
Abstract
Excitotoxicity and oxidative stress mediate neuronal death after hypoxic—ischemic brain injury. We examined the possibility that targeting both N-methyl-
Introduction
Interruption of blood supply to the brain by local thrombosis, embolic particles, or blood vessel rupture can cause primary neuronal death in the ischemic core, accompanied by secondary death in the ischemic penumbra as a result of activation of multiple death pathways. Therapeutically, thrombolytic and anticoagulant drugs are used to counteract these events by increasing reperfusion and modifying coagulation.
Another target for therapeutic intervention is neuronal death, which persists for hours and days after even a brief ischemic attack. Research findings have showed the involvement of glutamate and calcium in neuronal death after hypoxic—ischemic brain injury. Such injury causes glutamate release, excess activation of ionotropic glutamate receptors, and accumulation of intracellular calcium, which results in rapid and fulminant neuronal death (Choi and Rothman, 1990; Mattson and Kroemer, 2003). Increased calcium then triggers neuronal injury by activating cytotoxic proteins such as nitric oxide synthase, calpain, and endonuclease, and by impairing mitochondria and endoplasmic reticulum.
A number of N-methyl-
Acetylsalicylic acid (aspirin) is widely used to prevent recurrent ischemic stroke; its therapeutic effect is related to its role as an irreversible cyclooxygenase inhibitor, inhibiting conversion of arachidonic acid to thromboxane A2 and thereby inhibiting platelet aggregation (Botting, 1992). Recently, aspirin and salicylic acid were shown to prevent NMDA receptor-mediated excitotoxicity by blocking nuclear factor-κB and c-Jun N-terminal kinase (Grilli et al, 1996; Ko et al, 1998). Aspirin can protect against ischemic neuronal death by inhibiting voltage-gated Ca2+ channels, p44/42 mitogen-activated protein kinase, and glutamate release (Kim et al, 2001; De Cristobal et al, 2002; Vartiainen et al, 2003). However, the therapeutic potential of aspirin in the brain may be limited given that high doses (as much as 10 mmol/L) of the drug are needed to prevent NMDA and Zn2+ neurotoxicity in primary cortical cell cultures. The antiinflammatory drug sulfasalazine, a conjugate of 5-aminosalicylic acid and sulfapyridine, inhibits NMDA neurotoxicity more potently than aspirin and in addition offers neuroprotection against free radical injury (Gionchetti et al, 1990; Ryu et al, 2003).
We propose that the dual pharmacological actions of these salicylates on NMDA receptor-mediated excitotoxicity and free radical neurotoxicity offer the potential for improved neural protection after ischemic brain injury and that development of chemical derivatives of these compounds with greater potency, safety, and effectiveness might offer a novel, successful therapy.
Materials and methods
Primary Cortical Cell Cultures
Mouse cortical cell cultures were prepared as previously described (Noh and Gwag, 1997). Animals were handled in accordance with a protocol approved by our institutional animal care committee. Brains were removed from 14-day-old fetal mice, and the cerebral cortices were isolated, gently triturated, and plated on 24-well plates (5 hemispheres/plate, approximately 2.5 × 105 cells/plate) precoated with 100 μg/mL poly-
Determination of Neurotoxicity
Mixed cortical cell cultures (11 to 14 DIV) containing neurons and glia were briefly exposed to NMDA (300 μmol/L) or continuously exposed to α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (10 μmol/L), kainate (30 μmol/L), Fe2+ (50 μmol/L),
Electrophysiologic Recording
Whole-cell voltage-clamp recordings were performed on primary cultured cortical neurons (11 to 14 DIV) at room temperature (18°C to 23°C). Whole-cell currents were recorded and analyzed using an Axopatch 200A amplifier (Axon Instruments, Foster City, CA, USA) with Digidata-1322A A/D converter and pClamp 8.0 (Axon Instruments). The 2 to 3-MΩ-resistance recording pipettes were filled with an internal solution containing 135 mmol/L CsCl, 10 mmol/L N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 1.2 mmol/L MgCl2, 4 mmol/L ATP-Na2, 0.5 mmol/L CaCl2, and 11 mmol/L ethyleneglycol tetraacetate (pH adjusted to 7.3 with CsOH). The external solution was composed of 140 mmol/L NaCl, 2 mmol/L KCl, 2 mmol/L CaCl2, 10 mmol/L N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, and 0.01 mmol/L glycine (pH adjusted to 7.3 to 7.4 with NaOH). N-methyl-
Focal Cerebral Ischemia: Clip Model
Male Sprague—Dawley rats (260 to 300 g) were anesthetized with chloral hydrate (400 mg/kg, intraperitoneally) and subjected to transient focal cerebral ischemia by occlusion of the right middle cerebral artery and both common carotid arteries, as previously described (Chen et al, 1986). In brief, the common carotid arteries were exposed through a vertical midline neck incision. The right middle cerebral artery was exposed under surgical microscope and occluded with a microclip positioned inferior to the cerebral vein. Complete interruption of blood flow was visually confirmed under surgical microscope. Both common carotid arteries were then occluded with microclips. Body temperature was measured with a rectal probe and maintained at 37°C with a heating pad. The aneurysm clips were released 60 mins later, and restoration of blood flow was observed under a microscope.
Focal Cerebral Ischemia: Intraluminal Thread Model
Male Sprague—Dawley rats (260 to 300 g) were anesthetized and maintained with 2.5% isoflurane in 70% N2O and 30% O2 using a face mask. Body temperature was maintained at 37°C with a heating pad. The right common carotid artery, external carotid artery, and internal carotid artery were exposed. A length 4-0 monofilament nylon suture (17 mm, precoated with silicone) with its tip rounded by heating near a flame was advanced from the lumen of the internal carotid artery until it blocked the origin of the right middle cerebral artery. After 90 mins, the rats were reanesthetized with isoflurane and subjected to reperfusion by withdrawing the suture.
2,3,5-Triphenyltetrazolium Chloride Staining
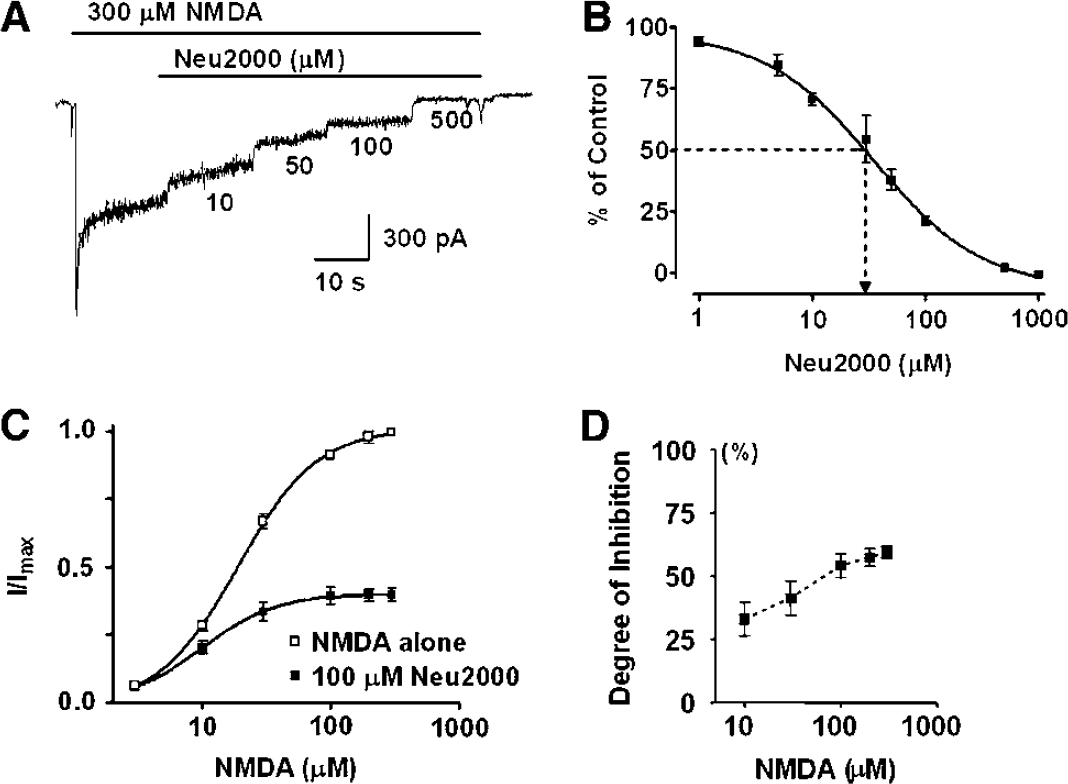
To analyze infarct volume, rats were euthanized by chloral hydrate 24 h after reperfusion, and brains were sectioned coronally into six 2-mm-thick slices in a rodent brain matrix (Harvard Instruments Incorporation, South Natick, MA, USA) and placed in 2% 2,3,5-triphenyltetrazolium at 37°C for 30 mins. Images of brain sections were captured by an image analysis system (Bio-Rad, Hercules, CA, USA). Infarct volume was determined by correcting tissue swelling according to the following equation to each coronal brain slice (Belayev et al, 2003):
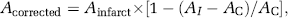
where AI is the ipsilateral hemisphere area and AC is the contralateral hemisphere area. Hemispheric infarct volume was then determined by adding the infarct volumes measured in each brain slice using TINA 2.0 (KAIST, Daejeon, Korea).
Determination of Mitochondrial Free Radicals in Neurons
Mitochondrial free radical generation was analyzed as described previously (Kim et al, 2002). Rats were subjected to clip occlusion of the rMCA and both common carotid arteries (common carotid arteries) for 60 mins, placed into a Kopf stereotaxic apparatus, and injected using a Hamilton syringe in the lateral ventricle (coordinate: 1.0 mm anterior to the bregma, 1.9 mm lateral to the midline, and 2.8 mm below the pial surface) with 0.4 nmol Mitotracker Red CM-H2XRos (Molecular Probes, Eugene, OR, USA) dissolved in an 8-ml mixture of dimethylsulfoxide and saline (1:1 v/v). Animals were euthanized at indicated points of time after reperfusion and perfused with phosphate-buffered saline followed by 3% paraformaldehyde. The brain was immediately removed, immersed in fixative for 2 to 4 h, incubated in 30% sucrose at 4°C for 2 days, and sectioned into 30 μm slices. Sections were stained with Hoechst 33258 for 5 mins and visualized under a fluorescent microscope (Ex = 466 nm, Em = 360 nm). Mitochondrial free radicals were determined by monitoring the oxidized fluorescence product (Ex = 554 nm, Em = 576 nm) of Mitotracker Red CM-H2XRos under a fluorescence microscope equipped with a cooled charged couple device system (Zeiss, Göttingen, Germany). Mitochondrial free radical intensity was determined by Image Gauge 3.12 (Fuji, Tokyo, Japan).
For neuronal staining, sections were further incubated in 0.3% H2O2 and 0.25% Triton X-100 for 30 mins at room temperature, and reacted for 1 h with 5% bovine serum albumin followed by mouse anti-NeuN monoclonal antibody (Chemicon, CA, USA) overnight at 4°C. Sections were washed with phosphate-buffered saline, incubated with fluorescein isothiocyanate-labeled anti-mouse immunoglobulin (Vector Laboratories, CA, USA) for 3 h, washed with phosphate-buffered saline, and visualized under the fluorescence microscope.
Analysis of White Matter Damage and N-Methyl-d-Aspartate Antagonist-Induced Neuronal Damage
For white matter staining, brain was sectioned at a thickness of 20 μm on a cryostat. Sections were mounted on gelatin-coated slides, dehydrated with graded alcohol, and stained with 0.1% Luxol fast blue solution and hematoxylin—eosin solution. White matter damage was analyzed by measuring decrease in optical densities (ODs) of myelin stained with Luxol fast blue as described (Schabitz et al, 2000). For the striatum, at a magnification of × 400, the ODs of 30 to 40 white matter fiber tracks were measured. The ODs of the external capsule were measured from 10 randomly chosen areas at a magnification of × 40. The OD ratio was calculated by dividing the ipsilateral OD by the contralateral OD.
N-methyl-
Behavioral Tests
The Modified Neurological Severity Score (mNSS) was calculated as a measure of motor, sensory, and reflex function, and balance using a modified version of the sensory tests, as described previously (Roof et al, 2001). The total mNSS was scaled to 0 to 28 (normal = 0, maximal deficit = 28). The mNSS was determined by measuring reflex and abnormal movements (subtotal score = 0 to 4: pinna reflex = 0 to 1, corneal reflex = 0 to 1, startle reflex = 0 to 1, and seizure, myoclonus, or myodystony = 0 to 1), responses to being raised by the tail (subtotal score = 0 to 3: forelimb flexion = 0 to 1, hindlimb flexion = 0 to 1, and head movement over 10° to vertical axis within 30s = 0 to 1), and results of sensory tests (subtotal score = 0 to 12: visual placement of forelimbs = 0 to 3, tactile placement of forelimbs = 0 to 3, proprioceptive adduction of hindlimbs = 0 to 3, tactile placement of hindlimbs = 0 to 3), and beam balance tests (subtotal score = 0 to 6: balances with steady posture = 0, grasps side of beam = 1, hugs beam and one limb falls down = 2, hugs beam and two limbs fall down or spins over 60s = 3, attempts to balance on beam but falls off over 40s = 4, attempts to balance on beam but falls off over 20s = 5, falls off and no attempt to balance or hang onto beam within 20s = 6).
The Rotarod test was performed by measuring the time animals remained on a Rotarod (Ugo Basile, Italy) that was gradually accelerated from 4 to 40 r.p.m. over 5 mins. The animals were pretrained for 3 days before middle cerebral artery occlusion (MCAO). Data are presented as the mean percent duration of three Rotarod measurements compared with presurgery.
The adhesive-removal somatosensory test was performed by measuring the time to removal of a small piece of adhesive rectangular tape (1 cm2, Fischer, PA, USA) applied to the radial aspect of each forelimb (Schabitz et al, 2004). The time to remove each tape was recorded on 3 trials per day, each with a 3-mins interval in dark and faintly illuminated by an infrared light. The animals were trained for 3 days before surgery. Once the rats were able to remove the tape within 10 secs, they were subjected to ischemia.
Statistical Analysis
For comparisons of parametric data, analysis of variance, followed by a two-tailed t-test, was used to determine statistical significance. Comparisons of nonparametric data were performed using Kruskal—Wallis test, followed by Mann—Whitney U-tests. A P < 0.05 was considered statistically significant.
Results
Blockade of NMDA Neurotoxicity by Neu2000 in Cortical Cell Cultures
After synthesizing structural derivatives of aspirin and sulfasalazine, we tested their ability to protect against NMDA neurotoxicity in cortical cell cultures. Of these derivatives, Neu2000 (Figure 1A) prevented NMDA neurotoxicity more potently than sulfasalazine or aspirin. Neu2000 showed apparent neuroprotection against 300 μmol/L NMDA at doses as low as 30 μmol/L, whereas aspirin and sulfasalazine showed neuroprotection at only much higher doses (Figure 1B). Neu2000 did not protect cortical neurons against α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid- or kainate-mediated excitotoxicity (data not shown).
Blockade of NMDA neurotoxicity by aspirin, sulfasalazine, and Neu2000. (
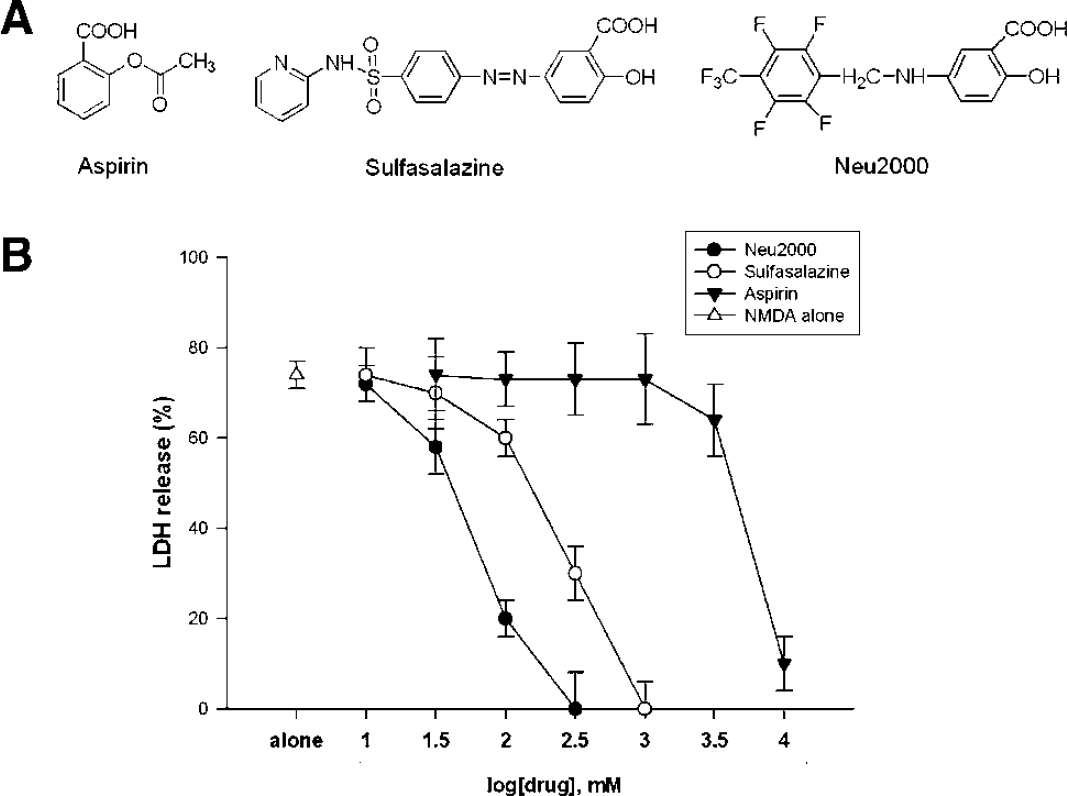
Additional experiments were performed to examine whether the neuroprotective effects of Neu2000 were attributable to NMDA receptor antagonism. Neu2000 inhibited the electrophysiologic response of cultured cortical neurons to 300 μmol/L NMDA in a concentration-dependent manner (Figure 2A), indicating that the effect was mediated by a specific action at NMDA receptors. The dose—response curve of Neu2000 was constructed and fitted to the following equation (Figure 2B):
Neu2000 blocks NMDA-induced currents. Whole-cell currents were elicited from cultured cortical neurons (11 to 14 DIV) at a holding potential of -60mV. (
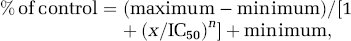
where n is Hill's coefficient and IC50 is the concentration of Neu2000 resulting in 50% blockade. The Neu2000 dose—response had an IC50 of 35.38 ± 5.94 μmol/L and Hill's coefficient of 0.91 (n = 8). This finding suggests that Neu2000 blocks NMDA-evoked currents more potently than does sulfasalazine (IC50 = 294 μmol/L) (Gionchetti et al, 1990) and may bind stoichiometrically to the NMDA receptor.
To elucidate the mode of action whereby Neu2000 blocks NMDA receptors, we tested for changes in the NMDA concentration—response with administration of Neu2000 (Figure 2C). Neu2000 (100 μmol/L) significantly reduced the maximal NMDA response by 58.31 ± 2.72% (n = 5) and the EC50 values of NMDA from 18.88 ± 1.85 to 9.92 ± 0.17 μmol/L (n = 5, P < 0.05), indicative that it is a noncompetitive antagonist. Moreover, the antagonistic effect of Neu2000 increased with increasing concentrations of NMDA, a common characteristic of noncompetitive inhibition (Figure 2D).
Blockade of Free Radical Injury by Neu2000 in Cortical Cell Cultures
In addition to the neuroprotective effects of Neu2000 against NMDA neurotoxicity, we showed blockade of free radical injury by Neu2000. Mixed cortical cell cultures of neurons and glia exposed to 50 μmol/L Fe2+ underwent widespread neuronal death 24 h later. Aspirin did not affect Fe2+-induced neurotoxicity, but sulfasalazine (30 to 100 μmol/L) produced a partial attenuation. Treatment with 10 to 100 μmol/L vitamin E or 3 to 10 μmol/L trolox, a membrane-permeable derivative of vitamin E, dose dependently reduced Fe2+-induced neuronal death. Edaravone, a free radical scavenger, partially reduced the free radical injury at a dose of 3 μmol/L, but its protective effect disappeared at higher doses. Administration of Neu2000 produced a marked reduction of Fe2+-induced neurotoxicity, even at doses of 0.1 to 0.3 μmol/L (Figure 3A). Neu2000 blocked the degeneration of neurons and glia in cortical cell cultures exposed to
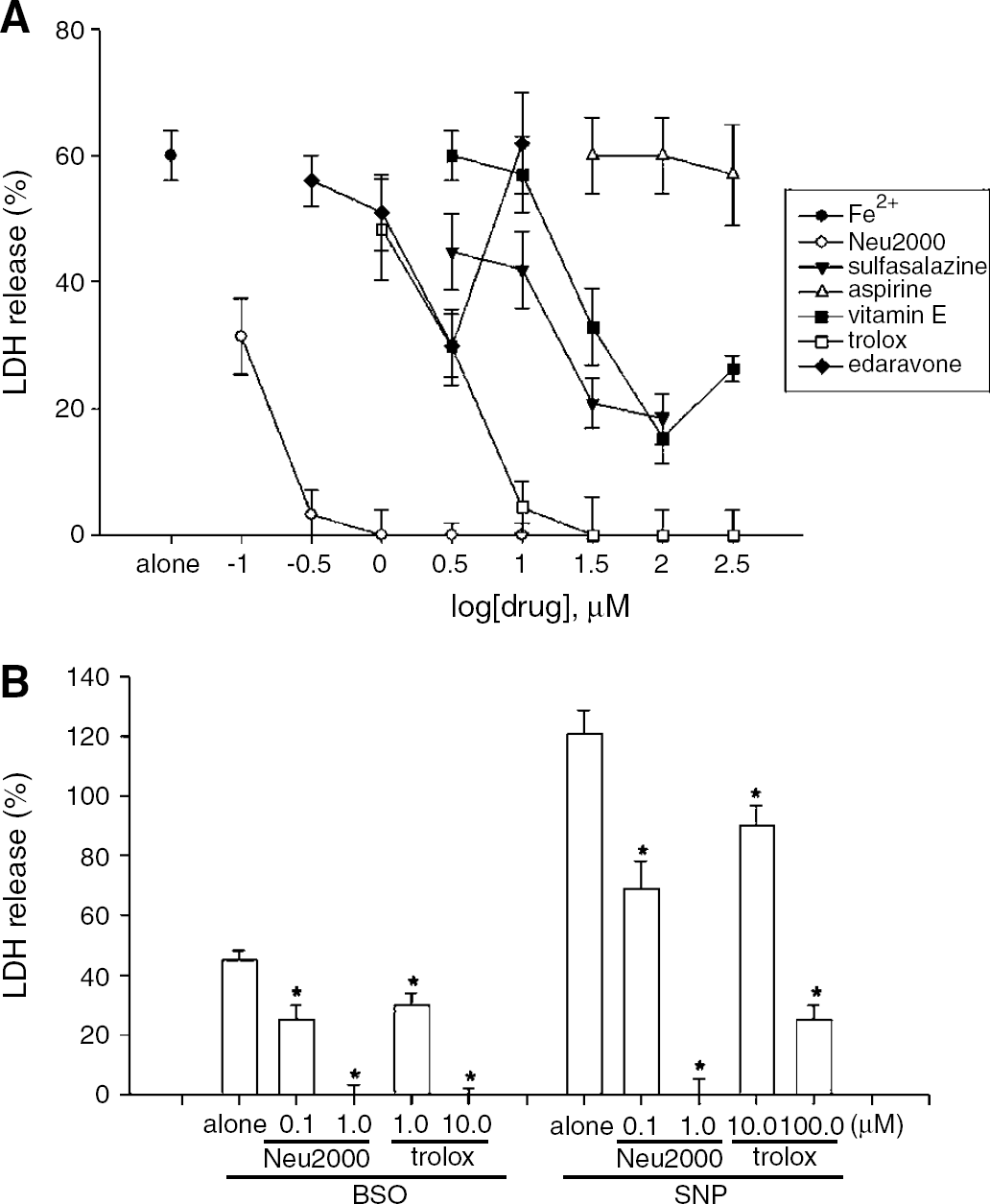
Blockade of free radical toxicity by Neu2000. (
Enhanced and Extended Neuroprotection Against Transient Focal Cerebral Ischemia by Neu2000: Clip Occlusion Model
We next investigated the neuroprotective effects of Neu2000 in an animal model of hypoxic—ischemia. Intravenous administration of Neu2000 5 mins after reperfusion substantially and dose dependently reduced cerebral infarct evolving 24 h after 60-mins occlusion of the MCA (Figure 4A). At doses of 2.5 to 5 mg/kg (i.v.), Neu2000 produced a large neuroprotective effect, with a maximal reduction in infarct volume of 66%. In comparison, MK-801 (3 mg/kg), an NMDA antagonist, and trolox (50 mg/kg) produced maximal neuroprotection of approximately 46% and 34%, respectively (Figure 4B). Deeply eosinophilic neurons with pyknotic nuclei were observed in the retrosplenial cortex at 3 days after administration of MK-801 in adult rat as reported previously (Fix et al, 1993). However, neuronal damage was not observed in the most vulnerable cortical area after administration of 5 mg/kg Neu2000 (i.v.), a dose showing maximal neuroprotection in the current MCAO model. Moreover, eosinophilia of neurons was not observed in the retrosplenial cortex at 3 days after administration of 30 mg/kg Neu2000 (Figure 4C). These findings suggest that Neu2000 safely and profoundly protects against ischemic brain injury, presumably by blockade of both NMDA and free radical neurotoxicity.
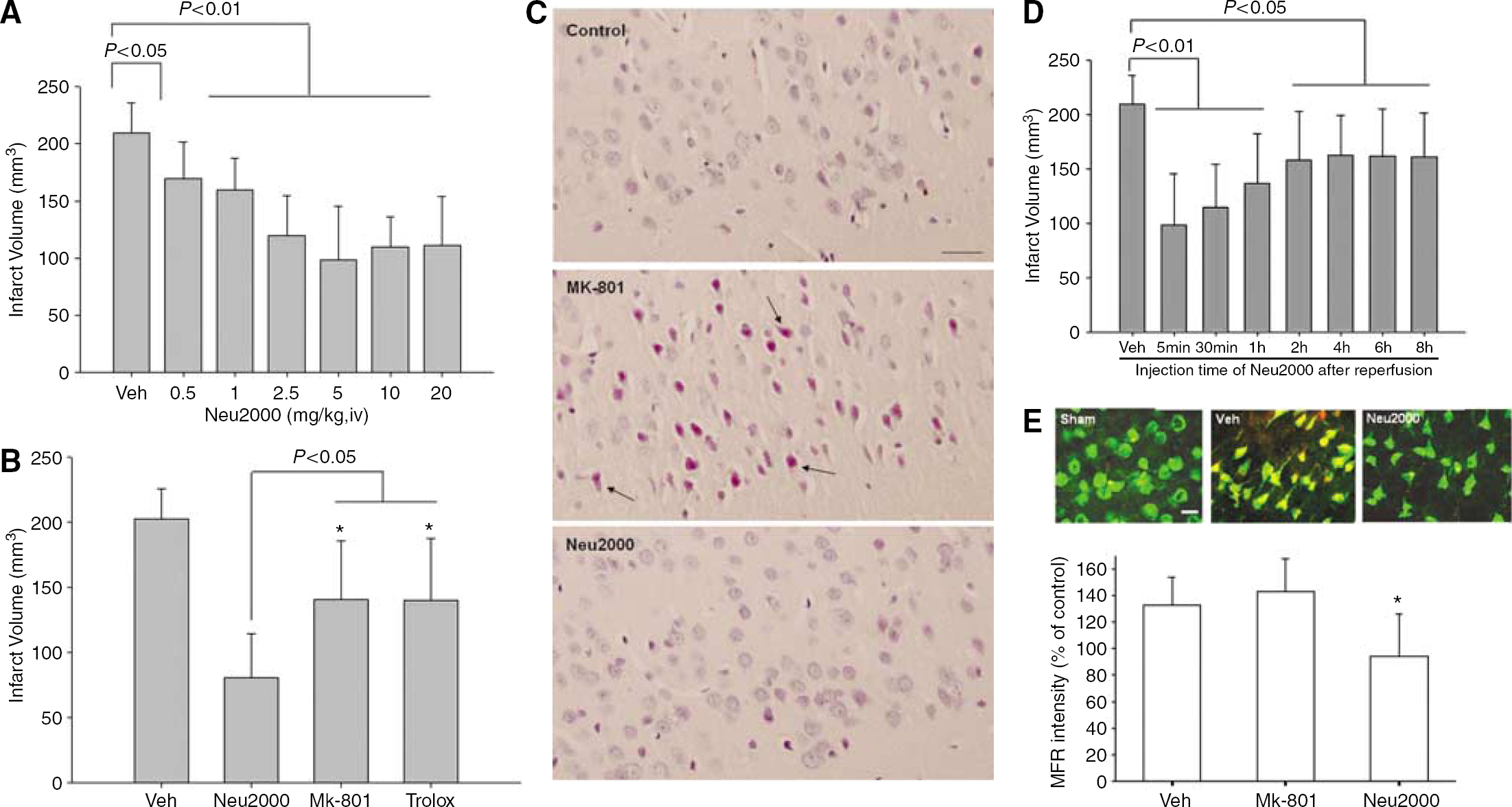
Profound and extended neuroprotective actions of Neu2000 in clip occlusion model of focal cerebral ischemia. (
In the adult rat, NMDA antagonists have been shown to decrease infarct volume after transient MCAO when administered just before ischemic (Margaill et al, 1996). Antioxidants such as NXY-059, ebselen, and edaravone appeared to extend the therapeutic window as shown by the attenuation of ischemic brain injury when administered within 3 h after reperfusion (Watanabe et al, 1994; Kuroda et al, 1999; Imai et al, 2001). In addition to the profound neuroprotective effect of Neu2000, we found that its therapeutic window was markedly extended compared with those of NMDA receptor antagonists and antioxidants. Neu2000 (5 mg/kg, i.v.) maximally reduced infarct volume 24 h after 60-mins MCAO when administered 5 mins after reperfusion. The neuroprotective effects were gradually reduced with increased delay in administration, but Neu2000's effects were still significant when delivered 8 h after reperfusion (Figure 4D).
It is conceivable that Neu2000 exerts its profound and extended therapeutic effects by preventing free radical injury as well as NMDA receptor-mediated excitotoxicity. To examine this possibility, we studied the temporal pattern of reactive oxygen species production 60 mins after MCAO. In cortical neurons, ROS were produced in the ischemic core region within 30 mins after reperfusion and in the penumbral neurons within 6 h after reperfusion (data not shown). Reactive oxygen species production in the penumbral area 8 h after reperfusion was nearly completely blocked by Neu2000, but not by MK-801, administered 4 h after reperfusion (Figure 4E and Figure 4F). Thus, the extended therapeutic window of Neu2000 seems to be attributable to its free radical scavenging action in part that blocks the slow production of reactive oxygen species in the penumbral areas.
Long-lasting Neuroprotection and Behavioral Sparing Against Transient Focal Cerebral Ischemia with Neu2000: Intraluminal Thread Occlusion Model
In consideration of the failure of numerous neuroprotective agents in human clinical trials of stroke versus their success in reducing infarct volume in animal models of stroke, the stroke therapy academic industry roundtable has recommended that at least two outcome measures, including functional response and infarct volume, be investigated in a randomized and masked fashion to evaluate neuroprotectant efficacy in animal models of stroke (STAIR, 1999). We performed additional randomized, blind experiments using the rat intraluminal thread occlusion model to test if Neu2000 would prevent white matter injury and behavioral deficits as well as reduce infarct volume. Administration of Neu2000 (5 mg/kg, i.v.) 30 mins after reperfusion did not change physiologic variables such as arterial pH, PCO2, PO2, and hematocrit after 90-mins MCAO (Table 1). Treatment with Neu2000 substantially reduced infarct volume evolving in the cortex and the striatum at 3 days of recirculation after 90-mins MCAO (Figure 5A). White matter damage in the striatum and external capsule was evident 3 days after MCAO and was markedly reduced by Neu2000 (Figure 5B), suggesting that Neu2000 protects white matter such as axons and myelin as well as gray matter from ischemic brain injury.
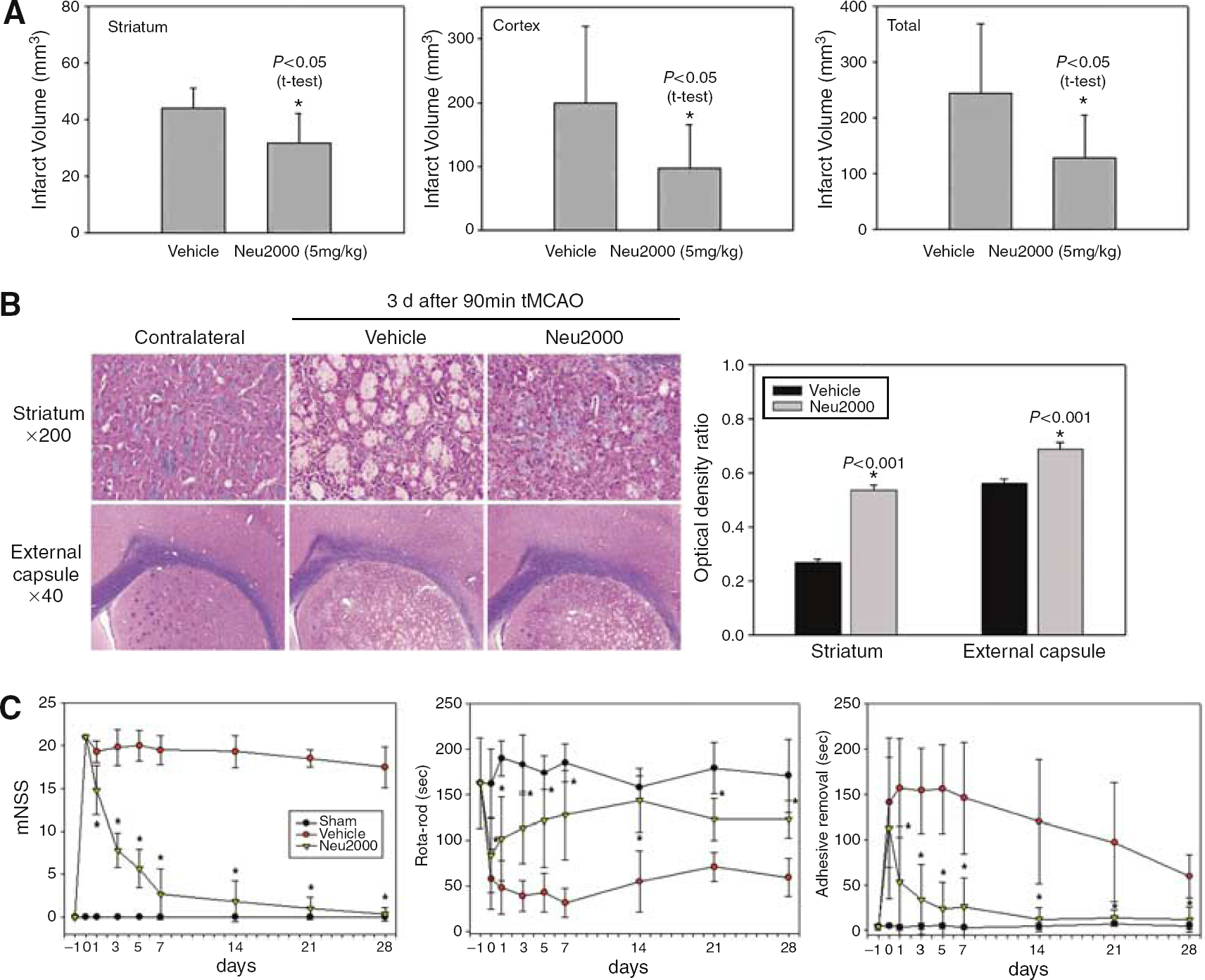
Neu2000 reduces neuronal death in gray and white matter and spares behavior in the intraluminal thread occlusion model of MCA. (
Arterial blood gases and hematocrit in rats treated with Neu2000 after 90-mins tMCAO
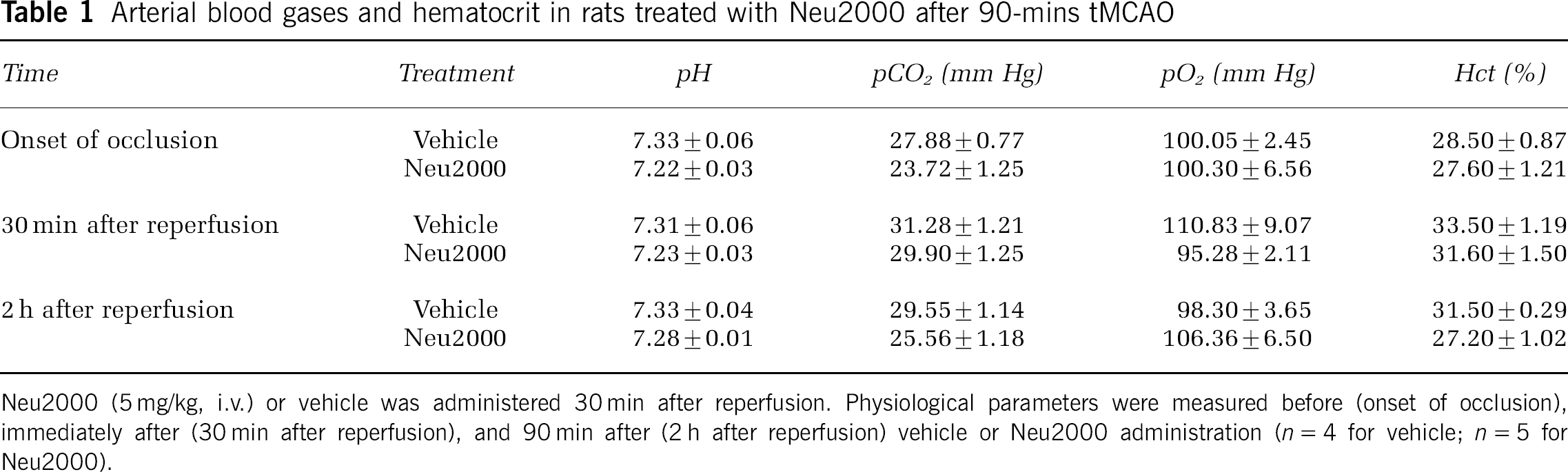
Neu2000 (5 mg/kg, i.v.) or vehicle was administered 30 min after reperfusion. Physiological parameters were measured before (onset of occlusion), immediately after (30 min after reperfusion), and 90 min after (2 h after reperfusion) vehicle or Neu2000 administration (n = 4 for vehicle; n = 5 for Neu2000).
Neurologic deficits were evaluated using the mNSS immediately after 90-mins MCAO (Figure 5C). The vehicle-treated control group showed similar levels of neurologic deficit 1 month after ischemic injury. Administration of Neu2000 appeared to reduce mNSS measured 1 day after 90-mins MCAO. Neu2000 further reduced the mNSS by up to 70% relative to the control group 3 days after MCAO. The behavioral improvement by Neu2000 lasted for 1 month after MCAO. Ischemic lesion in the striatum resulted in impaired coordination of motor skill as shown by decreased mean time spent on the Rotarod (Figure 5C). Motor function was significantly improved within 1 day in animals treated with Neu2000, and the improvement lasted for 1 month after ischemic injury. Administration of Neu2000 appeared to ameliorate hemineglect behavior within 1 day after ischemic injury, as shown by shortened latency to adhesive tape removal.
Discussion
We hypothesized that salicylic acid compounds, which have multiple neuroprotective actions, might be successfully used for prevention of ischemic brain injury if structural derivatives with improved potency and safety could be developed. We have provided evidence that Neu2000, a derivative of aspirin and sulfasalazine, exerts dual neuroprotective effects: blocking NMDA neurotoxicity as uncompetitive NMDA antagonist and blocking free radical neurotoxicity as a free radical scavenger. The dual neuroprotective actions of Neu2000 were associated with increased efficacy in a clip occlusion model of transient MCA occlusion compared with NMDA antagonists or antioxidants. Neu2000 also prolonged the therapeutic time window by up to 8 h in both the clip and thread models of transient MCA occlusion. Furthermore, single bolus injection of Neu2000 protected sensory and motor function for at least 28 days after ischemic insult.
The neuroprotective effects of NMDA antagonists have been well documented in animal models of ischemia and extensively investigated in clinical trials of human stroke patients. Selfotel, cerestat, and eliprodil, strong NMDA antagonists that act in a competitive, noncompetitive, and NR2B-selective manner, respectively, showed no efficacy in phase III trials for acute stroke therapy (Cheng et al, 2004). Psychomimetic, cardiovascular, and neurotoxic effects appear to limit the therapeutic potential of potent NMDA antagonists (Olney et al, 1991; Muir and Lees, 1995; Krystal et al, 2003). Moderate affinity, uncompetitive NMDA antagonists such as memantine, and NPS 1506 do not show undesirable side effects within the therapeutic range for neuroprotection (Parsons et al, 1999; Mueller et al, 2000; Labiche and Grotta, 2004). We have shown that Neu2000 is an uncompetitive NMDA antagonist that spares neurologic behaviors after ischemic injury and does not cause neurotoxicity, suggesting that it is a secure NMDA antagonist suitable for treating stroke. We showed the following pharmacological actions for Neu2000: (1) Neu2000 is a noncompetitive NMDA antagonist (IC50 = 35 μmol/L, 8.4-fold more potent than sulfasalazine), (2) Neu2000 has antioxidant effects (IC50 = 0.11 μmol/L for Fe2+-induced neuronal death in cortical cell cultures, 330-fold more potent than vitamin E), and (3) Neu2000 has antithrombotic effects (data not shown). The agonist-dependent antagonism by Neu2000, similar to memantine, Ro 8 to 4303, and ifenprodil (Kew et al, 1996; Chen and Lipton, 1997; Kew et al, 1998) may have some therapeutic advantages in that Neu2000 should block glutamate activation more efficiently in unhealthy neuronal systems where excessive glutamate release is expected. Based on these collective findings, we propose Neu2000 as a novel candidate for stroke treatment.
The lack of beneficial effects of NMDA antagonists in human clinical trials has been due primarily to their short therapeutic time window and unwanted side effects (Labiche and Grotta, 2004). Activation of NMDA receptors results in the generation of reactive oxygen and nitrogen species that partially mediate subsequent neuronal death (Monyer et al, 1990; Dawson et al, 1991). However, free radicals are produced mainly in the period of postischemic reperfusion. These are known to mediate ischemic cell necrosis in the intestine, kidney, and heart (McCord, 1985), and likely contribute to delayed neuronal death after hypoxic—ischemic brain injury, which is insensitive to NMDA antagonists.
Antioxidants have a longer therapeutic window than NMDA antagonists in animal models of stroke and have been also been investigated in clinical trials for acute stroke patients. The clinical trial of tirilazad, a lipid peroxidation inhibitor, at high doses tended to show some beneficial effects in mortality and behavior with 53 acute stroke patients within 4 h of the onset of symptoms (Haley, 1998). Systemic administration of ebselen, a seleno-organic compound with antioxidant activity, significantly reduced infarct volume and improved functional outcome in acute stroke patients who started treatment within 6 h of onset (Ogawa et al, 1999). Edaravone has been approved as a neuroprotective drug for stroke patients in Japan (EAISG, 2003). NXY-059, a free radical trapping agent, is the first neuroprotectant shown to produce a statistically significant reduction in the primary outcome of disability in a worldwide trial for acute ischemic stroke (Lees et al, 2006). The neuroprotective effects of Neu2000 against free radical injury are also attributable to free radical scavenging, and results with primary cortical cell cultures indicate greater efficacy and potency compared with edaravone and NXY-059. This suggests that the antioxidant property of Neu2000 provides additional neuroprotection against ischemic brain injury.
Neu2000 is a candidate molecule for preventing both NMDA and free radical neurotoxicity without toxic side effects and shows marked neuroprotection and an extended therapeutic window in animal models of focal cerebral ischemia. The present findings buttress a novel hypothesis that a multitarget strategy involving timely prevention of NMDA receptor-mediated excitotoxicity and free radical accumulation may provide effective intervention of multifaceted neuronal death and extend the therapeutic time window in stroke patients.