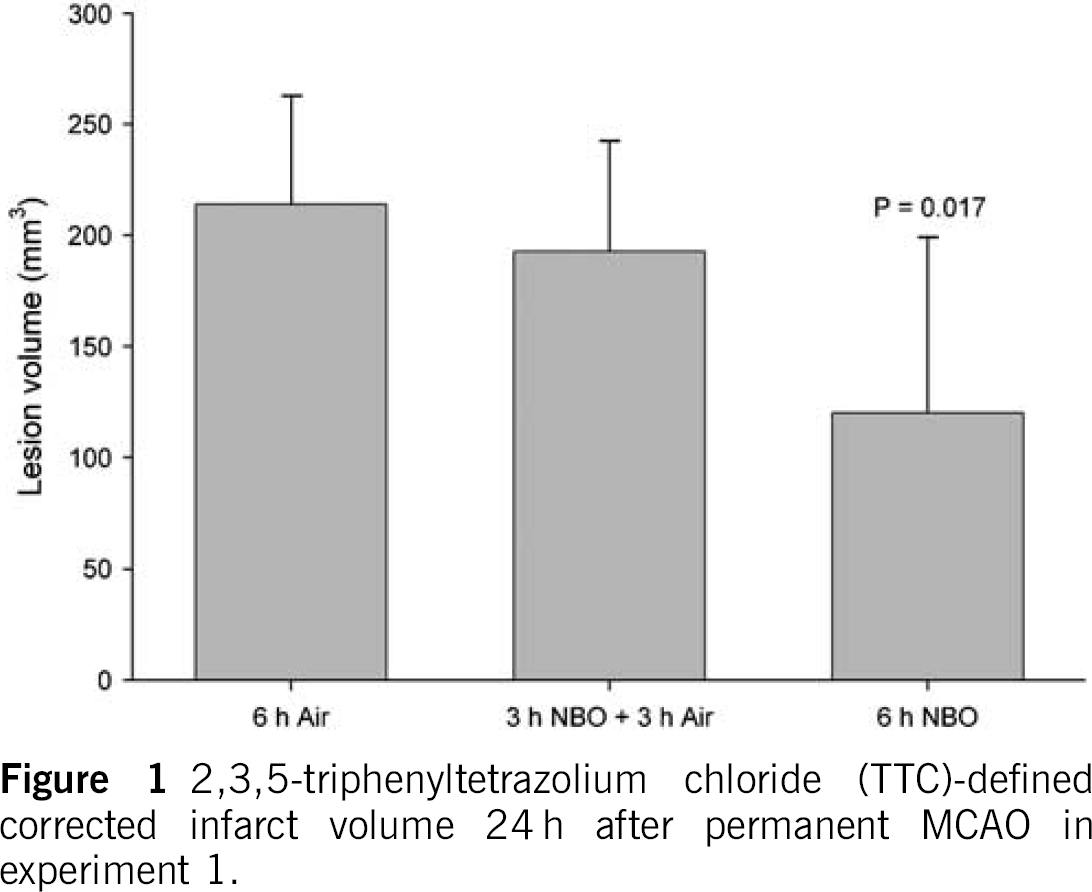
2,3,5-triphenyltetrazolium chloride (TTC)-defined corrected infarct volume 24 h after permanent MCAO in experiment 1.
Histology
Histological data from rats subjected to pMCAO (experiment 2) are shown in Table 3 and Figure 2. Standard H&E-staining showed lesions similar in location to those observed on MRI (experiment 3, data not shown).
Immunohistological results from experiment 2
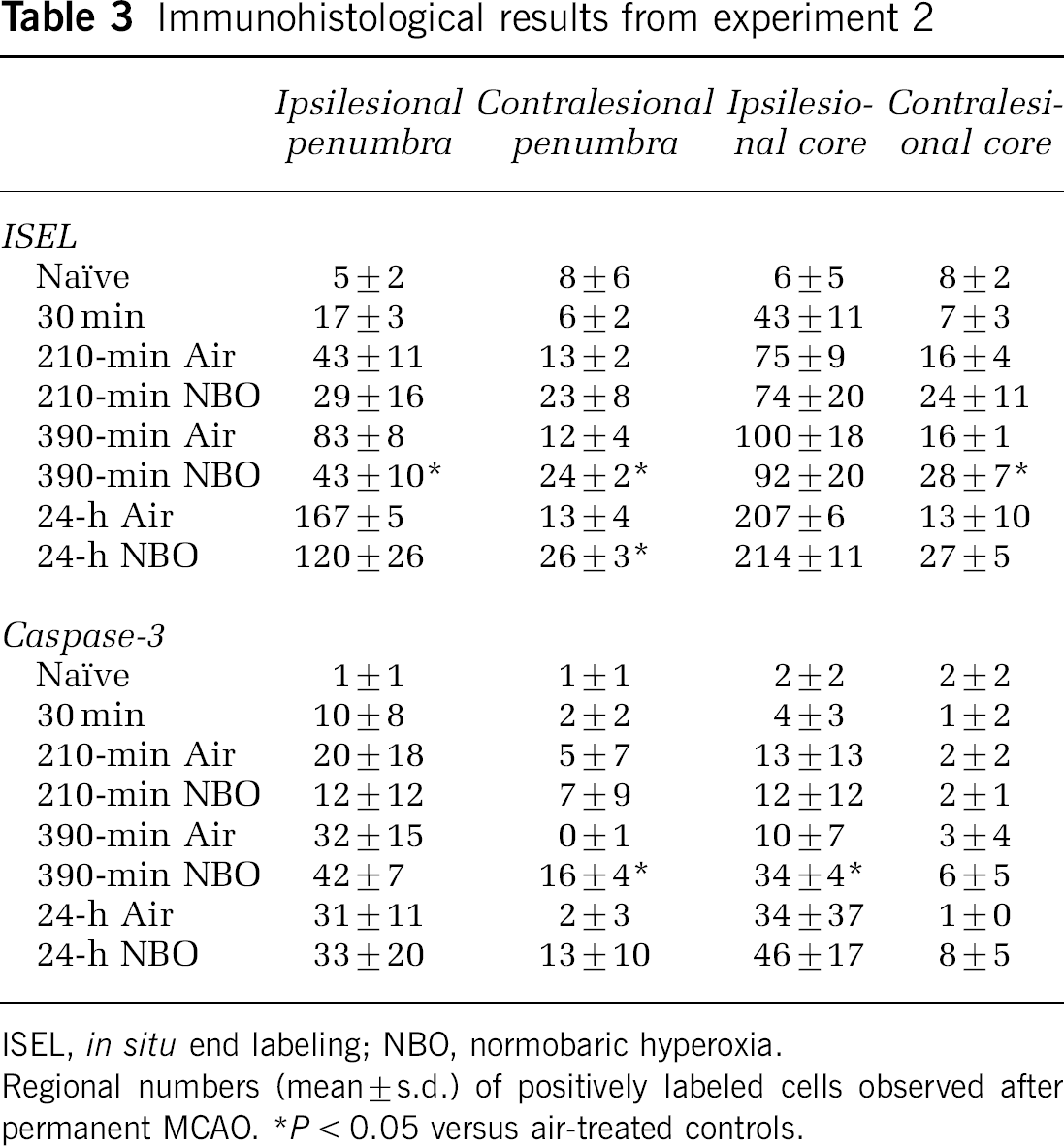
ISEL, in situ end labeling; NBO, normobaric hyperoxia.
Regional numbers (mean ± s.d.) of positively labeled cells observed after permanent MCAO.
P < 0.05 versus air-treated controls.
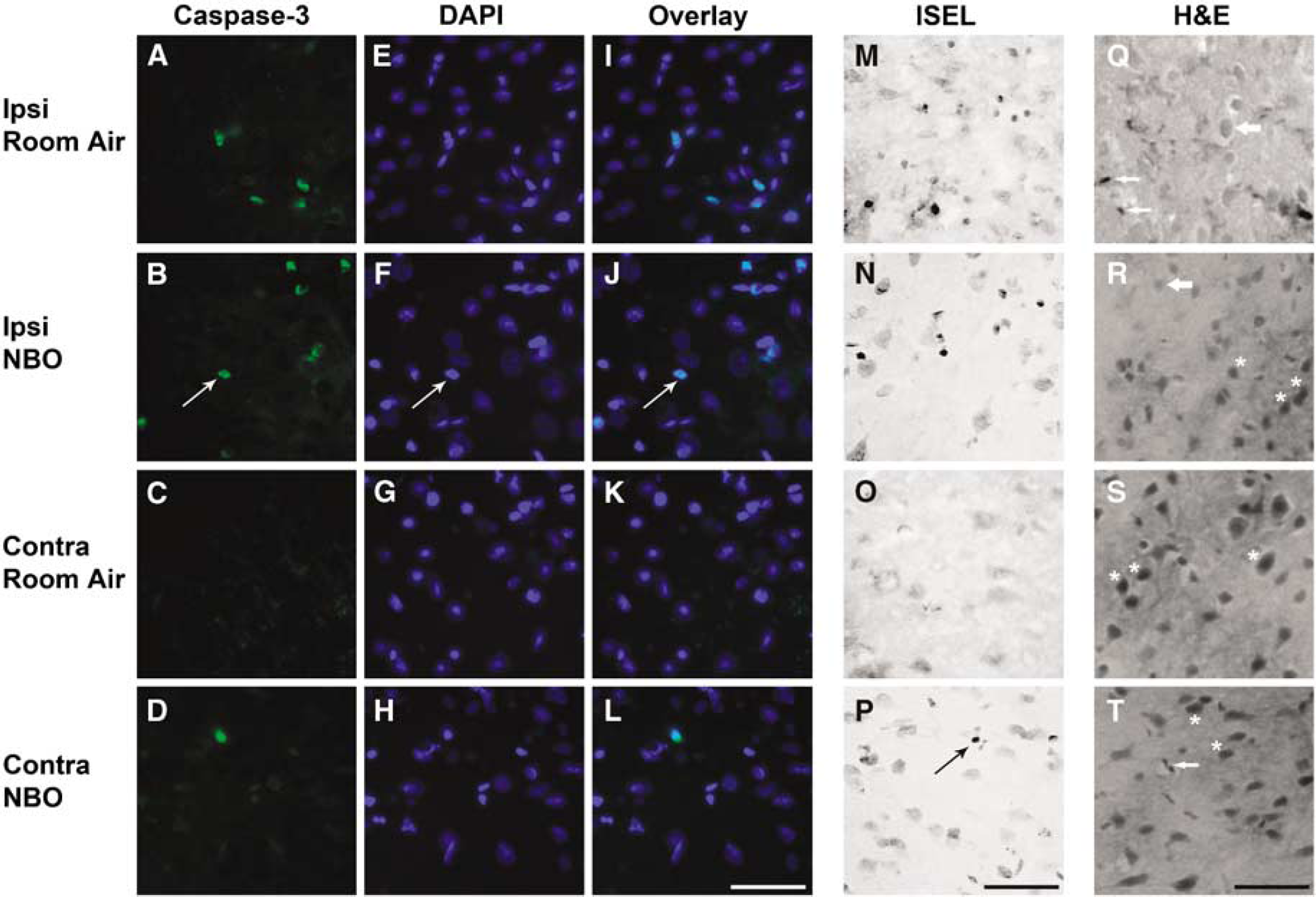
Cell death in penumbral ROIs at 390 mins after permanent middle cerebral artery occlusion (MCAO). Photomicrographs of cleaved caspase-3 (Caspase-3,
Relative to room air treatment, NBO acutely (210 and 390 mins) reduced the number of ISEL-positive cells in the ipsilesional penumbra (P < 0.05 at 390 mins) but not the ipsilesional core. However, at 210 mins to 24 h NBO-treated animals had approximately twice as many ISEL-positive cells as controls in the contralateral, nonischemic ROIs corresponding to penumbra (P < 0.05 at 390 mins and 24 h) and core (P < 0.05 at 390 mins). Importantly, in ipsilesional penumbral ROIs (NBO and control groups) as well as contralesional, nonischemic ROIs (NBO group only), H&E-staining showed isolated necrotic and/or apoptotic neurons that were surrounded by normal-appearing neuropil, glia, and endothelium, suggestive of selective neuronal death (i.e., incomplete infarction). The severity of these histopathological changes correlated positively with the number of ISEL-labeled cells (data not shown) suggesting that the latter stain correctly identified apoptotic and necrotic cell death.
The number of cleaved caspase-3-positive cells in ipsilesional ROIs did not differ significantly (with the exception of the core at 390 mins, P < 0.05) between treated and untreated animals. Compared with room air treatment, NBO increased caspase-3 positivity in the nonischemic hemisphere ROIs corresponding to the penumbra (P < 0.05 at 390 mins). Although a similar effect was observed in the nonischemic ROIs corresponding to the core, this effect did not reach statistical significance.
Spatiotemporal Evolution of Apparent Diffusion Coefficient and Cerebral Blood Flow-Derived Lesion Volumes and Correlation with 2,3,5-Triphenyl-tetrazolium Chloride-Derived Infarct Volumes
Figure 3 summarizes the spatiotemporal evolution of threshold-derived ADC and CBF lesion volumes. In both experiments (permanent and transient MCAO), the CBF lesion volume during occlusion did not differ between groups (NBO versus room air) and remained relatively constant over time. In contrast to control animals, ADC-derived lesion volume stopped growing after the start of NBO and did not increase during treatment. The DWI/PWI mismatch was significant at all time points in NBO-animals (P < 0.05) and up to 45 mins (tMCAO) and 60 mins (pMCAO) after stroke onset in controls (P < 0.05), respectively. The TTC-defined infarct size did not differ between permanent MCAO groups and approximated the 3-h CBF and ADC lesion volumes (P > 0.05).
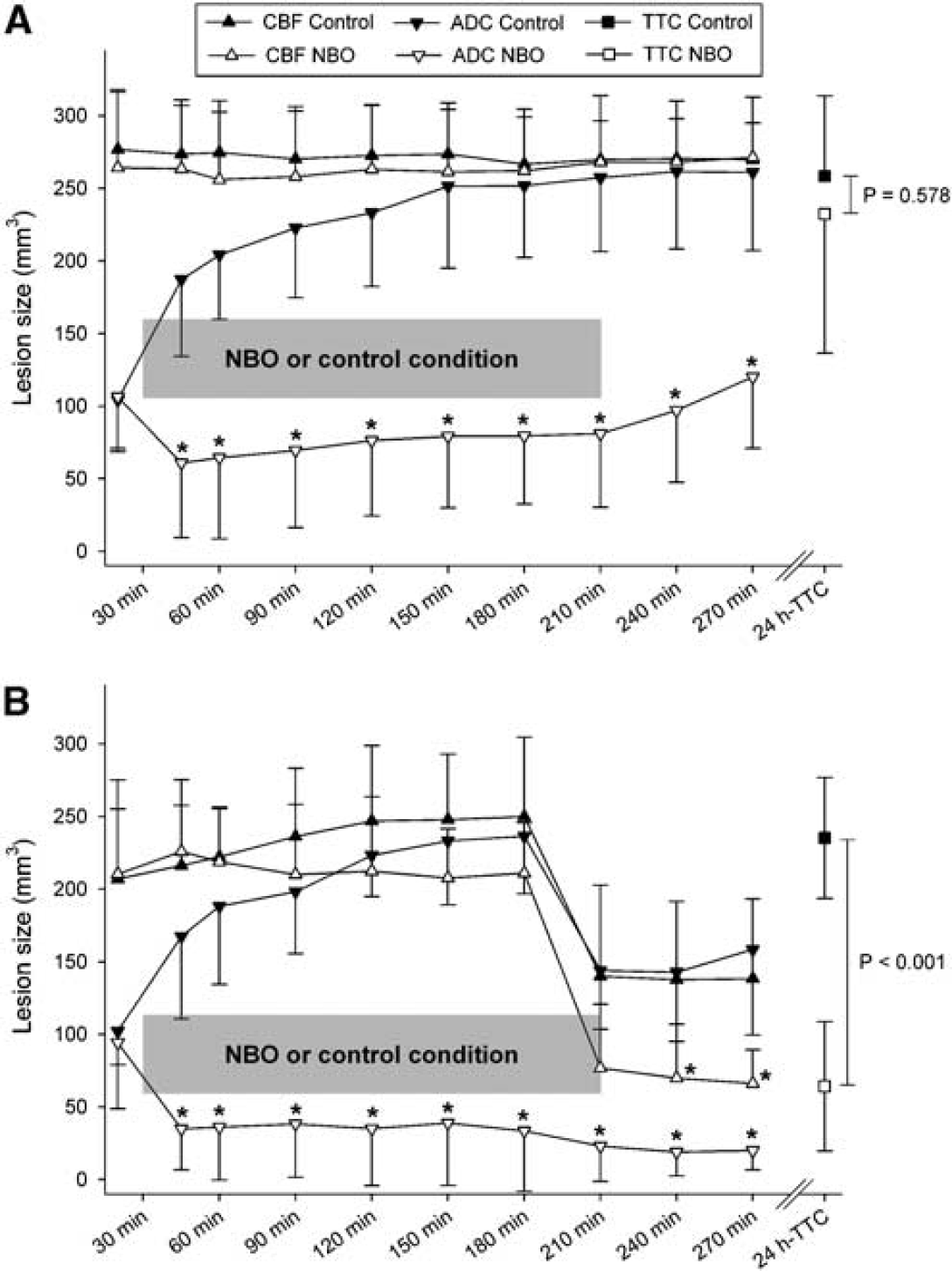
Spatiotemporal evolution of threshold-derived apparent diffusion coefficient (ADC) and cerebral blood flow (CBF) lesion volumes of normobaric hyperoxia-treated (NBO) and room air-treated control (control) rats subjected to (
During occlusion in experiment 3b (tMCAO), the CBF lesion volume did not differ between groups and remained relatively constant over time. In comparison to control animals, ADC-derived lesion volume stopped growing after the start of NBO and did not increase during treatment. The DWI/PWI mismatch was significant at all time points in NBO animals (P < 0.05) and up to 45 mins after stroke onset in controls (P < 0.05). MR angiography showed successful large vessel reperfusion of the MCA after suture withdrawal at 3 h after occlusion in both the NBO and control groups (data not shown). However, tissue reperfusion was incomplete with significantly better reperfusion in NBO-treated animals relative to room air-treated animals (P < 0.05 at 240 and 270 mins). Similar to pMCAO animals, the TTC-derived lesion volume almost matched the 3-h CBF/ADC lesion volumes in controls (P > 0.05). In contrast, combined with 3-h reperfusion, NBO appeared to salvage a substantial part of the mismatch and significantly reduced the final TTC-derived lesion volume relative to room air controls.
Quantitative Cerebral Blood Flow Values within Region of Interests
Figures 4A and 4B show CBF values within investigated ROIs of experiments 3a and 3b. In both experiments, there were no significant differences in region-specific contralesional CBF values between or within groups (P > 0.05, data not shown) and for clarity in the figures, were averaged over time into a single value (Contra).
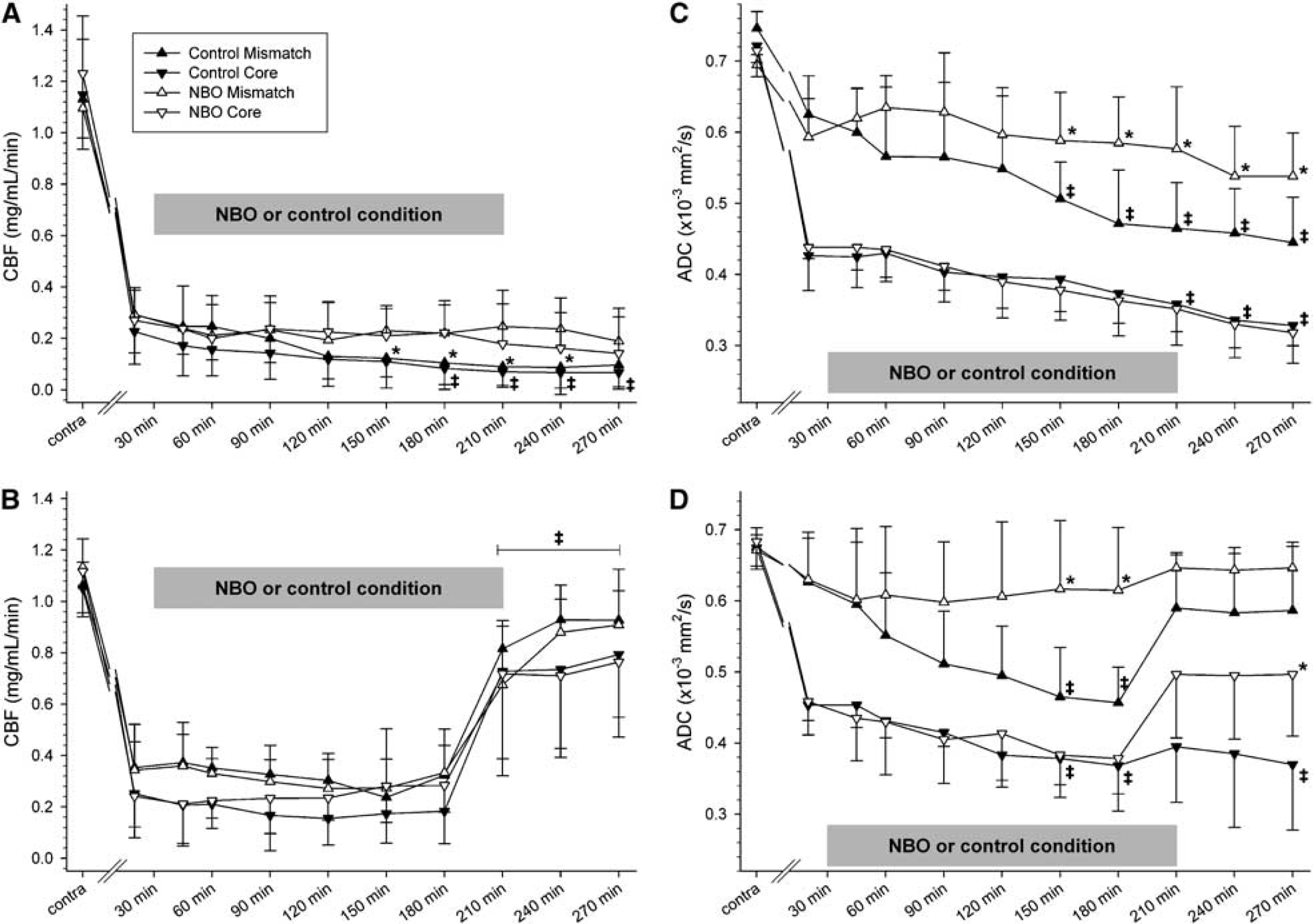
Regional evolution of (
In experiment 3a (pMCAO), postocclusion region-specific CBF did not change over time in NBO-treated animals (P > 0.05). In controls, core CBF was significantly lower by 180 mins relative to 20 mins values (P < 0.05). There were no significant intergroup differences in postocclusion core CBF (P > 0.05). However, by 150 mins after MCAO mismatch CBF values were significantly lower relative to NBO-treated animals (P < 0.05). Lastly, there were no significant differences in region-specific CBF between- or within-groups in the contralesional hemisphere (P > 0.05, data not shown).
In experiment 3b (tMCAO), there were no significant differences in region-specific CBF between- or within-groups in the contralesional hemisphere (P > 0.05, data not shown). Furthermore, no significant intergroup differences in ipsilesional region specific CBF (P > 0.05) were ascertained. After reperfusion, mismatch and core CBF significantly increased in both groups relative to 20 mins values.
Quantitative Apparent Diffusion Coefficient Values within Region of Interests
Figures 4C and 4D show ADC values within investigated ROIs of experiments 3a and 3b. In both experiments, there were no significant differences in region-specific contralesional ADC values between- or within-groups (P > 0.05, data not shown) and for clarity in the figures, were averaged over time into a single value (Contra).
In experiment 3a (Figure 4C), postocclusion mismatch ADC did not decrease during NBO in treated animals (P > 0.05). In controls, however, mismatch ADC values were significantly reduced by 150 mins after MCAO compared with 20 mins values as well as to NBO-treated animals (P < 0.05). In both groups, postocclusion core ADC values were significantly reduced by 210 mins after MCAO compared with 20 mins values (P < 0.05) without significant intergroup differences (P > 0.05).
In experiment 3b (Figure 4D), ipsilesional postocclusion core and mismatch ADC values did not change significantly over time in NBO in treated animals (P > 0.05). In controls, mismatch and core ADC values were significantly reduced at 150, 180, and 270 mins after MCAO compared with 20 mins values (P < 0.05). During occlusion, there were no significant intergroup differences in core ADC (P > 0.05); however, mismatch ADC was significantly higher in NBO-treated animals at 150 and 180 mins. After reperfusion, core ADC values of NBO animals significantly improved at 270 mins relative to controls.
Discussion
There is growing evidence that normobaric oxygen therapy (NBO) is a promising treatment strategy to improve outcome after acute ischemic stroke (Flynn and Auer, 2002; Kim et al, 2005; Singhal et al, 2002a, b). This study substantially extends these prior findings by evaluating different treatment durations, cell-death pathways as well as regional tissue susceptibility to ischemia in two different rat models of focal cerebral ischemia. Acute neuroprotection of NBO was shown in vivo by serial quantitative DWI and PWI, immunohistochemistry, neurological scoring, and post-mortem TTC staining in a clinically relevant setting. However, with prolonged ischemia the acute beneficial effects of short-term (3 h) NBO could not be sustained. Lastly, though NBO-attenuated cell death in the ipsilesional penumbra, neuronal death increased in the contralesional, normal hemisphere.
In vivo MR imaging showed that initial pretreatment ADC lesion volumes did not increase during NBO, whereas they progressively increased in room air-treated control animals. This effect of ‘ischemic lesion freezing’ occurred mainly in the cortex. Because the CBF-derived lesion volumes remained unchanged, NBO treatment resulted in a persistent CBF/ADC mismatch. Consistent with this mismatch persistence, the ADC in the mismatch zone remained relatively stable without a further decrease in NBO-treated rats, whereas in the control groups, regional ADC in the mismatch area shifted toward the core zone values as ischemia progressed. In contrast, NBO had no effect on the ADC characteristics in the core region, typically located in the striatum. These findings indicate that NBO essentially exhibited its neuroprotective effects on tissue with still normal diffusion characteristics (mismatch tissue), and such tissues were primarily in the cortex, as confirmed by a complete cessation of lesion growth in the cortex, corroborating previous experimental and clinical studies (Flynn and Auer, 2002; Kim et al, 2005; Singhal et al, 2005, 2002a, b). Because NBO has been shown to significantly improve penumbral but not core pO2 levels (Liu et al, 2004), it can be expected that NBO predominantly attenuates necrosis in the periinfarct areas rather than in the core, explaining the observed protection of the mismatch region. In addition to improving tissue oxygenation, NBO, particularly in combination with hypercarbia, has also been shown to inhibit cortical spreading depressions, thereby blunting generalized brain injury (Kraig et al, 1991). Indeed, NBO-induced hypercapnia has been reported clinically (New, 2006), and in the present study, significantly higher PaCO2 levels were observed in NBO-treated animals relative to air breathing controls, which may have contributed to tissue protection.
Aside from improving tissue oxygen delivery to areas of diminished blood flow (Liu et al, 2004), it has been suggested that oxygen may partially be neuroprotective by inducing cerebral vasoconstriction in normal brain tissue, with subsequent decreases in intracranial pressure and/or an inverse steal of blood into the mismatch area (Nighoghossian and Trouillas, 1997). However, no significant changes in CBF were observed in this study, corroborating previous studies that showed unchanged cerebral perfusion during or shortly after oxygen treatment in models of focal cerebral ischemia (Henninger et al, 2006a). The observed mild elevation of PaCO2 may have counterbalanced the vasoconstrictive effects associated with hyperoxia (Omae et al, 1998), explaining the relatively unchanged CBF values. Hence, our data suggest that CBF changes are unlikely to be a major mechanism of neuroprotection in the acute phase of ischemia in this model.
In accord with previous experimental studies showing reduced final lesion sizes in suture models of transient (< 3 h) MCAO, NBO significantly extended the reperfusion window in this study (Flynn and Auer, 2002; Kim et al, 2005; Liu et al, 2006; Singhal et al, 2002a, b). Similar to previously reported results, 3-h NBO did not decrease final lesion size after permanent suture occlusion (Kim et al, 2005; Singhal et al, 2002a; Veltkamp et al, 2006). However, as NBO has been shown to be protective in a milder model of permanent ischemia (Veltkamp et al, 2006), we tested the hypothesis that longer (> 3 h) treatment durations may be beneficial in the more severe suture model, and indeed, 6-h NBO treatment reduced final infarct size if initiated within 30 mins after pMCAO. In addition to blunting spreading depression (Kraig et al, 1991), oxygen may have provided lasting neuroprotection by reducing ischemia-induced cell-death pathway activation (Veltkamp et al, 2006).
In light of the conflicting data regarding oxygen's influence on cell survival and the involved pathways (Kent et al, 2001; Liu et al, 2006; Sugawara and Chan, 2003; Unal-Cevik et al, 2004), we specifically sought to elucidate different modes of cell death after 3-h NBO in the pMCAO model. In general, necrosis predominates in the severely stressed core area, whereas neuronal apoptosis is concentrated in the less severely inflicted border zone of the ischemic territory (Unal-Cevik et al, 2004), and it may be argued that peripheral areas of the ischemic lesion may benefit the most from NBO therapy. Indeed, in this study, a significant cytoprotective effect (i.e., decreased selective neuronal death) in the ischemic penumbra but not the core was observed as indicated by ISEL staining. Free radical formation is an essential component of pathological mechanisms responsible for ischemic stroke and has been shown to exacerbate ischemic brain injury (Kent et al, 2001; Kontos, 2001; Sugawara and Chan, 2003), particularly when combined with reperfusion (Aronowski et al, 1997; Kent et al, 2001; Kim et al, 2005). In contrast, this study shows that NBO could decrease infarction, indicating that the benefit of hyperoxia may outweigh the risk for enhanced oxidative stress in the ischemic brain has been suggested previously (Henninger et al, 2006a; Schabitz et al, 2004; Sunami et al, 2000). Interestingly, we found more ISEL-positive cells in nonischemic hemisphere ROIs of NBO-treated animals relative to air-treated controls. Different, potentially interacting pathophysiological mechanisms may be responsible for the observed contralesional neuronal damage, including hypotension and transsynaptic effects (Zhu and Auer, 1995) as well as direct cytotoxicity of oxygen such as hyperoxia-induced reactive oxygen species formation (Lee and Choi, 2003). The former two possibilities are remote given the apparently normal (i.e., > 80 mmHg) mean arterial blood pressure in all animals as well as the relatively milder insult in NBO-treated animals compared with controls (Zhu and Auer, 1995). However, the last remains a viable candidate. For instance, hyperbaric hyperoxia has been shown to induce neuronal cell death preferentially in the hemisphere opposite to unilateral carotid artery occlusion (Balentine, 1968). Although we did not observe the previously described extensive and localized tissue necrosis (Balentine, 1968), this is plausibly owing to an interstudy difference in (among other methodological considerations) oxygenation duration as well as the different pressure levels used (i.e., normobaric versus hyperbaric oxygenation) or such phenomena may simply occur after the end point of this study. Further studies are undoubtedly warranted to clarify this issue.
Because caspases are essential effector molecules for carrying out apoptosis in eukaryotic cells with caspase-3 being a specific downstream death effector causing cell death, particularly in the penumbra (Ferrer and Planas, 2003; Wang and Lenardo, 2000), we specifically sought to elucidate the contribution of caspase-mediated cell death in this study. Caspase-3 can be activated independent of the mitochondria or through a mitochondrial pathway that requires caspase-9 (Wang and Lenardo, 2000). In this study, no significant difference in ipsilesional, penumbral caspases-3 positivity was observed between NBO- and room air-treated groups. In contrast, a significant increase was seen in ipsilesional core areas as well as in the nonischemic hemisphere compared with room air-treated controls. These data indicate that the observed cytoprotective properties of NBO in the ischemic brain are not determined by caspase-3-mediated pathways of apoptosis. Normobaric hyperoxia treatment appeared to induce such apoptotic pathways in nonischemic brain tissue, corroborating previous studies that showed hyperoxia-induced cell death through caspase-3 activation (Gerstner et al, 2006; Lee and Choi, 2003). In light of decreased penumbral caspase-8 cleavage with NBO (Liu et al, 2006), it can be speculated that treatment with NBO only delays initiation of early stages of apoptosis but ultimately cannot prevent cell death (Wang and Lenardo, 2000). Potentially, reactive oxygen species-mediated mitochondrial damage could lead to caspase-9 activation, leading to cell death through effector caspases such as caspase-6 (Gerstner et al, 2006; Lee and Choi, 2003; Wang and Lenardo, 2000); however, further studies are necessary to confirm these hypotheses.
In conclusion, our results show that NBO (1) acutely preserves the CBF/ADC mismatch without altering CBF, (2) significantly extends the time window for reperfusion, (3) induces lasting tissue protection in permanent ischemia, and (4) although capable of reducing penumbral cell death, can also induce cell death in unaffected, nonischemic brain tissue. These observations may be clinically relevant as only a small minority of clinical stroke patients are currently eligible for reperfusion therapy (Wardlaw et al, 2003) and thrombolysis is often delayed and/or incomplete (Hacke et al, 2005). Nevertheless, because the greatest amount of tissue protection was observed with reperfusion, our data suggest that NBO could be a useful adjunct to thrombolytic therapy and further studies are warranted to determine interactions between NBO and recombinant tissue plasminogen-activator thrombolysis. In light of the observed induction of apoptotic cell-death pathways, further research is necessary to evaluate different treatment paradigms to further improve the risk/benefit ratio of NBO. Lastly, though previous studies demonstrated lasting neuroprotection after treatment with oxygen (Flynn and Auer, 2002; Henninger et al., 2006a), our findings warrant a lengthier observation period that may enhance the ability to show whether the tissue protection observed by MRI and brain pathology persists — or does not persist — during more chronic stages.
Footnotes
Acknowledgements
The authors report no conflicts of interest.