
Abstract
The purpose of this study was to examine what levels of hyperglycemia cause blood–brain barrier (BBB) disruption during permanent and transient middle cerebral artery occlusion in the rat and when the adverse effects of hyperglycemia occur. Cerebrovascular function was assessed by measuring the influx rate constant (Ki) for 3H-inulin and by measuring cerebral plasma (14C-inulin) and 51Cr-labeled red blood cell (RBC) volume. Different glucose protocols were used to produce mild sustained hyperglycemia (blood glucose ∼150mg/dL) or transient-severe hyperglycemia (with a spike in blood glucose of ∼ 400 mg/dL). As expected, transient-severe hyperglycemia at the time of occlusion induced marked BBB disruption in animals undergoing 2 h of ischemia with 2 h of reperfusion (25-fold increase in permeability compared with the contralateral core). However, the mild hyperglycemia model induced similar disruption. Similarly, after permanent occlusion, both hyperglycemia models enhanced disruption and they both produced marked (∼ 50%) reductions in cerebral plasma volume. Apparent cerebral RBC volume also decreased when measured during the final 5 mins of 2 h of ischemia with transient-severe hyperglycemia. However, there was no decrease if the 51Cr-labeled RBCs were circulated for the whole 2 h, indicating RBC trapping. The spike in blood glucose in the severe hyperglycemia model was used to examine when hyperglycemia induced BBB disruption. Hyperglycemia shortly after occlusion caused severe disruption. In contrast, hyperglycemia after 90 mins of occlusion caused little disruption. These results suggest that mild hyperglycemia has a profound effect on BBB function and that very early correction of hyperglycemia is necessary to prevent adverse effects.
Introduction
Hyperglycemia (HG) is a risk factor for augmented brain injury during human cerebral ischemia (Kushner et al, 1990; Pulsinelli et al, 1983). Higher admission glucose levels are associated with significantly lower odds of desirable clinical outcomes and significantly higher odds for symptomatic intracerebral hemorrhage, regardless of recombinant tissue plasminogen activator (rt-PA) treatment (Bruno et al, 1999). Acute, but not chronic, HG of greater than 158 mg/dL is also associated with a lower rate of recanalization in tPA-treated patients (Ribo et al, 2005), with tPA-treated, hyperglycemic patients having a worse outcome after early reperfusion in comparison to those with delayed or no reperfusion (Alvarez-Sabin et al, 2004).
Animal models of cerebral ischemia indicate that preischemic HG exacerbates brain injury after both global (Dietrich et al, 1993; Pulsinelli et al, 1982) and focal ischemia (de Courten-Myers et al, 1988; Nedergaard and Diemer, 1987) with reperfusion. Hyperglycemic-induced cerebral acidosis is generally considered the mechanism for enhanced brain injury (Chopp et al, 1988; Li and Siesjo, 1997), with most interest focused on injury to neurons and astrocytes. However, blood–brain barrier (BBB) opening during reperfusion is exacerbated by severe HG (Dietrich et al, 1993; Ginsberg et al, 1980; Siemkowicz, 1981) and can result in hemorrhagic conversion of the infarct (de Courten-Myers et al, 1992). We have proposed that damage to the brain endothelial cell during ischemia with HG, may have profound effects on the evolution of brain injury after reperfusion that could lead to hemorrhagic transformation during reperfusion (Kawai et al, 1997; Keep et al, 2005). Four hours of permanent occlusion of the middle cerebral artery (pMCAO) results in very significant decreases in the energy-dependent transport of taurine and glutamine and myoinositol (Keep et al, 1999) that are exacerbated by transient-severe HG, suggesting compromised endothelial cell function (Kawai et al, 1999). These effects of HG are associated with a reduction in plasma and red blood cell (RBC) volume during ischemia (Kawai et al, 1997; Kawai et al, 1999), perhaps indicative of a perfusion defect resulting from endothelial cell swelling (Keep et al, 2005).
Two unresolved issues in terms of the effects of hyperglycemia on BBB function during stroke are what levels of hyperglycemia are harmful and when are they harmful. The original NINDS rt-PA trial (Group NSS, 1995) used a serum glucose of > 400 mg/dL as an exclusion factor, but modest hyperglycemia is prevalent in stroke patients (Alvarez-Sabin et al, 2004). Most animal studies examining the effect of hyperglycemia on BBB function during cerebral ischemia have used acute severe hyperglycemia and not mild hyperglycemia. The question of when hyperglycemia has an adverse effect is important clinically because of difficulties in treating stroke patients soon after a stroke. The current GIST trial (Gray et al, 2004) aims to normalize blood glucose 8 to 24 h after stroke onset. The purpose of the current study was, therefore, to examine the effect of mild and severe hyperglycemia on BBB disruption in a rat model of focal cerebral ischemia (MCAO). We have used the fact that our severe model of ischemia causes a transient spike in blood glucose to examine when hyperglycemia has its most deleterious effects after an ischemic event.
Materials and methods
Animal Preparation and Middle Cerebral Artery Occlusion
The University of Michigan Committee on the Use and Care of Animals approved the protocol for these animal studies. Adult male Sprague–Dawley rats (Charles River Laboratories, Wilmington, MA, USA) weighing 275 to 350 g were used for all experiments. Rats were allowed free access to food and water before the experiment. Animals were anesthetized with pentobarbital (65 mg/kg, intraperitoneally) before any surgery. Body temperature was maintained at 37°C with a servo-controlled heating pad.
Rats underwent occlusion of the left MCA, with and without reperfusion, using the intraluminal thread method (Ennis and Keep, 2006; Longa et al, 1989) under pentobarbital anesthesia (65 mg/kg). The bifurcation of the common carotid artery was exposed and the external carotid artery was ligated distally. A 3–0 monofilament nylon suture, its tip rounded by heating, was introduced into the internal carotid artery lumen through the stump of the external carotid artery and gently advanced into the internal carotid artery 21 to 23 mm past the common carotid artery bifurcation to block the origin of MCA. Reperfusion was initiated through removal of the thread and tying off the distal external carotid artery.
Blood–Brain Barrier Disruption
Blood–brain barrier disruption was assessed by measurement of the influx rate constant (Ki) for 3H-inulin (New England Nuclear, Boston, MA, USA) using a method modified from Ohno et al (1978) with 14C-inulin (American Radiolabeled Chemicals, St Louis, MO, USA) as a marker for cerebral plasma volume (CPV). 3H-inulin (80 μCi) was injected through a femoral vein 15 mins before the end of the experiment, whereas 14C-inulin (15 μCi) was given as a second injection 0.5 mins before the end of the experiment. The arterial concentration of 3H-inulin was determined using a peristaltic pump (Gilson, Middleton, WI, USA) to create an artificial organ by continuous withdrawal of blood (0.1 mL/mins) into a polyethylene catheter (PE 205). At the end of the experiment, a terminal plasma sample was taken and the rat was killed by decapitation.
Brains were rapidly removed and the cortex ipsi- and contralateral to the MCA occlusion was sampled as described previously (Masada et al, 2001). In brief, the hemisphere was divided, and the brain stem, hippocampus, cerebellum, and thalamus were removed. The remaining telencephalic tissues ipsi- and contralateral to the MCAO were placed flat on a nonabsorbent surface and the portion inferior to the rhinal fissure was removed. Three samples (ipsi- or contralateral) were taken from the remaining tissue using 5- and 7-mm cork borers. The core region was taken from the cortex and striatum underlying the MCA immediately distal to the region of occlusion. The intermediate region was a ring of tissue surrounding the core zone, whereas the outer region consisted of the remainder of the cortical tissue. The results presented are from the core region unless otherwise stated.
Samples were immediately weighed and digested in methylbenzethonium hydroxide (Sigma-Aldrich, St Louis, MO, USA). Scintillation fluid (CytoScint, MP Biomedicals, Irvine, CA, USA) was added to the brain and plasma samples and radioactivity was counted by a Beckman 3801 liquid scintillation counter (Fullerton, CA, USA). The Ki was calculated from the formula
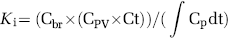
where Cbr is the counts per gram of brain, CPV is the plasma volume of the brain determined from the 14C-inulin space, Ct is the terminal plasma concentration of 3H-inulin, and ∫Cp dt is the integral of the 3H-inulin plasma concentration (Cp) for the experiment. The latter was calculated from the radioisotope content of the continuously withdrawn arterial blood sample. The 3H-inulin was purified using a G-25M Sephadex column (PD-10, Pharmacia, Piscataway, NJ, USA) before use.
Red Blood Cell Volume of the Brain
The effect of transient-severe HG on cerebral RBC volume during pMCAO was determined with RBCs tagged with 51Cr using a method described in detail previously (Kawai et al, 1999; Keep et al, 1995). Briefly, 1.5 mL of blood from a donor rat was washed and centrifuged, and the packed RBCs were incubated with 30 μCi of 51Cr for 1 h at 37°C. The labeled RBCs were then washed and kept on ice. Just before use, a 200-μL aliquot of cells was resuspended with 100 μL phosphate-buffered saline; this solution was injected into the femoral vein and allowed to circulate for either 5 mins or 2 h. The plasma volume was determined using 14C-inulin (15 μCi), given as a second injection 0.5 mins before the end of the experiment. At the end of the experiment, a terminal arterial blood sample was withdrawn and the animal was decapitated. Brain sampling and preparation were as described for measuring influx rate constants. Whole-blood samples were bleached with 30% H2O2 before counting. The cerebral RBC volume for each sample was calculated as (dpm/g brain) × (systemic hematocrit)/(dpm/mL whole blood).
Hyperglycemia and Experimental Groups
The present study contrasts the response of the BBB to mild and severe HG during permanent and transient cerebral ischemia. Mild HG was produced by intraperitoneal injection of 10 to 30 mL/kg of 300 mmol/L glucose, 30 mins before occlusion. Control animals received the same intraperitoneal dose of 300 mmol/L saline. Both control animals and those receiving 300 mmol/L glucose underwent ligation of the renal artery and vein to enhance the response to the glucose load. Owing to a variable blood glucose response to the injected glucose load, only animals with 60 mins blood glucose above 150 mg/dL were included in the study. Severe HG was induced by intraperitoneal injection of varying doses (range 2.5 to 10 mL/kg) of 2.8 mol/L glucose. Arabinose (2.8 mol/L) was injected as an osmotic control. The renal arteries and veins were not ligated in animals undergoing transient-severe HG.
The experiments were in five parts. The first part examined the effect of transient-severe and mild hyperglycemia on BBB disruption in rats undergoing transient MCAO. Animals undergoing severe HG received 10 mL/kg of 2.8 mol/L glucose, intraperitoneally, 30 mins before occlusion, with either saline- or 2.8 mol/L arabinose-injected animals serving as controls. For mild hyperglycemia, 300 mmol/L glucose (10 to 30 mL/kg) was injected intraperitoneally, 30 mins before occlusion. Saline was injected as a control. Rats underwent 2 h of ischemia followed by 2 h of reperfusion (2/2-I/R). BBB disruption (3H-inulin Ki) was determined over the last 15 mins of the experiment. Plasma volume was measured during the last 0.5 mins, using 14C-inulin to correct for intravascular content.
In the second part, the effect of different levels of blood glucose on the inulin Ki during 2/2-I/R was measured by injecting 2.5, 5.0, or 7.5 mL/kg of 2.8 mol/L glucose, 5 mins after MCAO.
The third part investigated the influence of the timing of the injection of glucose on the inulin Ki. Animals were treated 30 mins before or 5, 30, 90, or 125 mins (inulin Ki) after occlusion of the MCA with 5 mL/kg of 2.8 mol/L glucose. Arabinose (2.8 mol/L, 5 mL/kg), injected either 5 or 125 mins after MCAO, served as control.
The fourth part examined the effect of severe and mild hyperglycemia on BBB disruption in rats undergoing pMCAO. Rats underwent 2.5 h of pMCAO during mild HG or 2 and 4 h of pMCAO during severe HG (10 mL/kg glucose; intraperitoneally, 30 mins before occlusion). BBB disruption (3H-inulin Ki) was determined over the last 15 mins of the experiment. Plasma volume was measured during the last 0.5 mins, using 14C-inulin to correct for intravascular content.
The final part investigated a potential defect in cerebral perfusion caused by severe HG (10 mL/kg, 2.8 mol/L glucose, intraperitoneally, 30 min before occlusion) during pMCAO. Chromium-51-labeled RBCs were circulated either during the final 5 mins or during the entire 2 h of pMCAO. Cerebral plasma volume (CPV) was measured over the final 30 secs of each type of experiment.
Statistical Analysis
Data in this study are presented as mean ± s.e. Data were analyzed with Student's t-test except when multiple comparisons were made using ANOVA, which was followed by either a Newman–Keuls test or a Dunnetts test. Statistical significance was accepted at P ≤ 0.05.
Results
Physiological Parameters
Tables 1 and 2 show the terminal physiological parameters for the 2/2-I/R and pMCAO groups, respectively. Apart from some minor changes in arterial pH, the only differences between vehicle, mild HG, and severe HG were an increase in hematocrit in the latter. As expected, the glucose injection protocols induced HG. Figure 1 shows the time course of blood glucose in the 2/2-I/R animals. During mild HG, blood glucose reached a stable plateau between 60 and 90 mins after intraperitoneal administration, and remained almost constant throughout the remainder of the experiment. Severe HG, with no kidney ligation, produced a sharp spike in blood glucose (359 ± 12mg/dL) that peaked around 15 mins after injection. Because of the nature of this glucose response, we will henceforth call that model a transient-severe HG. Vehicle-injected animals displayed a stable blood glucose of approximately 125 mg/dL. Animals undergoing pMCAO, rather than 2/2-I/R, showed similar glucose concentrations.
Physiological parameters for animals undergoing 2/2-I/R

Values are means ± s.e. measured at the end of the experiment.
* Saline versus hyperglycemia
mild versus severe hyperglycemia. One, two, or three symbols indicate a significant difference at the P ≤ 0.05, P ≤ 0.01, or P ≤ 0.001 levels, respectively.
Physiological parameters for animals undergoing pMCAO
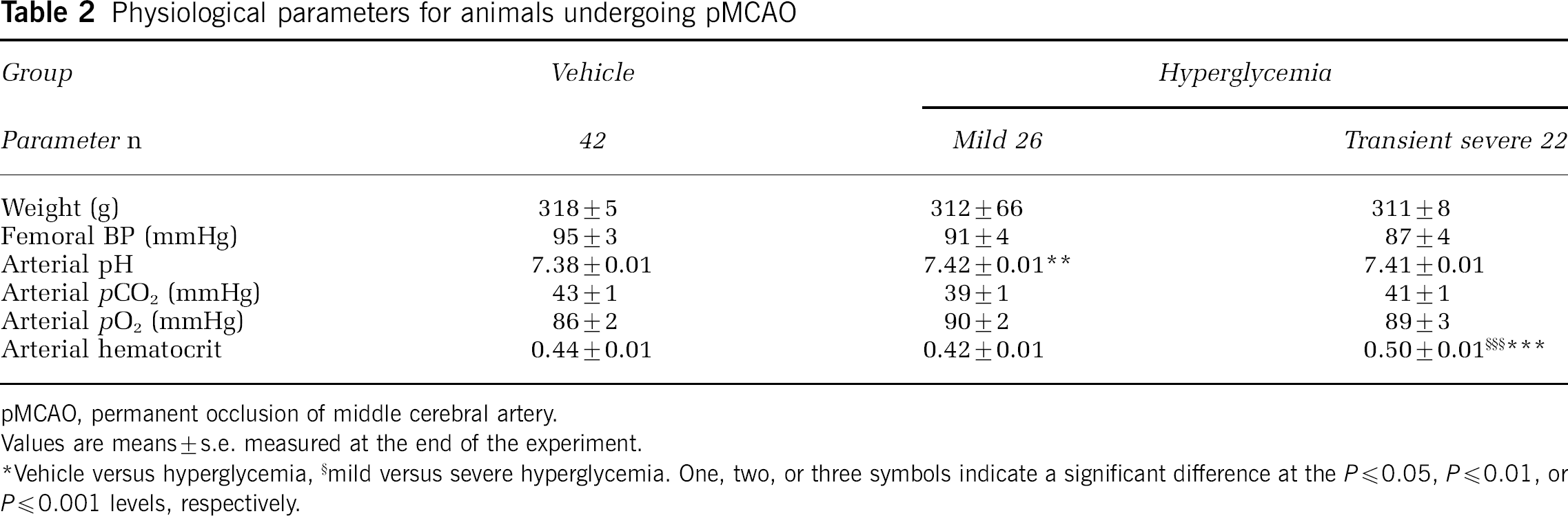
pMCAO, permanent occlusion of middle cerebral artery.
Values are means ± s.e. measured at the end of the experiment.
* Vehicle versus hyperglycemia
mild versus severe hyperglycemia. One, two, or three symbols indicate a significant difference at the P ≤ 0.05, P ≤ 0.01, or P ≤ 0.001 levels, respectively.
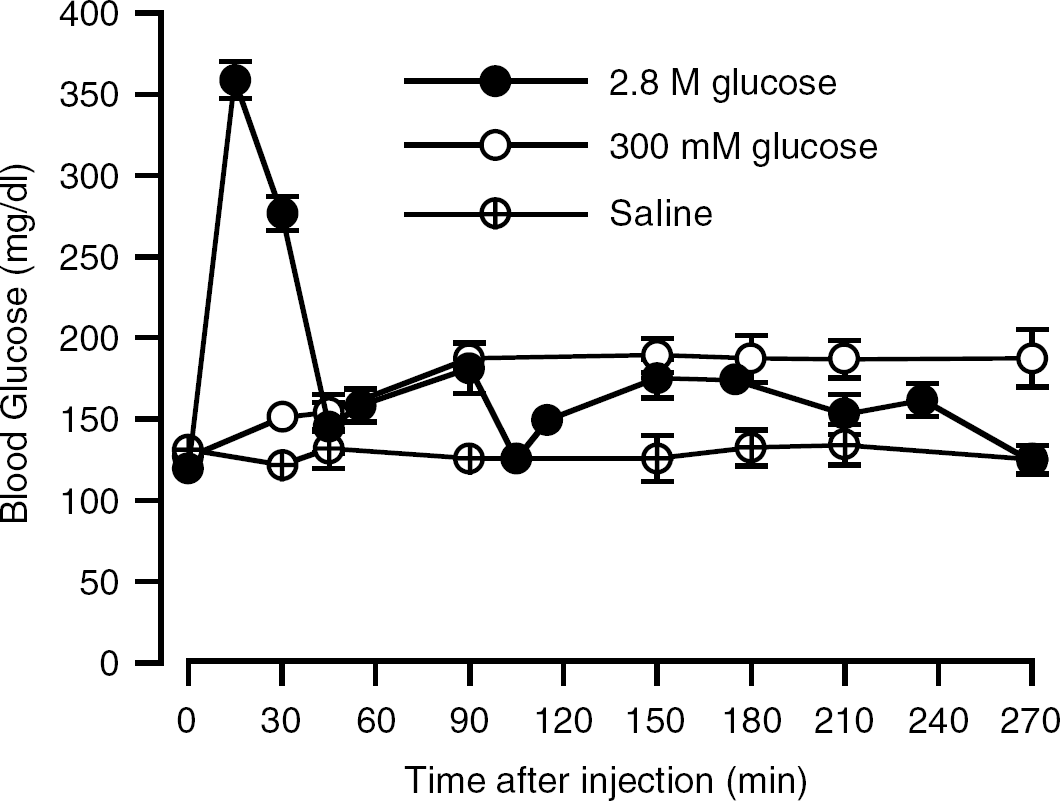
Blood glucose after intraperitoneal glucose injection in the mild and transient-severe HG models. Animals undergoing mild HG received glucose (300 mmol/L, 10–30 mL/kg) 30 mins before occlusion of the MCA. Transient-severe HG was produced using injection of 5 mL/kg of 2.8 mol/L glucose at various times before or after MCAO. All animals were reperfused at 120 mins. The glucose time course did not differ between various injection times and the results are pooled. Values are means ± s.e.
2 h Ischemia followed by 2 h Reperfusion of the MCA
Rats undergoing 2/2-I/R with transient-severe HG (10 mL/kg, 2.8 mol/L glucose) had a very marked increase in the influx rate constant (inulin Ki) for 3H-inulin in the ischemic core (1.67 ± 0.15 μL/g/min) compared with that found in either saline-(0.21 ± 0.05 μL/g/min) or arabinose-treated rats (Figure 2A). All of the animals in the glucose-injected group also presented with hemorrhagic conversion in the core and intermediate regions at the time of killing. Mild HG during 2/2-I/R (Figure 2B) also produced a large increase in the 3H-inulin Ki in the ischemic core (1.36 ± 0.15 versus from 0.22 ± 0.05 μL/g/min in saline-injected controls; P < 0.001). Unlike transient-severe HG, mild HG during 2/2-I/R did not result in a macroscopic hemorrhagic conversion. The increase in inulin permeability during 2/2-I/R produced by mild or transient-severe HG was not significantly different as shown by ANOVA (P ≥ 0.5).
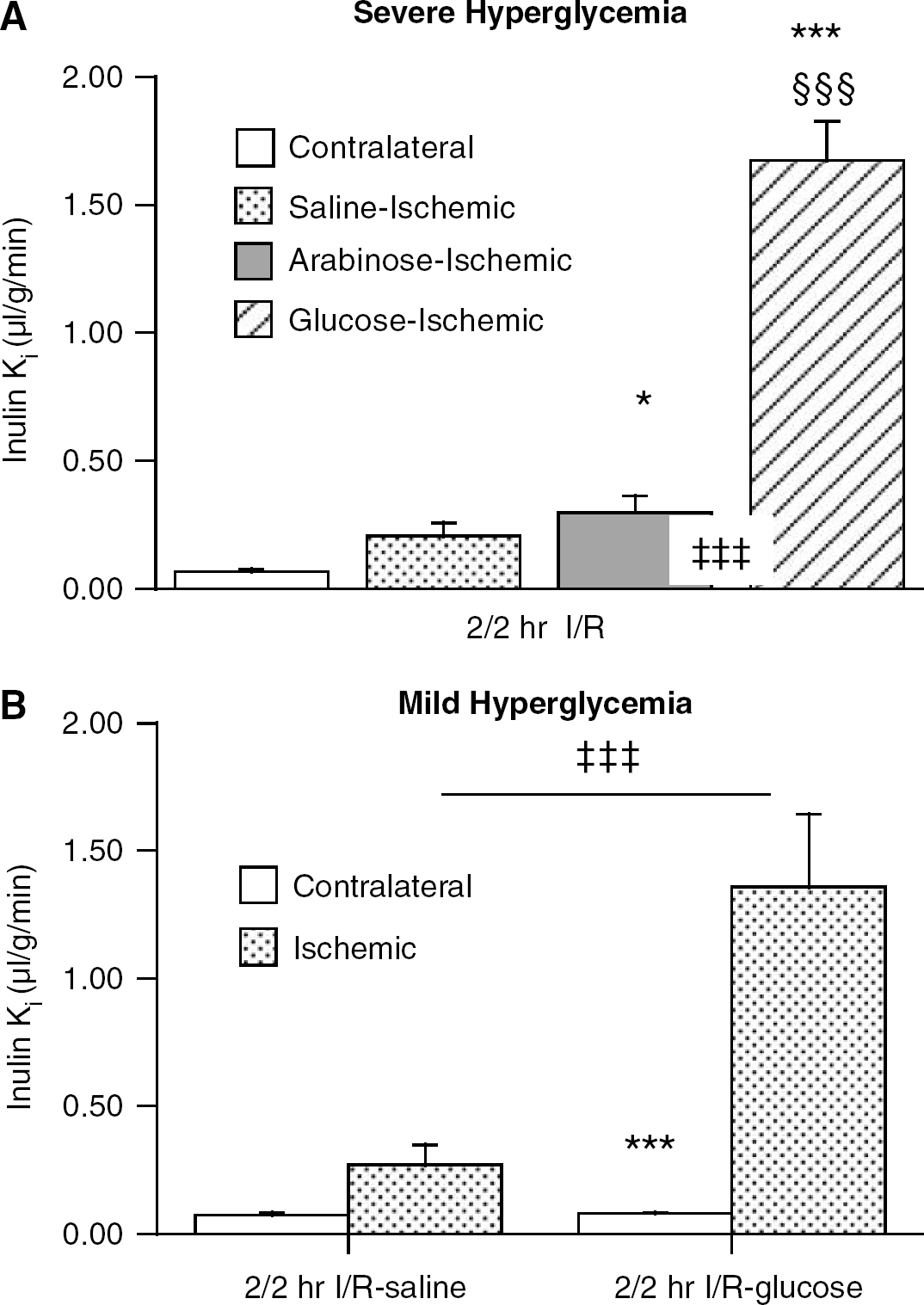
The effect of transient -severe (
The importance of blood glucose concentration during early ischemia was tested in rats that were injected 5 mins after occlusion of the MCA with 2.5, 5.0, or 7.5 mL/kg of 2.8 mol/L glucose or arabinose (5.0 mL/kg). The Ki for 3H-inulin (Figure 3A) increased in the core of the ischemic hemisphere after 2/2-IR from 0.19 ± 0.03 μL/g/min in arabinose-treated animals to 0.59 ± 0.23, 2.72 ± 0.94, and 2.45 ± 0.59 μL/g/min in animals receiving 2.5, 5.0, and 7.5 mL/kg of 2.8 mol/L glucose, respectively (Figure 3A). The blood glucose concentrations (measured 15 mins after injection) in each of these groups were 246 ± 18, 370 ± 16, and 447 ± 15 mg/dL, respectively.
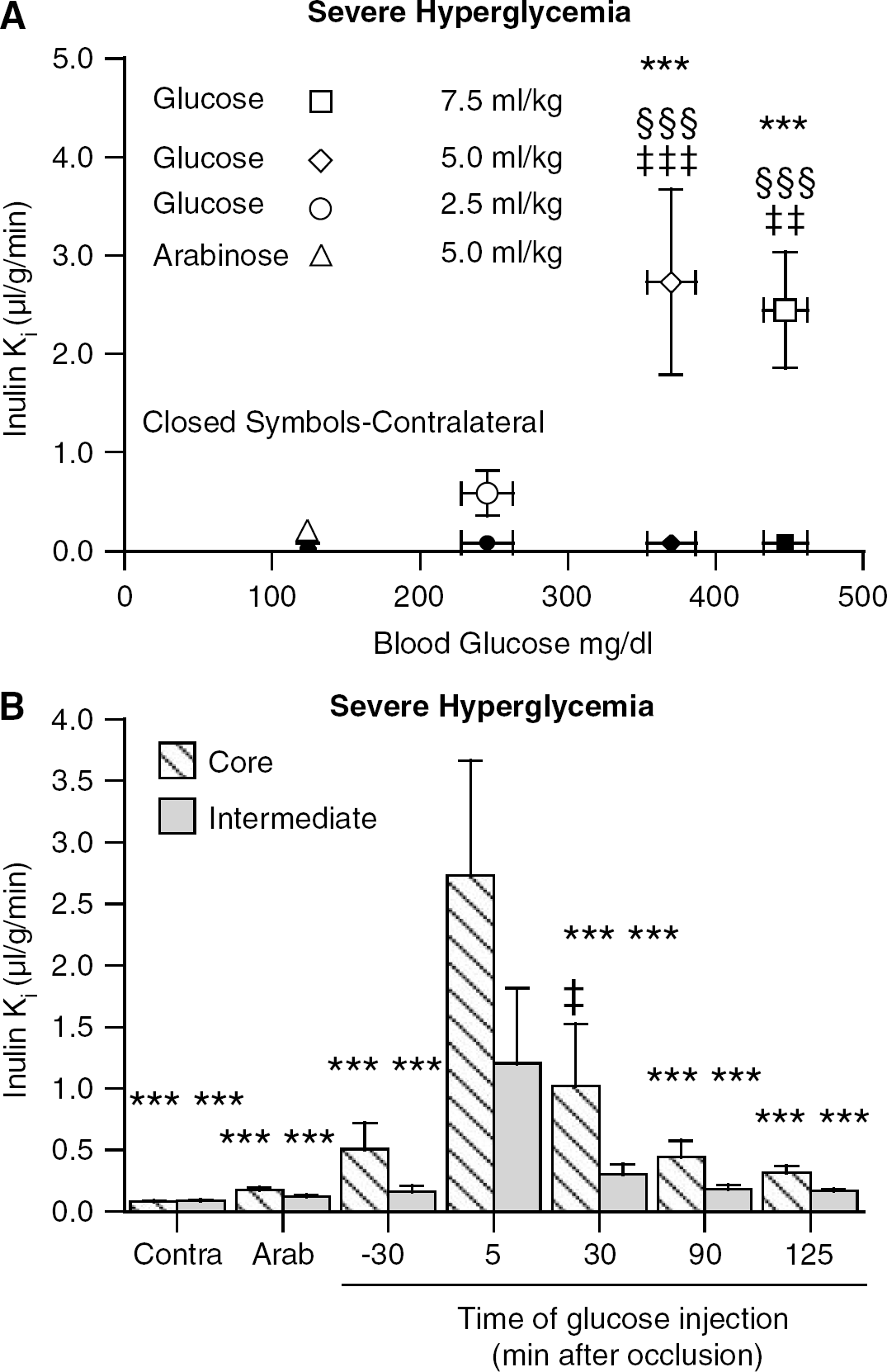
(
The sharp spike in glucose in the severe hyperglycemia model allowed assessment of the importance of the timing of hyperglycemia relative to MCAO in inducing BBB disruption during 2/2-IR. Rats received 5.0 mL/kg of 2.8 mol/L glucose, 30 mins before or 5, 30, 90, or 125 mins after occlusion of the MCA (Figure 3B). Arabinose (2.8 mol/L, 5 mL/kg), injected either 5 or 125 min after MCAO, served as control. In this instance, glucose administered 30 mins before MCAO produced a modest increase in inulin Ki in the ischemic core (0.51 ± 0.21 versus 0.18 ± 0.02 μL/g/min in arabinose-treated animals). Glucose given 5 mins after MCAO produced the largest increase in the inulin Ki to 2.72 ± 0.93 μL/g/min. The increase in the inulin Ki showed a progressive decline to 0.32 ± 0.05 μL/g/min as the time of administration of glucose increased to 125 mins after MCAO (i.e. 5 mins after reperfusion). Interestingly, the inulin Ki in the intermediate region of the stroke was also markedly increased (1.21 ± 0.61 μL/g/min) when glucose was given 5 mins after the stroke, but not at any of the other time points. The intermediate zone in our sampling scheme corresponds to the ischemic penumbra identified by Belayev et al (1997) as surrounding the core in this model of MCAO.
Permanent Middle Cerebral Artery Occlusion
In saline-treated controls, permanent MCAO (2.5 h) caused a modest 20% increase in inulin Ki in the ischemic core (0.088 ± 0.003 versus 0.074 ± 0.004 μL/g/min in the contralateral hemisphere; p < 0.05; Figure 4A). As with transient ischemia, mild HG significantly exacerbated that disruption (P ≤ 0.001) causing an 86% increase in inulin Ki (Figure 4A).
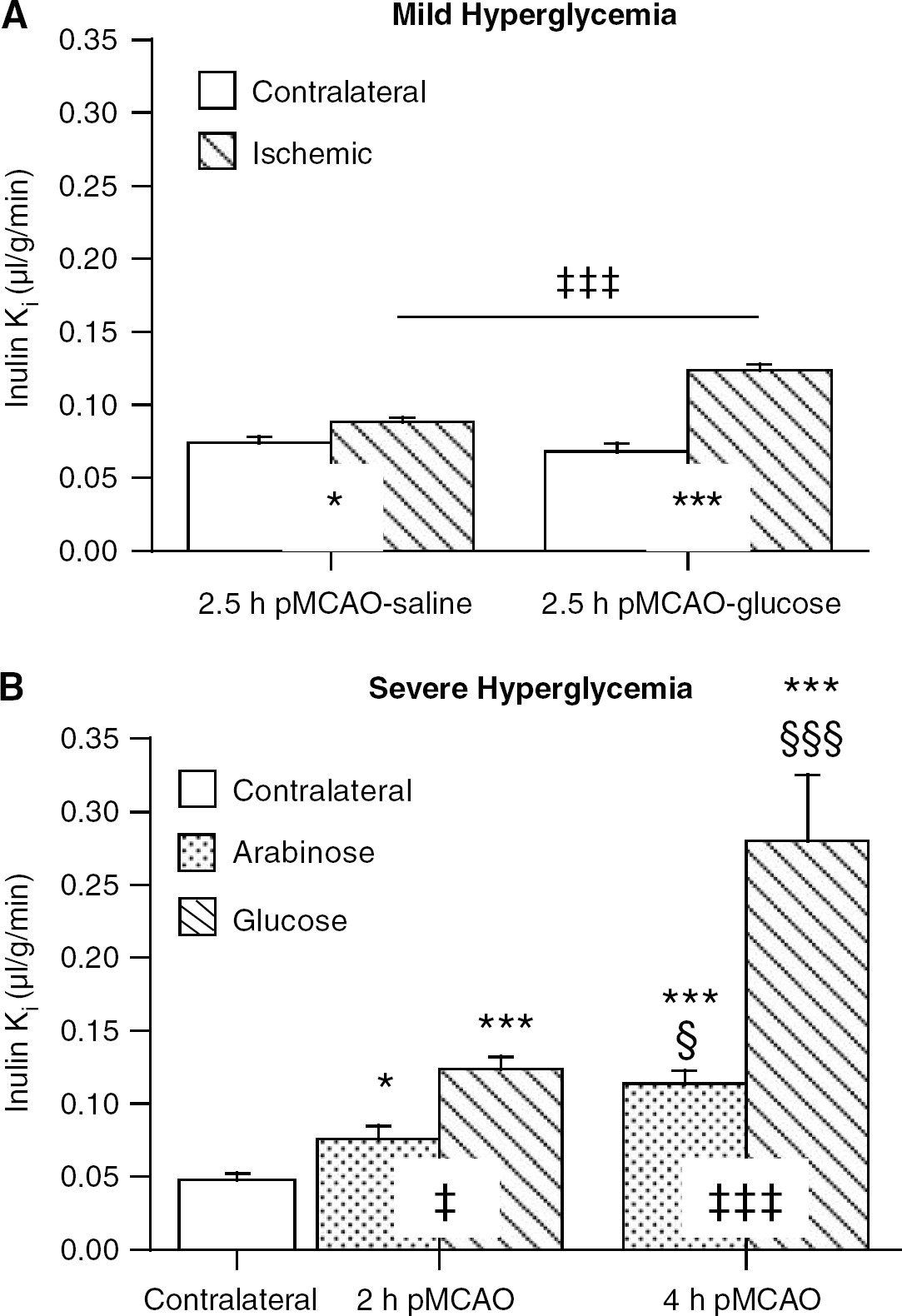
The effect of mild (
Transient-severe HG also significantly increased the inulin Ki in the ischemic core after either 2 or 4 h of pMCAO (Figure 4A). Animals were treated 30 mins before occlusion with either arabinose or glucose (both 2.8 mol/L, 5 mL/kg). In arabinose-treated rats, 2 and 4 h of pMCAO increased the inulin Ki in the ischemic core by 60% and 138% (compared with contralateral). In glucose-treated rats, the transient-severe HG resulted in a 2.5- and 6-fold increase (compared with the contralateral core) in inulin Ki at 2 and 4 h, respectively. Both increases were significantly greater than their respective arabinose-treated controls (Figure 4A). It should be noted that the increase in inulin Ki in the ischemic core during 2 h of transient-severe HG (0.124 ± 0.008 μL/g/min) was very similar to that during 2.5 h of mild HG (0.123 ± 0.004 μL/g/min).
We have previously found that transient-severe HG causes a 45 ± 13% reduction in cerebral plasma volume in the ischemic core during pMCAO (Kawai et al, 1999). We speculated that this large decrease in plasma volume might reflect either endothelial cell swelling or plugging of the microvasculature. Figure 5 contrasts the effect of mild (A) and transient-severe HG (B) on the plasma volume. Permanent MCAO resulted in very small reductions (≈12%) in the plasma volume of control animals (Figure 5A and 5B). Both mild and transient-severe hyperglycemic groups experienced approximately 50% reductions in comparison to their respective ischemic controls (P ≤ 0.001).
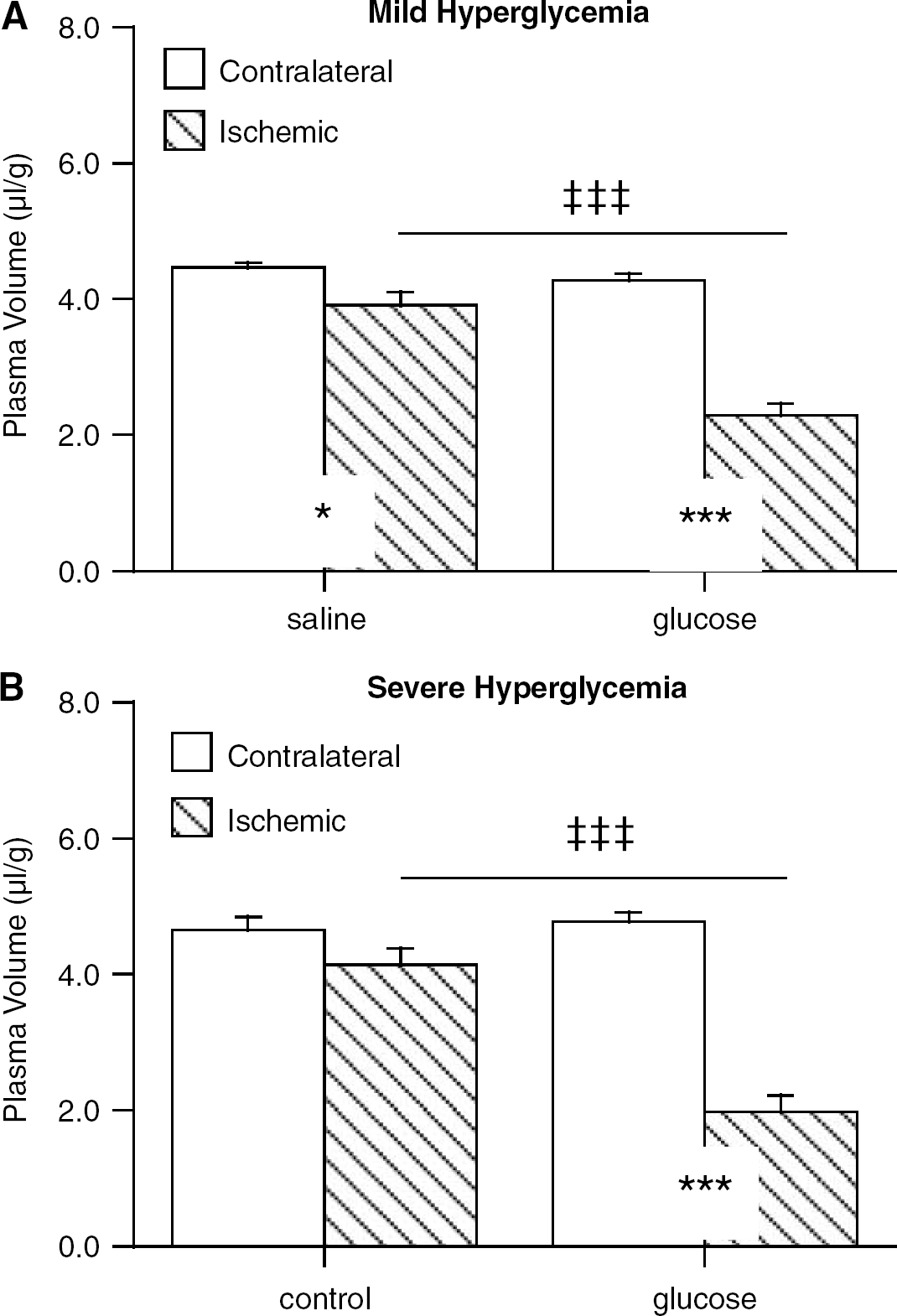
The effect of mild (
The defect in perfusion in the core of transient — severe hyperglycemic animals during pMCAO was further investigated using 51Cr-labeled RBCs. When the RBCs were circulated during the last 5 mins of 2 h of ischemia, a marked decrease in the cerebral RBC volume in the core of the ischemic core was noted (0.50 versus 2.48 μL/g in the contralateral hemisphere; P ≤ 0.001; Figure 6A). In contrast, when labeled RBCs were circulated for 2 h (i.e. from the time of occlusion), there was no difference in cerebral RBC volume between ischemic core and the contralateral hemisphere (P ≥ 0.2). These data suggest that RBCs are trapped in the ischemic brain as circulation time increases. Interestingly, the reductions in the plasma volume (Figure 6B), measured with a 30 secs circulation at the end of the 2 h of pMCAO, were very similar in both groups of rats.
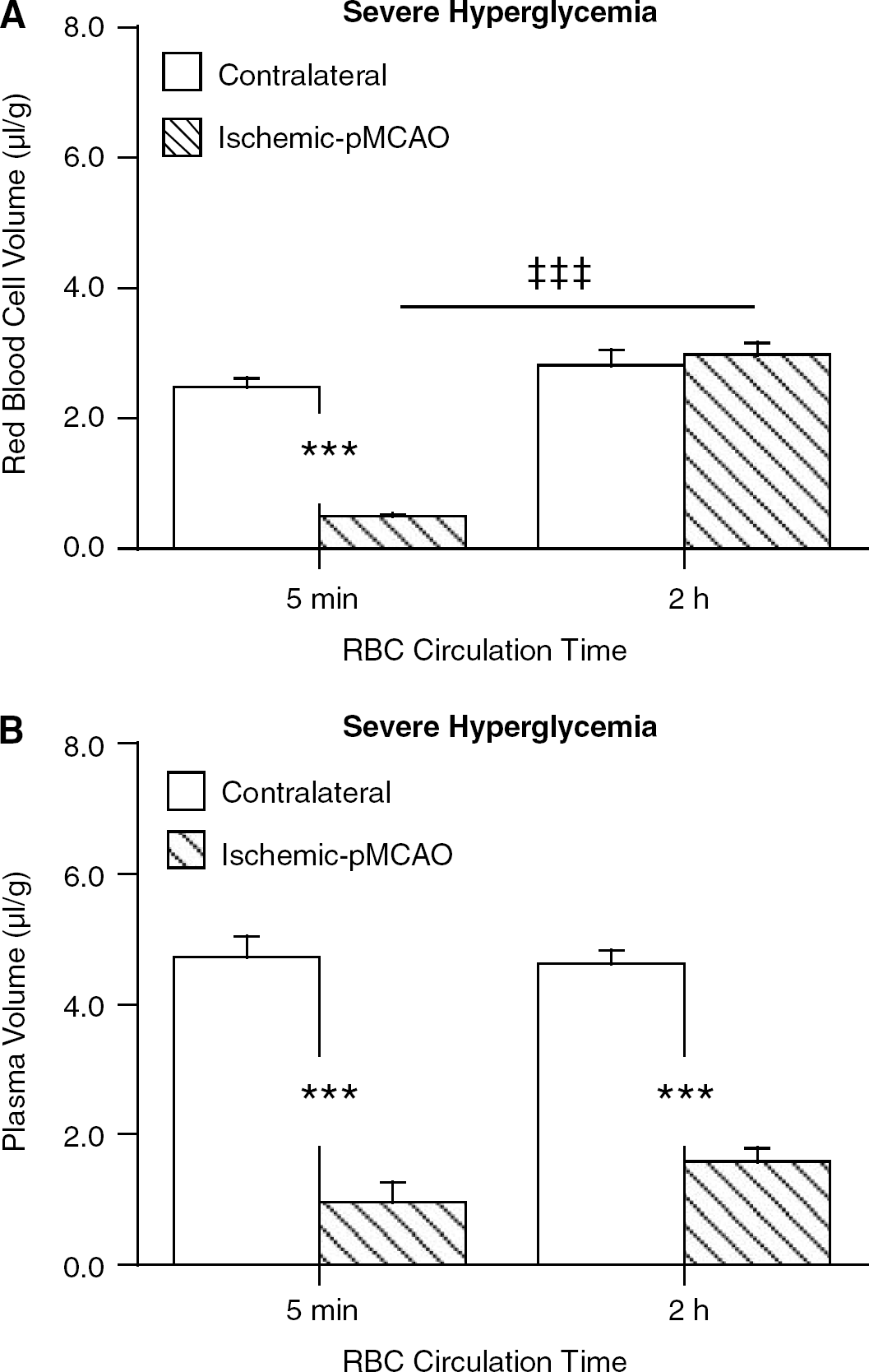
Red blood cell (RBC) volume (
Discussion
This study compares the effects of acute mild and transient-severe HG on the cerebral vasculature during permanent MCAO and during ischemia/reperfusion, and examines the effects of HG on BBB disruption at different stages of ischemia. The results show that even mild hyperglycemia (∼ 150 mg/dL) can have profound effects on cerebrovascular function and that much of the adverse effects of HG occur soon after the onset of ischemia, although these adverse effects become most evident when the animal is reperfused.
The current study used the influx rate constant for 3H-inulin to assess BBB disruption. Under the appropriate conditions of unidirectional uptake and a low rate of transfer relative to blood flow, the influx rate constant approaches the permeability–surface area product (PS) for that compound (Fenstermacher et al, 1981). Under normal circumstances, BBB permeability of most passively distributed molecules is determined by lipid solubility (Fenstermacker and Rapoport, 1984). However, ischemia results in the appearance of a few, relatively large water-filled pores (Ewing et al, 2003; Knight et al, 2005; Preston and Foster, 1997) probably reflecting tight junction disruption (Brown and Davis, 2002; Brown et al, 2004). Preston and Webster (2002) measured the differential movement of sucrose (MW = 342) and inulin (MW ≈ 5000) and found that sucrose showed a larger increase in Ki than for inulin and that the ratio of the change in Ki values (Δsucrose/Δinulin) approximated the ratio of their free diffusion coefficients in water, strongly suggesting the formation of water-filled pores. It should be noted, however, that because of relative permeabilities at the normal BBB, large molecular weight compounds such as inulin undergo a greater percentage change in Ki than smaller compounds, hence our choice of 3H-inulin for the current study. It should also be noted that because Ki is a measure of the PS product, changes in vascular surface area (S) would alter Ki. A reduction in the area of perfused vessels may offset the effect of an increase in permeability (P) on Ki during ischemia.
The advent of t-PA therapy to induce recanalization after embolic stroke, with the increased risk of hemorrhagic transformation (Dijkhuizen et al, 2002), has highlighted the need for a better understanding of endothelial cell dysfunction during the first hours of ischemia, before reperfusion (Keep et al, 2005). Hyperglycemia is one of the known risk factors for intracerebral hemorrhage, regardless of t-PA treatment (Bruno et al, 1999), which is certainly a result of massive opening of the BBB. The first important observation of the current study is the devastating effect of mild hyperglycemia (averaging only ∼ 150 mg/dL) on the integrity of the BBB during ischemia/reperfusion. Most experimental studies of hyperglycemia during cerebral ischemia have induced blood glucose values above 250 mg/dL. Here, mild HG (151 ± 8 mg/dL) induced about a 16.5-fold increase in inulin Ki in the ischemic compared with the contralateral core during 2/2-I/R. This increase in permeability was similar to that induced by acute transient-severe HG. In support of the current findings that mild hyperglycemia can have adverse effects, there is evidence in humans that acute but not chronic HG greater than 158 mg/dL results in worse outcome for t-PA patients experiencing early reperfusion in comparison with those with delayed or no reperfusion (Alvarez-Sabin et al, 2004). Such results suggest that insulin therapy in stroke patients should probably target blood glucose below 140 mg/dL to reduce the risk of hyperglycemic-induced opening of the BBB.
During permanent MCAO, mild HG also increased BBB permeability. Although that increase (1.8-fold) was less remarkable, it was again similar to that found with transient-severe HG. Our results also provide other evidence that mild HG has major adverse effects on cerebrovascular function during permanent MCAO in that there was an approximate 50% reduction in cerebral plasma volume in ischemic core of such animals. This reduction was again similar to that found in the transient-severe HG model used.
We have previously noted a decrease in cerebral plasma volume in permanent MCAO rats subjected to severe hyperglycemia (Kawai et al, 1997) and have suggested that this is due to endothelial cell dysfunction and swelling (Keep et al, 2005). In the current study, we report that the reduction in cerebral plasma volume is also associated with a reduction in apparent cerebral RBC volume as assessed with 51Cr-labeled RBCs circulated for 5 mins. However, when the tagged RBCs were circulated for the whole 2 h of ischemia, there was no difference in cerebral RBC volume between ischemic and non-ischemic hemispheres. This indicates that in hyperglycemic rats, tagged RBCs cease to be able to penetrate, or only penetrate very slowly, the ischemic core as ischemia progresses (as indicated by the 5 mins cerebral 51Cr-RBC volumes) and that RBCs become trapped within the ischemic vasculature of such rats (as indicated by the difference between 5 and 120 mins cerebral 51Cr-RBC volumes).
If, in the hyperglycemic brain, RBCs become trapped within areas of the ischemic vasculature, this may result in areas of zero oxygen delivery and marked vascular damage. As those areas would not be perfused, this would not be reflected by marked increases in BBB permeability. However, if blood flow is restored to those areas, marked BBB disruption and even hemorrhagic transformation may occur. Thus, it is our hypothesis that adverse effects of hyperglycemia occur during ischemia (see below) but that the adverse effects on BBB permeability may only become most evident during reperfusion. If the trapping of RBC is an important cause of cerebrovascular dysfunction or if such trapping is a marker of dysfunction, it should be possible to image such trapping with magnetic imaging resonance (MRI) (Singer et al, 2003). An experimental study in hyperglycemic rats has documented a decrease in cerebral blood volume in hyperglycemic rats using MRI (Quast et al, 1997). It is interesting that all blood vessels appear to be perfused after reperfusion in hyperglycemic rats (Li et al, 1998), suggesting that reperfusion can restore RBC flow.
It should be noted that we did not find marked reductions in cerebral plasma volume in normoglycemic rats during MCAO. Indeed there is good evidence that with normoglycemia, almost all blood vessels are continuously perfused during MCAO (Vogel et al, 1999). However, we have found decreased cerebral plasma volume in control rats subjected to MCAO when xylazine/ketamine is used as the anesthetic. That anesthetic and several others induce acute hyperglycemia (Kawai et al, 1997). Those results led us to use pentobarbital in the current experiments, as it does not affect blood glucose. However, it is possible that the effects of pentobarbital on tissue glucose handling (Gjedde and Rasmussen, 1980) could also modify the effects of hyperglycemia.
An alternate approach to examining RBC trapping as a measure of cerebrovascular dysfunction that might lead to hemorrhage upon reperfusion is to examine BBB disruption. The ability to quantify the change in the influx rate constant for gadolinium-diethylene triamine pentaacetic acid (GD-DTPA) using MRI in patients experiencing cerebral ischemia may allow for a more precise determination of when the passive permeability of the BBB has increased and passed a point when t-PA therapy is no longer advisable (Ewing et al, 2003; Knight et al, 2005). The relative sensitivity of these measures remains to be elucidated.
It is not possible to directly compare the effects of the mild and transient-severe HG models in this study. The doses of glucose used were very different (0.7 mmol/kg for the mild HG model and 7 to 28 mmol/kg in the transient-severe model), but the time profiles of the two models were also very different. Thus, although the peak hyperglycemia was much greater in the transient-severe model compared with the mild model, the longer term glucose concentrations were not (Figure 1). Thus, we examined the effects of different glucose loads using the same injection protocol (Figure 3A). The results indicate that the degree of hyperglycemia does affect BBB disruption, with animals that received 5.0 rather than 2.5 mL/kg of 2.8 mol/L glucose having an approximate fivefold greater increase in inulin permeability during 2/2-I/R. There was, however, evidence of a plateau effect with disruption by 5.0 and 7.5 mL/kg of 2.8 mol/L glucose not being significantly different.
The acute spike in blood glucose after the intraperitoneal injection of 2.8 mol/L glucose allowed an examination of when during stroke glucose has adverse effects. The results clearly demonstrated that HG at the time of occlusion or soon after occlusion had the worse effects on BBB disruption. Injection of glucose after 90 mins of ischemia or after 120 mins of ischemia and 5 mins of reperfusion had very little effect on BBB disruption. The current GIST trial is evaluating the utility of insulin treatment to reduce post-ischemic hyperglycemia and poor outcome from stroke (Gray et al, 2004). The protocol was designed to reduce the blood glucose to 4 to 7 kg/L, 8 to 24 h after admission. The results of the current study suggest that delayed insulin treatment may not prevent most of the adverse effects of HG on the BBB.
In conclusion, this study shows that cerebrovascular function is compromised by both mild and transient-severe HG early during ischemia and that this probably leads to very large increases in BBB disruption during reperfusion. The ischemic phase of this model of hyperglycemic aggravated stroke is characterized by a perfusion defect that includes both a decrease in cerebral plasma volume and trapping of RBCs in the microvasculature. Finally, hyperglycemia exacerbated BBB disruption when present before or just after the onset of ischemia, but it had much less effect when hyperglycemia began after reperfusion. This suggests that therapies directed at ameliorating the effects of hyperglycemia should be given as soon as possible after stroke onset.