
Abstract
Patients with traumatic brain injury (TBI) routinely exhibit cerebral glucose uptake in excess of that expected by the low levels of oxygen consumption and lactate production. This brings into question the metabolic fate of glucose. Prior studies have shown increased flux through the pentose phosphate cycle (PPC) during cellular stress. This study assessed the PPC after TBI in humans. [1,2-13C2]glucose was infused for 60 mins in six consented, severe-TBI patients (GCS < 9) and six control subjects. Arterial and jugular bulb blood sampled during infusion was analyzed for 13C-labeled isotopomers of lactate by gas chromatography/mass spectroscopy. The product of lactate concentration and fractional abundance of isotopomers was used to determine blood concentration of each isotopomer. The difference of jugular and arterial concentrations determined cerebral contribution. The formula PPC = (m1/m2)/(3 + (m1/m2)) was used to calculate PPC flux relative to glycolysis. There was enrichment of [1,2-13C2]glucose in arterial-venous blood (enrichment averaged 16.6% in TBI subjects and 28.2% in controls) and incorporation of 13C-label into lactate, showing metabolism of labeled substrate. The PPC was increased in TBI patients relative to controls (19.6 versus 6.9%, respectively; P = 0.002) and was excellent for distinguishing the groups (AUC = 0.944, P < 0.0001). No correlations were found between PPC and other clinical parameters, although PPC was highest in patients studied within 48 h of injury (averaging 33% versus 13% in others; P = 0.0006). This elevation in the PPC in the acute period after severe TBI likely represents a shunting of substrate into alternative biochemical pathways that may be critical for preventing secondary injury and initiating recovery.
Introduction
After traumatic brain injury (TBI), the brain experiences a period of metabolic perturbation during which energy and substrate demands are increased from normal resting levels (Bergsneider et al, 1997, 2000; Glenn et al, 2003; Hovda, 1996; Hovda et al, 1992). During this critical postinjury period, there are initially high levels of cerebral glucose consumption, followed by a variable period of normal to mildly depressed glucose metabolism (Bergsneider et al, 1997, 2000). However, during the same period, many studies have demonstrated a low level of cerebral oxygen consumption relative to the unexpectedly preserved glucose metabolism (Glenn et al, 2003; Jaggi et al, 1990). In fact, the degree to which oxygen metabolism is depressed correlates with poorer outcomes. This paradoxically preserved glucose consumption in the presence of depressed oxygen metabolism was initially termed ‘relative hyperglycolysis.’ If this change in metabolism was solely owing to a decrease in tricarboxylic acid (TCA) cycle activity and oxidative phosphorylation, then we would expect an increase in lactate production from unopposed glycolytic activity. However, the expected cerebral lactate production that should accompany relative or absolute hyperglycolysis is uncommon; instead, it appears that cerebral lactate uptake is more common acutely after TBI and that lactate may be used as an alternative fuel (Glenn et al, 2003; Soustiel et al, 2005; Vespa et al, 2003).
This discrepancy brings into question the metabolic fate of the extra glucose that is not accounted for by oxidative metabolism or anaerobic glycolysis. Correction of ionic fluxes and glutamate depletion both require large amounts of ATP, and thus ultimately glucose and other substrates, to restore electrical and physiological equilibrium (Hovda, 1996). Additional metabolic needs that may be important for either the prevention of secondary injury or recovery include scavenging of free radicals, or repair and synthesis of macromolecules, such as cell membrane phospholipids, DNA, and proteins. In fact, inability to reduce free radicals may be an important source of further secondary injury in the traumatized brain (Arundine and Tymianski, 2004; Lewen et al, 2000). Multiple downstream products of glucose metabolism help supply the substrates for these purposes, including acetyl-CoA for fatty acid synthesis, ribose for nucleotide synthesis and repair, and pyruvate, as well as many TCA cycle intermediates for amino-acid synthesis.
The pentose phosphate cycle (PPC; also termed the ‘pentose phosphate shunt’ or ‘hexosemonophosphate shunt’) is a potentially important pathway that supplies the ribose and reducing equivalents in the form of NADPH for scavenging of free radicals in conjugation with glutathione, and may be particularly critical for secondary injury prevention. The PPC is also important for fatty acid synthesis, cholesterol synthesis, and neurotransmitter synthesis and degradation (Baquer et al, 1988; Lehninger, 2005). The PPC, however, has not been studied acutely after human TBI, and in fact, there have been few studies of the normal resting cerebral PPC in humans. Therefore, we proposed that alterations in cerebral metabolism after TBI would result in increased glucose consumption by the PPC relative to the normal state. This hypothesis was tested using [1,2-13C2]glucose labeling techniques to detect metabolites of glycolysis and the PPC, and to estimate the relative flux through both pathways.
Carbon labeling techniques to detect flux through various metabolic pathways, including glycolysis, the PPC, the TCA cycle, and others, evolved from 14C-labeling in the 1960s (Katz and Wood, 1960). More recent techniques use glucose labeled with 13C, a nonradioactive isotope of carbon with low natural abundance (∼1%). Labeled-glucose administration is followed by detection of metabolites by nuclear magnetic resonance spectrometry or gas chromatography coupled to mass spectrometry (GC/MS). This technique has been described by many investigators in brain (Ben-Yoseph et al, 1995; Gruetter, 2002; Kaibara et al, 1999; Ross et al, 2003) and astrocyte culture (Lee et al, 1996) as well as many other organ systems and cell cultures. By administering glucose labeled with 13C in the first and second carbon, the label can be effectively followed through its downstream metabolic products. Different metabolic pathways result in the label's presence in different positions of the metabolic products. By comparing the relative abundances of these isotopomers (13C-containing metabolites varying in position and number of 13C-carbon atoms), it is possible to determine the relative fluxes through these pathways (Figure 1). Additional studies of hypothermia and cellular transformation and normal rat brain have used [13C] Glucose labeling technologies to show increased flow of substrate through the PPC in times of cellular stress (Ben-Yoseph et al, 1995; Boros et al, 2002; Kaibara et al, 1999; Lee et al, 1998).
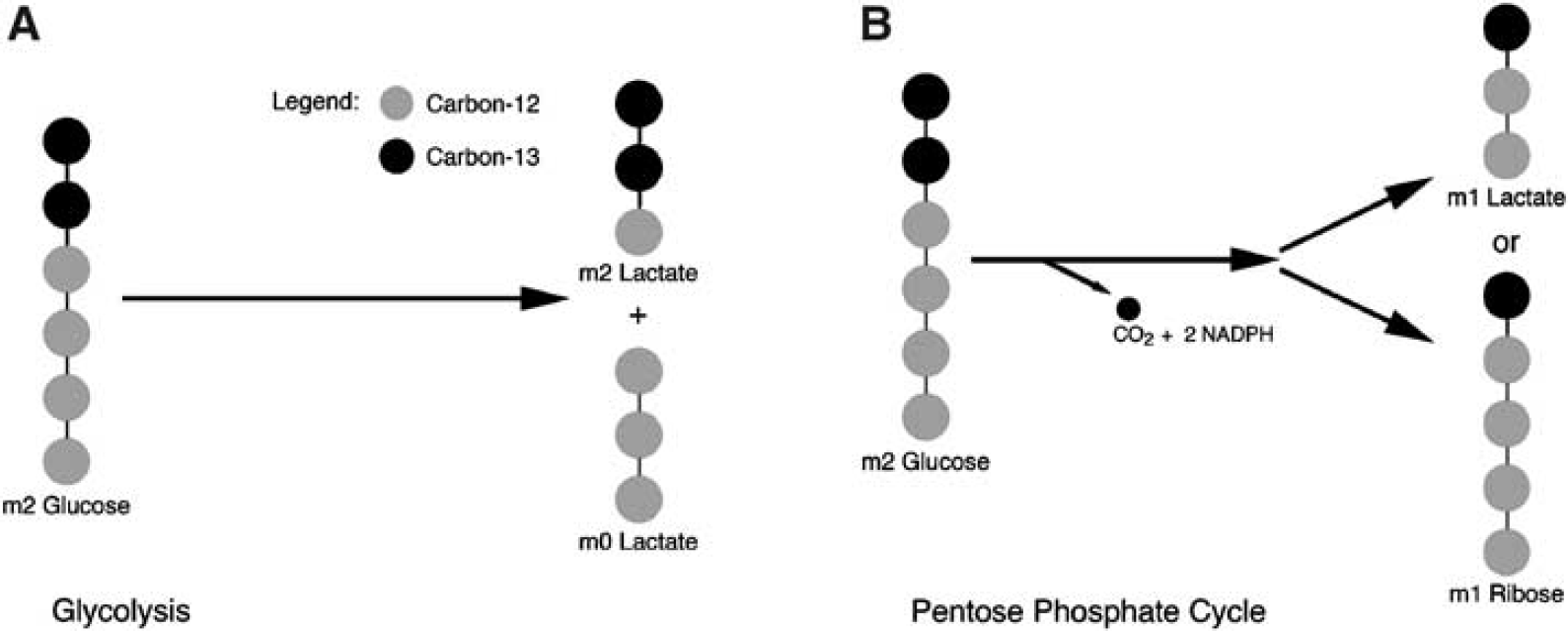
Metabolic schematic demonstrating differential labeling of lactate derived from (
Materials and methods
Patient Selection
Mechanically ventilated severe head-injured patients, aged 14 years and older, admitted to UCLA Medical Center within 24 h of injury were eligible for inclusion in the study. Severe TBI was defined as closed or penetrating injury, including gunshot and stab wounds, with a postresuscitation Glasgow Coma Scale (GCS) ≤ 8, or deterioration to a GCS ≤ 8 within 24 h of admission. All patients required mechanical ventilation and intracranial pressure monitoring. Exclusion criteria included the following: (1) terminal illness (e.g., advanced cancer, AIDS); (2) severe previous neurologic disease; and (3) acute complete spinal cord injury. These inclusion and exclusion criteria are consistent with those used for previous publications from the UCLA Brain Injury Research Center (Bergsneider et al, 1997, 2000; Glenn et al, 2003; Vespa et al, 2003). UCLA's Institutional Review Board approved the protocol, and informed consent was obtained from the patients' legal representatives.
One TBI patient studied using the 13C-labeling technique was later excluded because their PPC flux could not be calculated. At all time points in the infusion, there was a net consumption of lactate by the brain. Therefore, subtracting the arterial from venous concentrations resulted in a negative number. A negative number in the PPC formula is uninterpretable and gives no information about cerebral metabolism because we cannot separate the cerebral contribution to lactate production from systemic metabolism.
General Management Protocol
After initial stabilization and/or surgical evacuation of intracranial hematomas, patients were admitted to the intensive care unit and treated in accordance with a Level I Trauma Center protocol. The standard management goals included maintenance of intracranial pressure less than 20 mm Hg and cerebral perfusion pressure above 70 mm Hg in accordance with the recommendations set forth by The Brain Trauma Foundation and the Joint Section on Neurotrauma and Critical Care. A jugular bulb catheter and radial arterial catheter were inserted in all patients as soon as possible after admission to allow determination of arterio-venous differences for glucose (AVDglucose), arterio-venous differences for lactate (AVDlactate), and arterio-venous differences for oxygen (AVDO2), and for sampling of 13C-labeled glucose and lactate.
Jugular Bulb Catheterization
The dominant jugular vein was selected based on the dominant jugular foramen as visualized on admission computerized tomography scan. A 5 Fr Cordis and a 4 Fr Oxymetric catheter (Baxter Critical care, Baxter Health Care, Deerfield, IL, USA) were inserted to approximately 15 cm until resistance was encountered. After confirmation of placement by lateral skull X-ray, the catheter was calibrated in vivo. Calibration was repeated every 12 h.
Cerebral Metabolism and Blood Flow Measurements
Measurements of AVDglucose, arterial lactate, AVDlactate, AVDO2, and cerebral blood flow (CBF) were made simultaneously during the first 10 days after injury in all patients. Arterial and venous blood samples were collected every 12 h. Cerebral blood flow was assessed by the intravenous 133Xenon clearance technique once daily. The collection and processing techniques for CBF, cerebral metabolic rate for glucose (CMRglucose), cerebral metabolic rate for lactate (CMRlactate), and cerebral metabolic rate for oxygen (CMRO2) followed the same protocol as described previously by Glenn et al (2003), and will not be repeated in detail here.
Infusion Protocol and Sample Collection
All TBI patients were studied within 7 days of injury. The UCLA Pharmaceutical Services Department prepared 60 mL bags of 10% 13C-labeled glucose ([1,2-13C2]glucose; Cambridge Isotope Laboratories Inc., Andover, MA, USA) in physiological saline. Samples were tested for sterility and pyrogenicity. The solution was then set to infuse through an automated pump (Colleague 3 CX; Baxter, Deerfield, IL, USA) over 60 mins (1 mL/min) into a central venous catheter. Blood glucose levels were monitored closely during the infusion to avoid hyperglycemia. At multiple time points (0, 15, 30, 45, 60, 90, and 120 mins), arterial and jugular bulb venous blood sampled were collected. One millimeter of each blood sample was drawn in a 3-mL syringe before being dispensed into two chilled and labeled 400-μL microcentrifuge tubes coated with heparin and lithium fluoride to prevent coagulation and glycolysis. The tubes were immediately placed on ice. The chilled tubes then underwent refrigerated (4°C) centrifugation at 1200g for 3 mins. The supernatant (plasma) was then transferred to labeled microcentrifuge tubes for storage at −80°C before sample analysis. Arterial and venous blood samples were also collected for determination of plasma concentrations of glucose and lactate.
Control Subjects
A cohort of six healthy, consented volunteer subjects was studied in the awaken state to determine normal cerebral PPC flux. Interventional radiology placed jugular bulb catheters under fluoroscopic guidance into proper position in the jugular bulb by a femoral vein approach. A radial arterial line and peripheral venous line were placed by a neurointensivist. Single measurements of AVDglucose, AVDlactate, and AVDO2 were made by taking simultaneous samples of arterial and venous blood as previously described by Glenn et al (2003). A CBF measurement was performed before [13C] Glucose studies using the intravenous 133Xe clearance technique. [13C] Glucose infusion and blood sampling were carried out with the same protocol as for trauma patients with the exception that the solution was infused through the peripheral venous catheter.
Sample Analysis
Glucose from plasma is derivatized as its aldonitrile pentaacetate derivative for GC/MS analysis (Katz et al, 1989). Chemical ionization (CI) was used to give the molecular ion (C1–C6) of glucose molecules at m/z 328 for 12C-glucose and m/z 331 for [1,2-13C2] Glucose. Lactate was extracted from 0.2 mL of plasma by ethyl acetate after acidification with HCl. Lactate was then derivatized to its propylamine-heptafluorobutyrate (HFB) form, and the m/z 328 to m/z 331 (carbons 1 to 3 of lactate, chemical ionization) was monitored for the detection of m1 (recycled lactate through the PPC) and m2 (lactate produced by glycolysis) for the estimation of pentose cycle activity (Tserng et al, 1984). Mass spectral data were obtained on the HP5973 mass selective detector connected to an HP6890 gas chromatograph. The settings were as follows: GC inlet 230°C, transfer line 280°C, MS source 230°C, and MS Quad 150°C. An HP-5 capillary column (30 m length, 0.25 mm diameter, and 0.25 μm film thickness) was used for glucose and lactate analysis. Results of the mass isotopomers in glucose and lactate are reported as molar fractions of m0, m1, m2, etc., where m0, m1, m2, etc. indicate the number of 13C atoms in the molecule (Lee et al, 1991).
Data Analysis
Lactate formed directly from glycolysis can show m0 labeling, whose origin is the bottom of the [1,2-13C2] D-glucose molecule, and m2 labeling, derived from the top of the same molecule. By the action of the PPC, one carbon is lost in the form of CO2 resulting in m1 labeling (Lee et al, 1998).
The separately determined levels of lactate in arterial and jugular plasma were then multiplied by the respective isotopomers' relative abundances (molar fractions) obtained by GC/MS. This yields an absolute blood concentration for each of the isotopomers. Based on the Kety–Schmidt theory of CBF, the contribution of each isotopomer by the brain was determined by subtracting the arterial concentration from the jugular concentration:
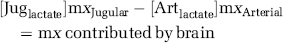
Using these m1 and m2 values derived by arterio-venous differences, the PPC was quantified relative to glycolysis by the previously described formula (Lee et al, 1998):
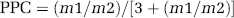
The average enrichment curves for labeled glucose as well as labeled lactate, derived from metabolism of the labeled glucose, were used to determine the time points at which enrichment was maximal and most stable (Figures 2 to 4). For all subjects, this plateau was reached toward the end of the infusion at the 45-, 60-, and 90-mins time points. The PPC values for these time points were averaged for each subject to determine their individual PPC flux.
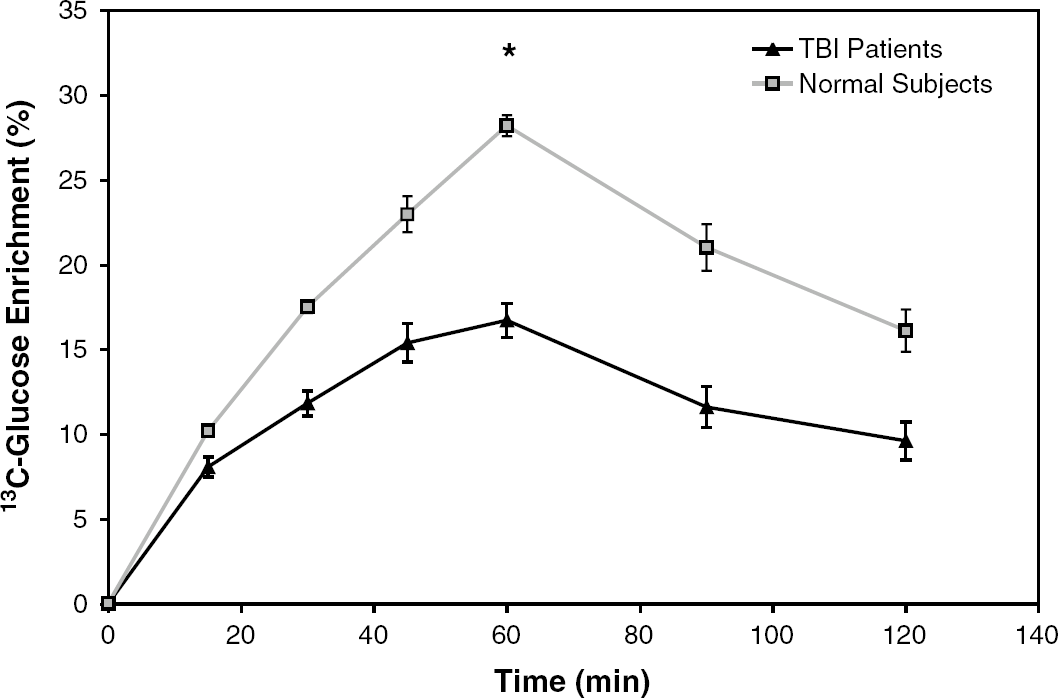
Average enrichment of 13C-labeled glucose in jugular blood for control subjects and TBI patients. Error bars represent s.e. of the mean. *Average maximum enrichment was 16.6% ± 1.3% in TBI patients and 28.2% ± 0.6% in control subjects (P = 0.004).
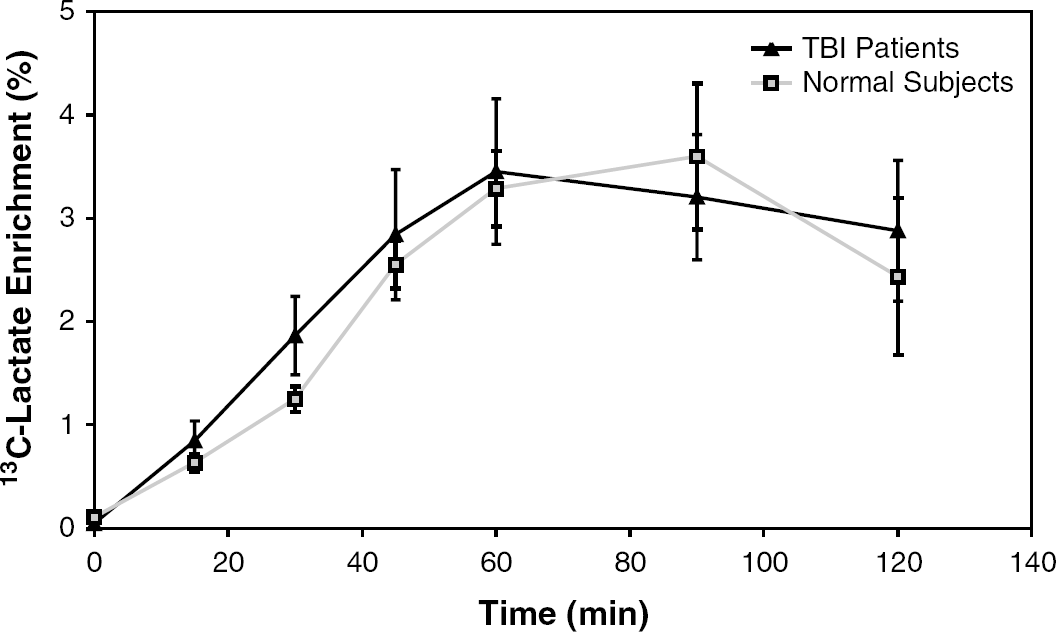
Average enrichment of 13C-labeled lactate produced by glycolysis in jugular blood. Error bars represent s.e. of the mean.
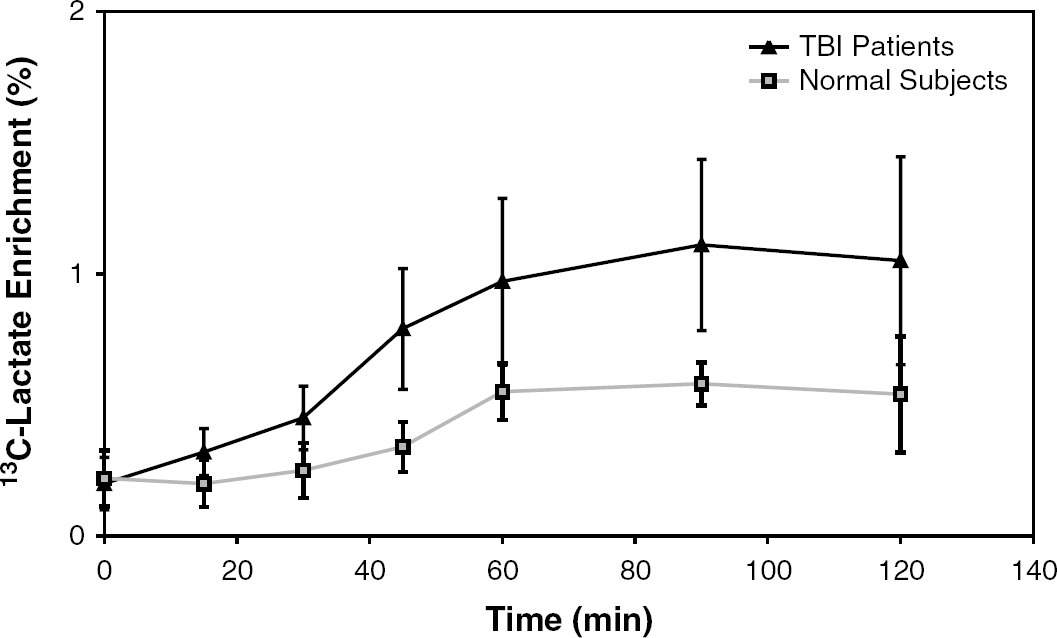
Average enrichment of 13C-labeled lactate produced by the PPC.
Statistical Analysis
The average PPC values at the peak glucose and lactate enrichment (45-, 60-, and 90-mins time points) for both trauma patients and normal subjects were compared by the Wilcoxon rank-sum test. The PPC values between control and TBI subjects were also compared using an area-under-the-curve method (MedCalc Software, Belgium). The Wilcoxon rank-sum test was also used to compare variables between the TBI subjects and control subjects (age, maximum [13C] Glucose enrichment, jugular glucose, arterial glucose, etc.). A Spearman rank-order correlation test was used to determine any correlations between PPC values and other metabolic (CMRO2, CMRlactate, CMRO2, AVDO2, etc.), physiologic (CBF, transcranial Dopple, etc.), clinical (GCS, CT grade, intracranial pressure), and Glasgow Outcome Scale parameters.
Results
Over a period of 2 years, six patients with severe TBI were studied, ranging from 17 to 79 years of age (mean = 44 years). Initial GCS varied from 3 to 9, and all patients had deteriorated to a GCS of 8 or less within 24 h of injury. Traumatic brain injury patients were studied on postinjury days 1 to 6 (median postinjury = day 3). Additionally, six control subjects were studied, aged 22 to 56 years (mean = 37) (Table 1).
Subject demographics and results: (a) TBI patients (b) normal subjects
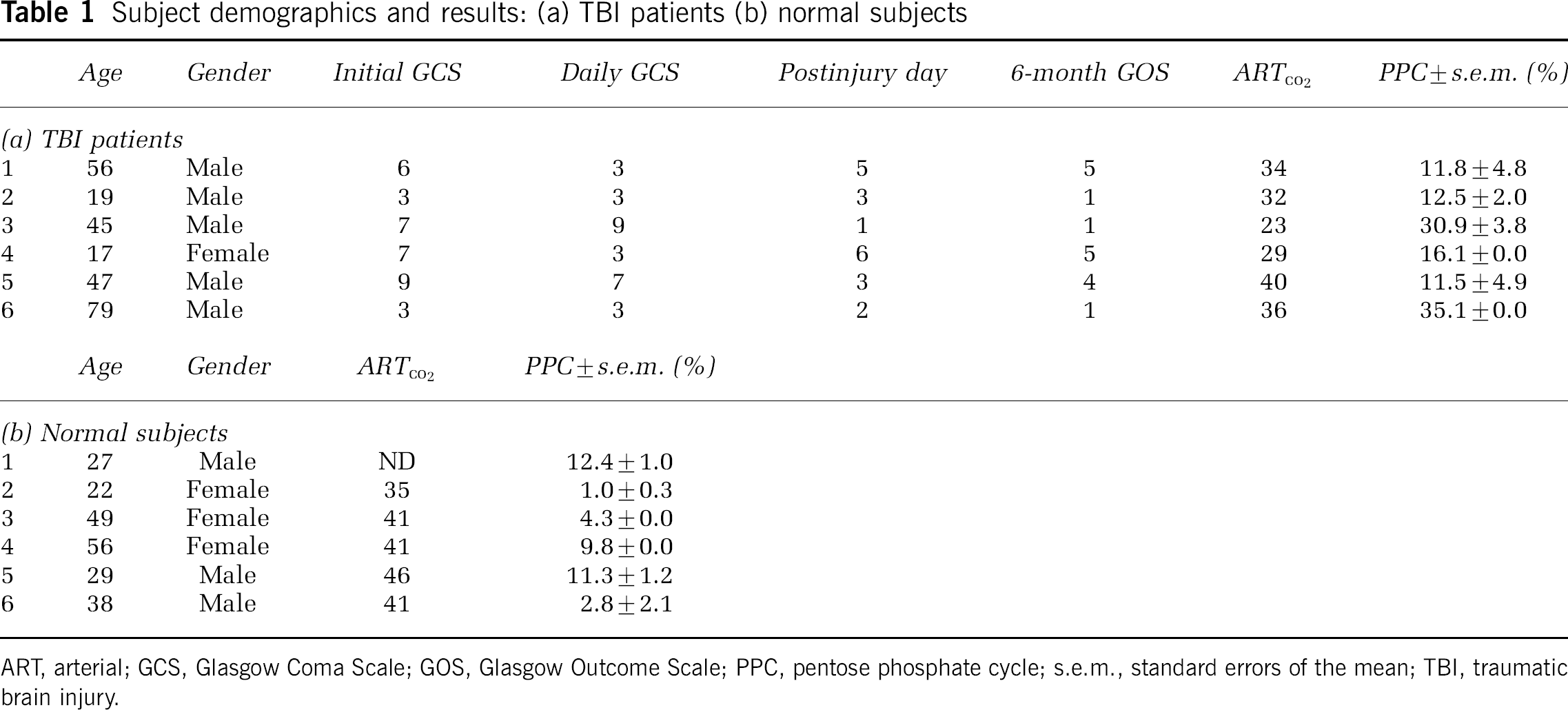
ART, arterial; GCS, Glasgow Coma Scale; GOS, Glasgow Outcome Scale; PPC, pentose phosphate cycle; s.e.m., standard errors of the mean; TBI, traumatic brain injury.
The maximum [1,2-13C2]glucose enrichment during the infusion varied between groups. In the TBI patients, the average maximum enrichment was 16.6% ± 1.3%, whereas in control subjects it was 28.2% ± 0.6% (P = 0.004) (Table 2; Figure 2). The whole blood jugular glucose showed a tendency to be higher in the TBI patients (average 98.9 ± 9.9 mg/dL) than the control subjects (average 75.4 ± 6.4 mg/dL; P = 0.13) (Table 2). This differential enrichment between groups did not impact the PPC calculation, as there was no correlation in either group between enrichment and PPC flux (Table 3).
Results and comparisons between groups

PPC, pentose phosphate cycle; TBI, traumatic brain injury.
P-values marked in italics are less than 0.05.
Correlations with PPC flux
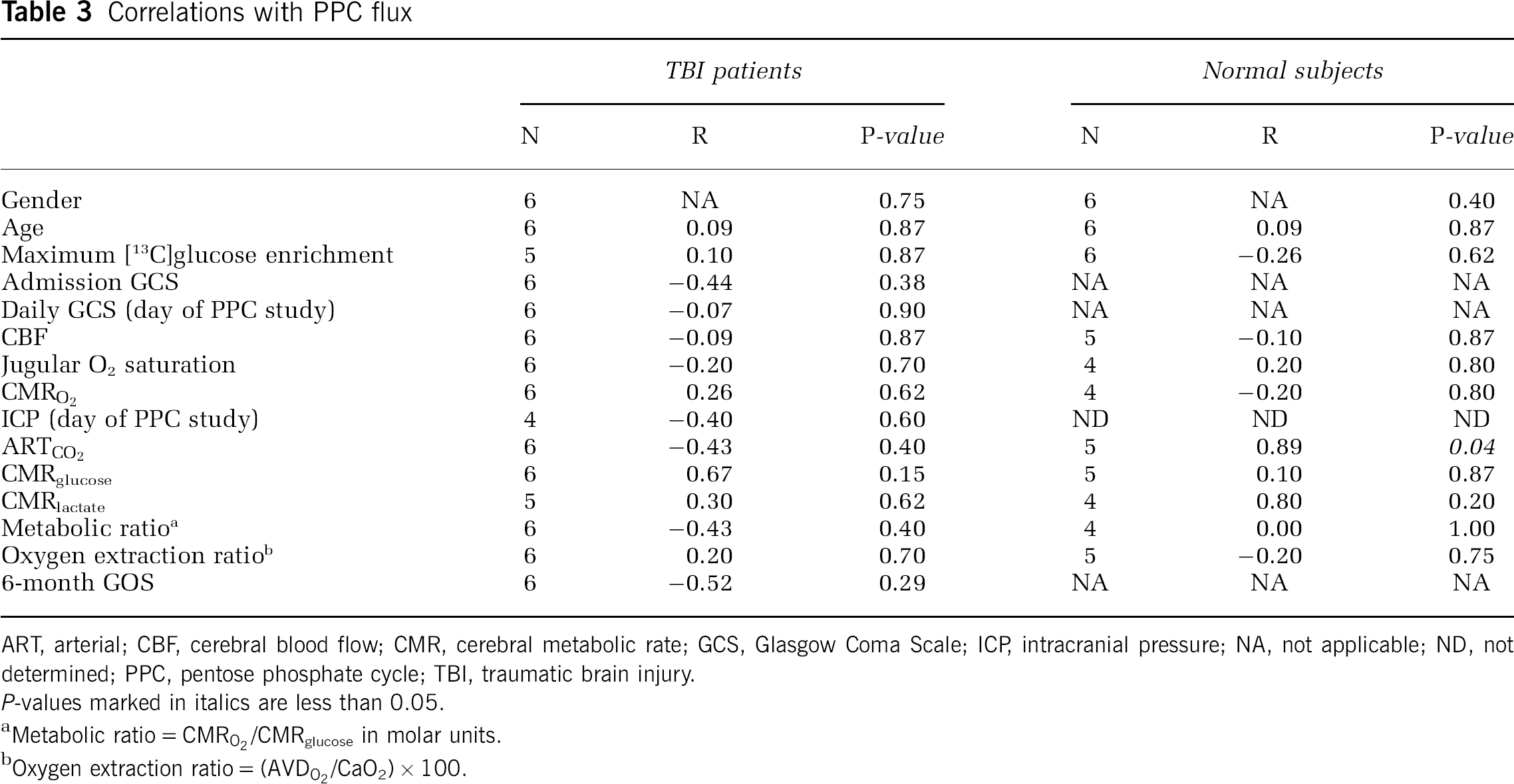
ART, arterial; CBF, cerebral blood flow; CMR, cerebral metabolic rate; GCS, Glasgow Coma Scale; ICP, intracranial pressure; NA, not applicable; ND, not determined; PPC, pentose phosphate cycle; TBI, traumatic brain injury.
P-values marked in italics are less than 0.05.
Metabolic ratio = CMRO2/CMRglucose in molar units.
Oxygen extraction ratio = (AVDO2/CaO2) × 100.
The enrichment of label in lactate, formed by metabolism of labeled glucose, followed a similar time course to glucose with the exception that the enrichments tended to plateau at the end of the infusion (45-, 60-, and 90-mins samples). (Figures 3 and 4). At all time points throughout the infusion and sampling period, the labeled lactate derived from the action of the PPC was higher in TBI patients than control subjects (Figure 4).
The average PPC for TBI patients was 19.6% ± 4.3%, compared with 6.9% ± 2.0% for control subjects (P = 0.002) (Figure 5; Tables 1 and 2). Area under the curve (AUC) analysis confirmed that the PPC value was excellent for distinguishing between control and TBI subjects (AUC = 0.944, s.e. = 0.073, 95% CI = 0.651 to 0.980, P < 0.0001, positive likelihood ratio (+ LR) if PPC > 11.255 = 6.00). There were no significant differences between the groups in terms of age (P = 0.93) or gender (P = 0.55).
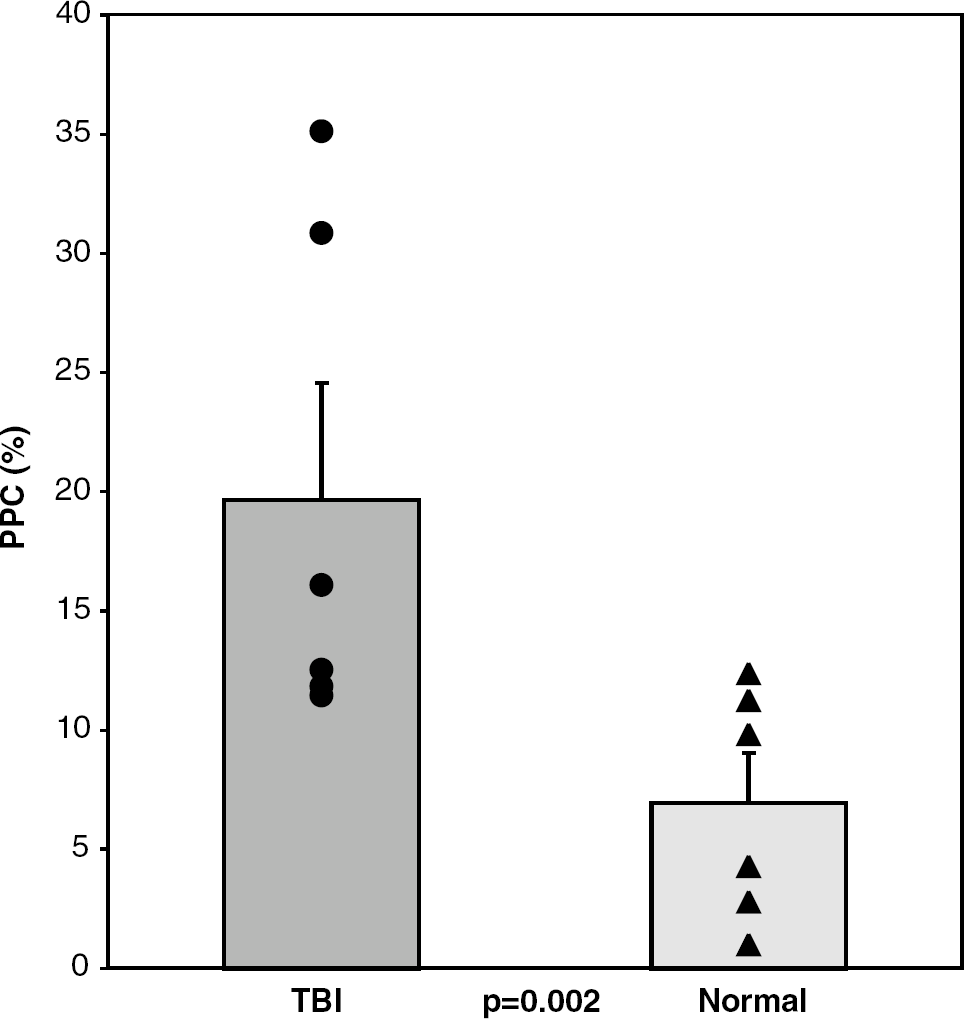
Pentose phosphate cycle fluxes in TBI patients and control subjects (P = 0.002). Bars indicate mean value with s.e. of the mean. Circles (TBI) and triangles (normal) indicate individual subjects.
There were no definitive significant correlations found between PPC and other demographic, metabolic, physiological, clinical, and outcome parameters tested, with the exception of a weak association with arterial levels of CO2 in control subjects (n = 5, R = 0.89, P = 0.04). Parameters tested included age, sex, GCS, intracranial pressure, CBF, TCD velocities, maximum labeled-glucose enrichment during infusion, postinjury day of study, and Glasgow Outcome Scale (Table 3). However, the average PPC for the two patients studied in the first 48 h after injury (33%) was significantly higher than that studied after 48 h (13%; P = 0.0006).
Discussion
Overview of Findings
To our knowledge, this study is the first to use 13C-labeling technology to quantitatively detect cerebral PPC activity in human controls. It is also the first to apply this technology to patients with acute severe head injury in the intensive care unit to define the PPC activity in those subjects. Whereas the cerebral PPC flux averaged 6.9% in six healthy control subjects, it was significantly higher, averaging 19.6%, in six severe TBI subjects studied within 7 days of injury (P = 0.002). Despite the small sample size, this difference appears to be robust statistically, particularly for the patients studied in the first 48 h after injury, who had the highest PPC activities measured (averaging 33%; P = 0.0006 compared with patients studied after 48 h).
Normal Cerebral Pentose Phosphate Flux and Increased Flux after Traumatic Brain Injury
The metabolic uncoupling of glucose and oxygen metabolism after TBI, along with the fact that lactate production by the brain may at times be negligible, leads to the conclusion that more glucose is being redirected toward nonglycolytic metabolic pathways than previously thought. There are at least two possibilities to explain this increased flux through the PPC. These changes could be categorized as dysfunctional, resulting from a pathologic block in the glycolytic pathway. Alternatively, they may represent a cellular compensatory upregulation in response to increased metabolic needs of the injured brain to prevent secondary injury.
A cell under stress faces demands for energy and substrates. More specifically, injured cells demand protection against oxidative damage, substrates for macromolecular anabolism (DNA, RNA, proteins, fatty acids, etc.), and energy for correction of electrolyte disturbances and repair of damaged DNA (for review see Hovda, 1996). Whereas glycolysis and oxidative metabolism primarily supply energy in the form of ATP, the PPC helps contribute to many other various demands. The PPC has a large reserve capacity to be upregulated in response to oxidative stress, acting as an electron donor in the redox recycling of glutathione (Hothersall et al, 1982). The PPC produces reducing equivalents in the form of NADPH, which, through glutathione, is important in the scavenging of free radicals (Arundine and Tymianski, 2004; Hall et al, 2004; Lewen et al, 2000). Experimental studies have demonstrated that free radical-mediated damage may be prevented by increasing glutathione production (Ben-Yoseph et al, 1996). NADPH is also used in some synthetic reactions such as fatty acid synthesis. The PPC produces ribose, important for DNA repair and replication and mRNA synthesis leading to protein synthesis. Additionally, not all lactate/pyruvate that ultimately enters the TCA cycle is destined to complete oxidative metabolism. Several intermediates in the cycle are used for amino acid and neurotransmitter synthesis. These changes and demands on a cell's metabolism have been demonstrated in other forms of cellular stress (transformation and hypothermia), and thus may also be important after TBI (Ben-Yoseph et al, 1995; Boros et al, 2002; Kaibara et al, 1999; Lee et al, 1998).
Our findings of control subjects' PPC values are comparable to the range of PPC activity previously reported in mammalian brains (range 0% to 8% in isolated rat or guinea pig brains) (Beaconsfield and Liuzzi, 1963; Ben-Yoseph et al, 1995; DiPietro and Weinhouse, 1959; Gaitonde et al, 1983; Guerra et al, 1967; Hostetler and Landau, 1967; Hostetler et al, 1970; Krass and Labella, 1967). Higher levels (16%) have been found in peripheral nerve tissue in the superior cervical ganglion in rats (Harkonen and Kauffman, 1974). An appropriate comparison for our study used adult monkey brains and found PPC activity of 5.6% in the cerebral hemispheres, 5.4% in brainstem, 6.4% in hypothalamus, and 8% in cerebellum (Hostetler et al, 1970). Recent studies from our laboratory of brain tissue extracts in a rat brain injury model confirm the increase in PPC flux after TBI (Bartnik et al, 2005).
Limitations
The PPC activity values obtained for control and TBI subjects had a large amount of intersubject variability. This variability may be related to (1) the true individual variability related to the pathophysiology of acute TBI, (2) the small sample size, (3) differences in postinjury day of study, or (4) the intrinsic error of this technique. Although we have not found the normal PPC flux to correlate significantly with any demographic or physiologic variables that we collected except arterial CO2 levels, it may normally vary depending on diet, environmental exposure (e.g., smoking, stress, temperature), emotional stimuli, or level of cognitive activity. In trauma patients, the PPC activity may also be related to parameters, such as oxidative stress level (free radicals), protein synthesis, cell membrane turnover and synthesis, or DNA synthesis, all of which may vary after injury in a time-dependant manner. The physiologic basis for the correlation with arterial CO2 levels is not clear and might due to the small sample size (Table 1b). Further studies with analysis of the level of oxidative stress are planned to further define clinical predictors, time course, and outcome correlated with PPC activity.
The primary shortcoming of this study technique is that the method does not sample brain tissue directly. Differences of lactate isotopomers in arterial and jugular venous blood are used as a surrogate marker to extrapolate brain metabolism. Although this method has proven useful in determining CMRs and blood flow by the Kety–Schmidt technique, it is prone to error. At some time points, the arterial levels of isotopomers of lactate are higher than the jugular, indicating cerebral consumption of lactate. At these time points, we are unable to separate the brain's contribution to the metabolism from the background of systemic metabolism. Thus, the AV difference (jugular minus arterial) is negative and the PPC calculation is uninterpretable. For this reason, the PPC value for the seventh TBI subject could not be determined and that patient was excluded from the study, as explained in the methods section.
The arterio-venous blood sampling technique is a global technique that only allows estimation of total brain activity. In contrast, TBI is typically marked by regional heterogeneity of hemorrhage, CBF, and metabolism. For example, whereas some tissue directly in or adjacent to contusions or hematomas may be dysfunctional or even necrotic, other adjacent areas may simply be stressed by the injury and distant sites may be normal. This study does not attempt to address this regionalism of brain metabolism; so it is possible the PPC may be greatly elevated in some areas, whereas normal in others. Pentose phosphate cycle activity may likewise vary by cell type (glial versus neural) and may vary over time; since only one PPC determination was made in each patient, its fluctuation over time remains unknown. This study also assumed that lactate derived from the PPC and glycolysis was intermixed and equally able to appear in jugular blood. It is possible that in fact there is some compartmentalization and that lactate derived from one pathway is more likely to leave the brain. However, the final enzymatic steps in the production of pyruvate and lactate are common to both glycolysis and the PPC, likely sharing enzymatic machinery. Lastly, PPC flux was determined relative to glycolytic activity, not necessarily accounting for all the excess glucose that was consumed by nonoxidative pathways in the brain, such as glycogen synthesis. Other studies have suggested that these fluxes may be increased after injury as well (Otori et al, 2004).
An additional important finding was that the labeled-glucose enrichment in the control subjects was on average greater than the enrichment obtained in the TBI patients (Figure 2). Trauma patients in the intensive care unit tend to have higher blood glucose levels than control subjects and their glucose homeostasis is impaired. Therefore, it is possible that the lower enrichments seen in these subjects is simply due to greater dilution in a larger pool of whole blood glucose and poor compensation for the extra glucose load. One may be concerned that this differential enrichment could drive the observed difference in PPC activity. However, the PPC calculation is dependent on the ratio of isotopomers derived from glycolysis versus the PPC, not on the absolute values. Therefore, the level of enrichment does not affect the results. Convincingly, within each of the groups, the percent enrichment does not correlate with the PPC calculated and therefore does not impact the final result (Tables 2 and 3).
Implications and Future Direction
Numerous studies of neuroprotective agents, after showing benefit in animal TBI models, have failed to yield a beneficial agent for human TBI (Maas et al, 1999). One possible explanation for this failure is that experimental TBI is very homogeneous, whereas human TBI is very heterogeneous in terms of patient age, comorbidities, diet, prior health, presenting symptoms, mechanism of injury, and type of intracranial injury. Although some neuroprotective agents may be beneficial in a subset of patients with precisely defined characteristics, when studied in aggregate, no benefit is seen. Combination treatments tailored specifically to each patient's specific metabolic derangement over time may be more effective. For example, oxidative stress is a known mechanism of secondary injury after TBI (Hall et al, 1994, 2004; Shohami et al, 1997; Smith et al, 1994). Therefore, the shunting of glucose metabolism toward the PPC may represent a compensatory response attempting to limit further oxidative damage. Hypothetically, those patients with the greatest oxidative burden may have the most elevated PPC as a compensatory response to help deal with the stress and prevent secondary injury. A further area of study would be to determine if the elevation in PPC flux truly correlates with the level of free radicals. If this were shown to be the case, then perhaps the PPC flux could be used to predict which patients would benefit from free radical scavenger-based neuroprotective agents (such as polyethylene glycol-conjugated bovine superoxide dismutase, vitamin E, and tirilazad mesylate; for review see Maas et al, 1999).
Improved techniques are clearly needed to measure quantitatively and directly metabolic fluxes in the brain after injury. [13C]-MR spectroscopy could make in situ brain studies possible (Gruetter, 2002; Rothman et al, 2003), although this technique is available at only a few centers currently. An injection of 13C-labeled glucose would be required, but it would obviate the need for arterial or jugular blood samples or brain samples. Magnetic resonance spectroscopy (MRS) would also provide a more regional analysis of metabolism. Therefore, various areas of the brain could be compared (white versus gray matter, ipsilateral versus contralateral to injury, pericontusional versus normal appearing brain, etc.). Finally, with higher enrichment of labeled glucose and use of additional labeled compounds such as 13C-labeled lactate, it would be possible to evaluate other pathways. For example, comparisons of glutamate and glutamine isotopomers can give information about fluxes through the TCA cycle, anaplerosis, and the relative activity of the pyruvate dehydrogenase complex versus the pyruvate carboxylase enzyme (Hyder et al, 1996; Lee et al, 1996; Rothman et al, 2003). Detection of 13C-label in proteins could show production of amino acids from glucose by way of TCA cycle intermediates.
Conclusions
Metabolic dysfunction and rearrangements after TBI may lead to an energy crisis and shunting of normal metabolic substrates from glycolysis and glucose oxidation to other pathways important for cellular repair, macromolecular synthesis, and secondary injury prevention, such as the PPC. This study using [13C]glucose labeling has both defined the normal resting range of PPC activity in healthy humans and demonstrated a significant increase in PPC flux after severe TBI. To our knowledge, this is the first study to define the PPC activity in normal human brain; it is also the first time these techniques have been applied to acute TBI patients in the intensive care unit to define these subjects PPC activity. Although this increased flux through the PPC did not correlate with clinical and metabolic indices, it possibly represents a shunting of substrates into intracellular pathways critical for preventing secondary injury. This finding indicates that no longer should it be assumed that after TBI the CMR of glucose measures glycolysis alone. Future studies to elucidate the time course, regionality, predictive factors, and impact on the outcome of this increased PPC activity in the acute period after severe TBI may help with the development of novel neuroprotective strategies that minimize secondary injury and maximize patient recovery.
Footnotes
Acknowledgements
We thank Maria Etchepare, RN and Jill Hutchinson, RN for their tireless work in recruiting and consenting subjects as well as running the clinical experiments. We thank John Boscardin, PhD for his help with biostatistics, Sara Bassilian for her expertise with the GC/MS technique and equipment, and Norman Lee for his help in processing the samples.