
Abstract
We hypothesized that heme oxygenase (HO)-1, the inducible form of HO, represents an important defense against early oxidative injury in the traumatized spinal cord by stabilizing the blood—spinal cord barrier and limiting the infiltration of leukocytes. To test this hypothesis, we first examined the immunoexpression of HO-1 and compared barrier permeability and leukocyte infiltration in spinal cord-injured HO-1-deficient (+/–) and wild-type (WT, +/+) mice. Heme oxygenase was expressed in both endothelial cells and glia of the injured cord. Barrier disruption to luciferase and infiltration of neutrophils were significantly greater in the HO-1 +/– than WT mice at 24 h postinjury (P ≤ 0.019 and = 0.049, respectively). We next examined by Western immunoblots the generation of 4-hydroxynoneal (HNE) and malondialdehyde (MDA), major products of lipid peroxidation, in the injured epicenter. There was a significant increase in 10 kDa HNE- and MDA-modified proteins in the HO-1 +/– as compared with WT mice (P = 0.037 and 0.043, respectively). Finally, we compared the degradation of myelin basic protein (MBP), an indicator of white matter damage, in the HO-1 +/– and WT mice by Western immunoblots. There was significantly greater degradation of MBP in the HO-1 +/– compared with WT mice (P = 0.049). Together, these findings show that HO-1 modulates oxidative stress and white matter injury in the acutely injured spinal cord. This modulation may be partially attributed to the ability of HO-1 to stabilize the blood—spinal cord barrier and limit neutrophil infiltration.
Introduction
The functional deficit that occurs as a consequence of spinal cord injury is not solely attributed to the initial mechanical damage to tissue, but is also a consequence of secondary events that promote additional tissue destruction. The development of early secondary damage is mediated by a variety of factors including those that are related to disruption of the blood—spinal cord barrier, the infiltration of leukocytes, and oxidative stress (Blight, 2002; Jones et al, 2005).
In this study, we examined the role of the inducible heme oxygenase (HO) isozyme, HO-1, in modulating barrier function and inflammation in the acutely injured spinal cord. Heme oxygenase is the rate-limiting enzyme that catalyzes the conversion of the pro-oxidant heme to equimolar quantities of the potent antioxidants biliverdin, which is rapidly converted to bilirubin, as well as carbon monoxide (CO) and pro-oxidant free iron (Maines, 2000). Two isoforms of HO have been most studied: HO-1 and HO-2. Heme oxygenase-1 is the inducible form with relatively low levels of HO-1 reported in the uninjured spinal cord (Mautes et al, 2000). However, HO-1 is markedly induced in response to a variety of injury and disease states in the central nervous system (Schipper, 2004), including traumatic spinal cord injury (Liu et al, 2002; Mautes et al, 2000).
Heme oxygenase-1 may play an important role in limiting early secondary tissue damage by modulating vascular function, including the infiltration of inflammatory cells after spinal cord injury. We have recently shown that vascular induction of HO-1 by systemic administration of heme stabilizes the blood—spinal cord barrier and limits early leukocyte infiltration in the injured spinal cord (Yamauchi et al, 2004). This immunomodulatory role of HO-1 has also been reported by others (Lee et al, 2004). Recent analyses of HO-1-knockout (–/–) mice as well as the HO-1 deficient human have also strengthened the hypothesis that HO-1 has potent anti-inflammatory properties (Kapturczak et al, 2004; Poss and Tonegawa, 1997; Yachie et al, 1999). Poss and Tonegawa (1997) have reported that HO-1-knockout mice accumulate iron in the kidney and liver, have chronic inflammation, and have increased levels of oxidized proteins and lipid peroxidation. There is also evidence for mild inflammatory changes in the liver of patients who are deficient in HO-1 (Yachie et al, 1999).
The early infiltration of leukocytes contributes to oxidative damage and limits functional recovery after spinal cord injury. We have shown that pharmacologic blockade of matrix metalloproteinases (MMPs) within the first 3 days after spinal cord injury reduces leukocyte infiltration, and results in improved motor recovery (Noble et al, 2002). Similar findings have been reported by Weaver and colleagues who effectively reduced early leukocyte infiltration by treating spinal cord-injured animals with a monoclonal antibody against CD11d/CD18 integrin expressed on leukocytes (Bao et al, 2004; Gris et al, 2004; Mabon et al, 2000). Treatment with this antibody for the first 2 days postinjury reduced oxidative stress and improved functional recovery. Collectively, these findings are quite exciting as they show that early blockade of infiltrating leukocytes may be a promising strategy for the treatment of the spinal cord-injured patient.
In this study, we hypothesized that HO-1 represents an important defense against acute secondary injury in the traumatized spinal cord by stabilizing the blood—spinal cord barrier and restricting the infiltration of leukocytes. Here, we show that barrier disruption and neutrophil infiltration are significantly increased in spinal cord-injured, HO-1 heterozygote (+/–) mice as compared with wild-type (WT, +/+) littermates. Moreover, these changes correlate with an increase in lipid peroxidation and white matter damage. Together, these findings implicate HO-1 in neuroprotection, a function that may be in part attributed to its ability to modulate early inflammation and barrier function.
Materials and methods
All procedures were approved by the University of California at San Francisco Institutional Animal Care and Use Committee and in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. HO-1 +/– and WT littermates, bred on a C57BL6 background in excess of 10 generations, as well as WT C57BL6 mice, ordered from Charles River Laboratories (Wilmington, MA, USA), were used in the study. There were no overt differences in the purchased and bred WT populations. Animals were housed in a temperature-(25°C) and light-(12 h light—dark cycle) controlled environment. HO-1 +/– mice exhibit no overt abnormalities and have a normal litter size. All studies described below were conducted in a masked manner.
Spinal Cord Injury Model
Adult male mice, weighing 25 to 35 g, were anesthetized with 2.5% Avertin (0.02 mL/g body weight) given intraperitoneally (i.p.), which is subsequently metabolized to CO (Min et al, 2003). With few exceptions, avertin administration was standardized across animals. Animals were maintained at 37°C on a heating pad during surgery. A contusive spinal cord injury was performed as previously reported by Noble et al (2002). Briefly, a dorsal midline incision was made and the dorsal face of T8 was exposed. The spinous process and lamina were removed and a circular region of dura, approximately 2.5 mm in diameter, was exposed. After stabilization of the vertebral column, a 3-g weight was dropped 5.0 cm onto the exposed dura. After injury, the overlying skin was closed with wound clips. Postoperative care included manual expression of each animal's bladder. All mice were euthanized 24 h after injury. The spinal cords were either quickly removed and prepared for biochemical analyses (Western immunoblots and measurements of HO activity and luciferase content) or perfused with fixative for subsequent anatomic studies.
Immunocytochemistry
Additional animals were re-anesthetized 24 h postinjury and perfused with 4% paraformaldehyde in phosphate-buffered saline (PBS). Spinal cords were postfixed in 4% paraformaldehyde at 4°C for 4 h, cryoprotected in 20% sucrose at 4°C for 3 days. A 1.0-cm length of spinal cord, centered over the injured site, was sectioned, 20 μm in thickness, on a cryostat at −20°C. Sections were mounted on slides and dried for 1 h at 37°C.
Co-Localization of Heme Oxygenase-1 with Blood Vessels
Two methods were used to confirm the localization of HO-1 in blood vessels. In the first method, HO-1 was immunolocalized in lectin-labeled vessels. At 24 h postinjury, anesthetized animals were injected intravenously (i.v.) with 100 μL of 50% fluorescein-labeled Lycopersicon esculentum (Tomato) lectin (Vector Laboratories, Burlingame, CA, USA), diluted in 0.9% saline, via the jugular vein. This fluorescent lectin has a high affinity for the endothelial glyocalyx and has been shown to label blood vessels (Thurston et al, 1996). After 5 mins, animals were perfused and cords were postfixed, cryoprotected, and sectioned as described above. To immunolocalize HO-1, 20-μm thick sections were rinsed in PBS for 5 mins and incubated in: (1) 2% goat serum/0.2% Triton X-100/0.1% bovine serum albumin (GS/TX/BSA), 5 mins; (2) 10% GS/TX/BSA, 20 mins; (3) rabbit anti-HO-1 polyclonal antibody (1:20,000 in 10% GS/TX/BSA, StressGen, Victoria, BC, Canada), 24 h at room temperature (RT); (4) PBS, 3 × 5 mins; (5) Cy3-conjugated goat anti-rabbit IgG (1:400 in 10% GS/TX/BSA, Jackson Laboratories, West Grove, PA, USA), 1 h at RT; and (6) PBS, 3 × 5 mins. Sections were coverslipped with fluorescein-mounting medium (Vectashield). Staining was evaluated using a Nikon Optiphot microscope equipped with a CCD camera (SPOT software, Model 1.3.0; Diagnostic Instruments, Inc., Sterling Heights, MI, USA), and imaged using PhotoShop 6.0 (Adobe Systems, San Jose, CA, USA). Controls consisted of the same reaction procedures described above, but in the absence of primary antibody.
In the second method, HO-1 was co-localized with platelet endothelial cell adhesion molecule-1 (PECAM-1, CD31). Platelet endothelial cell adhesion molecule-1 is a 130-kDa surface protein mainly expressed by endothelial cells. Fixed sections were immunostained with HO-1 antibody and Cy3-conjugated goat anti-rabbit IgG as described above. Sections were then incubated in the following: (1) rat anti-PECAM-1 (1:100 in 10% GS/TX/BSA, PharMingen, San Diego, CA, USA) at RT for 2 h; (2) PBS, 3 × 5 mins; (3) Alexa Fluor 488 goat anti-rat IgG (H + L) (1:200 in 10% GS/TX/BSA, Molecular Probes, Eugene, OR, USA) at RT for 1 h; and (4) PBS, 3 × 5 mins. Sections were coverslipped and visualized using a fluorescence microscope as described above.
Immunolocalization of Neutrophils
Sections were incubated as follows: (1) rat antineutrophil monoclonal antibody (1:1600 in 10% GS/TX/BSA, Caltag Laboratories, Burlingame, CA, USA) at RT, overnight; (2) PBS, 3 × 5 mins; Alexa Fluor 488 goat anti-rat IgG (H + L) (1:200 in 10% GS/TX/BSA, Molecular Probes) at RT for 1 h; and (3) PBS, 3 × 5 mins. Sections were coverslipped and prepared for quantification of neutrophils.
The neutrophil ratio was determined as we have previously described (Noble et al, 2002). Briefly, the total number of neutrophils was determined at the lesioned epicenter and at 1- and 2-mm rostral and caudal to epicenter, respectively. One representative section was selected for analysis from each region. The neutrophil ratio is defined as the total number of neutrophils 2-mm rostral and caudal to the lesioned site divided by the total number of neutrophils at the lesioned epicenter. Animals were excluded from this analysis if they showed marked asymmetry in hindlimb movement, if there was evidence of a dural tear, or if artifact precluded quantification throughout the entire section.
In some sections, double immunolabeling was used to determine the extent to which leukocytes express HO-1. Antibodies, methods, and controls were as described in the preceding paragraphs.
Immunolocalization of 4-Hydroxynoneal and Malondialdehyde
Animals were re-anesthetized and perfused with the nonaldehyde fixative Methacarn (methanol/chloroform/acetic acid, 6:3:1 by volume). Spinal cords were taken, postfixed in Methacarn at 4°C for 4 h, and cryoprotected in 20% (weight/volume) sucrose at 4°C for 3 days. Sections were cut and mounted on the slides as described above. Sections, 20 μm in thickness, were immunostained using the following: (1) rabbit anti-4-hydroxynoneal (HNE) or MDA antibody; (2) PBS, 3 × 5 mins; (3) Cy3-conjugated goat anti-rabbit IgG; and (4) PBS, 3 × 5 mins. Sections were coverslipped and analyzed as described above. Controls consisted of the same reaction procedures described above in the absence of primary antibody.
Total Heme Oxygenase Activity
Spinal cords were removed, weighed, and prepared for the quantitation of total HO activity. Heme oxygenase activity was measured as CO production as quantitated by gas chromatography as previously described (Vreman and Stevenson, 1999). Spinal cord samples (approximately 15 mg in wet weight) were pulse-sonicated to homogeneity in 9 volumes of ice-cold 0.1 mol/L potassium phosphate buffer (KPO4, pH 7.4) at 50% power with an ultrasonic cell disruptor with a one-eighth inch microprobe (Model XL2000, Misonics Inc., Farmingdale, NY, USA) in an ice bath. Sonicates were kept on ice and used immediately. Tissue sonicates were incubated with equal (20 μL) volumes of NADPH (4.5 μmol/L) and methemalbumin (150/11.2 μmol/L) for 15 mins at 37°C in 2-mL CO-purged septum-sealed vials. Reactions were terminated by addition of 2-μL 60% (w/v) sulfosalicylic acid and vials were placed in ice. The amount of CO generated into the vial headspace was determined by gas chromatography using a 60 × 0.53 cm2 (internal diameter) stainless-steel column packed with 5A molecular sieve, 60 to 80 mesh, at a temperature of 150°C and a reduction gas detector (RGA-2, Trace Analytical Inc., formerly of Menlo Park, CA, USA. Similar equipment is presently sold and serviced by Ametek, Inc., Newark, DE, USA and Peak Laboratories, LLC, Mountain View, CA, USA) operated at 270°C. HO activity was expressed as pmoles of CO generated per h per mg protein.
Methemalbumin was prepared daily by dissolving heme (9.9 mg) in 2.5 mL 0.4 mol/L trisodium phosphate and 100 mg of BSA. After addition of water to 8.0 mL, pH was gradually titrated to 7.4 with 1 mol/L HC1. The volume was then adjusted to 10.0 mL to yield a stock solution of 1.5 mmol/L/112 μmol/L. A working solution of 150 μmol/L methemalbumin was prepared by diluting the stock methemalbumin solution 1:10 with phosphate buffer.
Western Immunoblots
Samples of spinal cords at 24 h postinjury were quickly removed, frozen on dry ice, and stored at −70°C. A 4-mm segment of cord, centered over the impact site, was prepared for subsequent analysis. The epicenter segment was homogenized in lysis buffer, containing 50 mmol/L Tris-HCl, 150 mmol/L NaCl, 1% NP-40, 0.5% deoxycholate, and 0.1% SDS (pH 8.0), centrifuged at 16,000g for 30 mins. The protein concentration of supernatant was measured using the BCA protein assay kit (Pierce, Rockford, IL, USA). Equal amounts (10 μg) of total protein was loaded and separated on a 12% SDS-PAGE gel, and transferred onto a nitrocellulose membrane. The membrane was blocked with 5% nonfat dry milk in 0.1 mol/L PBS (pH 7.4), containing 0.05% Tween-20 (PBS-T) at 4°C overnight. The primary antibody to HO-1 (StressGen), myelin basic protein (MBP) (Chemicon International, Inc., Temecula, CA, USA), HNE, and MDA (Alpha Diagnostic International, Inc., San Antonio, TX, USA) were first diluted in blocking buffer at RT for 1 h. After three 10-min washes in PBS-T, the membrane was incubated with 1:1,200 diluted biotinylated goat anti-rabbit IgG (Vector Laboratories) at RT for 1 h. After another round of PBS-T washing, the membrane was incubated in an avidin—biotin—peroxidase complex (dilution 1:200, Vectastain ABC kit, Vector Laboratories) for 30 mins. Finally, the antigen was detected using the standard chemical luminescence method (ECL, Amersham Biosciences Corp, Piscataway, NJ, USA). Densitometric analysis was performed using the NIH Image Program. Analysis for MBP is expressed as a ratio of integrated optical density of degraded MBP relative to the value of an internal control (second band) within the same lane. Analysis for HNE and MDA is indicated as the integrated optical density of modified protein.
Blood—Spinal Cord Barrier Permeability Studies
The permeability of the barrier was quantitatively evaluated using luciferase as a marker of barrier integrity, as previously described (Noble et al, 2002). Briefly, recombinant luciferase was diluted to 1 mg/mL in luciferase storage buffer (Promega, Madison, WI, USA) and stored at −70°C. HO-1+/– and WT mice were re-anesthetized at 24 h after spinal cord injury and then placed on a warming blanket to maintain body temperature. Fifty percent of recombinant luciferase (100 μL/30 g body weight), diluted 1:1 in PBS/0.001% BSA was injected into the jugular vein of injured mice. Blood samples were collected 25 mins after injection of luciferase and then diluted 1:5 in 0.1 mol/L PBS. At 30 mins, each animal was perfused with PBS, and the spinal cord was quickly removed and stored at −70°C.
The cord was divided into five 3 mm-length segments: two rostral, one epicenter, and two caudal. The segments were homogenized in a 1:50 dilution by weight of 1 × cell lysis buffer (25 mmol/L Tris, pH 7.8, 2 mmol/L trans-1,2-diaminocyclohexane N,N,N′,N′-tetra-acetate monohydrate, 2 mmol/L dithiothreitol, 10% glycerol, and 1% Triton X-100). Lysates were centrifuged at 16,000g for 30 mins. The supernatants were transferred onto clean tubes and brought to a final dilution of 1:5000. The blood samples were finally diluted 1:1000. Enzyme activities, measured in 10-μL aliquots of the dilution, were based on luminescence using a luciferase assay kit (Promega) and a luminometer (TD-20/20; Turner Designs, Sunnyvale, CA, USA) with a 10-secs measuring time. Values are expressed as the ratio of activity within the injured segment relative to samples of blood.
Statistical Analyses
Data are expressed as mean ± s.d. Differences between two groups were compared using Student's t-test. Two-way analysis of variance was used to compare barrier permeability to luciferase along the axis of the cord between each of the genotypes. One-way analysis of variance followed by Bonferroni's Multiple Comparison Test was used to compare HO activity between groups and to analyze the Western immunoblots for HNE and MDA. Statistical significance was defined at P ≤ 0.05.
Results
Heme Oxygenase-1 is Expressed in the Vascular Compartment and in Recruited Neutrophils after Spinal Cord Injury
We first determined using WT mice if HO-1 was localized in blood vessels previously labeled with Lycopersicon (tomato) lectin, which binds selectively to the endothelial glycocalyx. Heme Oxygenase-1 expression was noted in the vasculature at the epicenter (data not shown) as well as at 4-, 3-, and 2-mm rostral to the epicenter (Figure 1). This localization was evident within both the gray and white matter. A similar pattern of expression was demonstrated in blood vessels that had been immunolabeled for PECAM (data not shown).
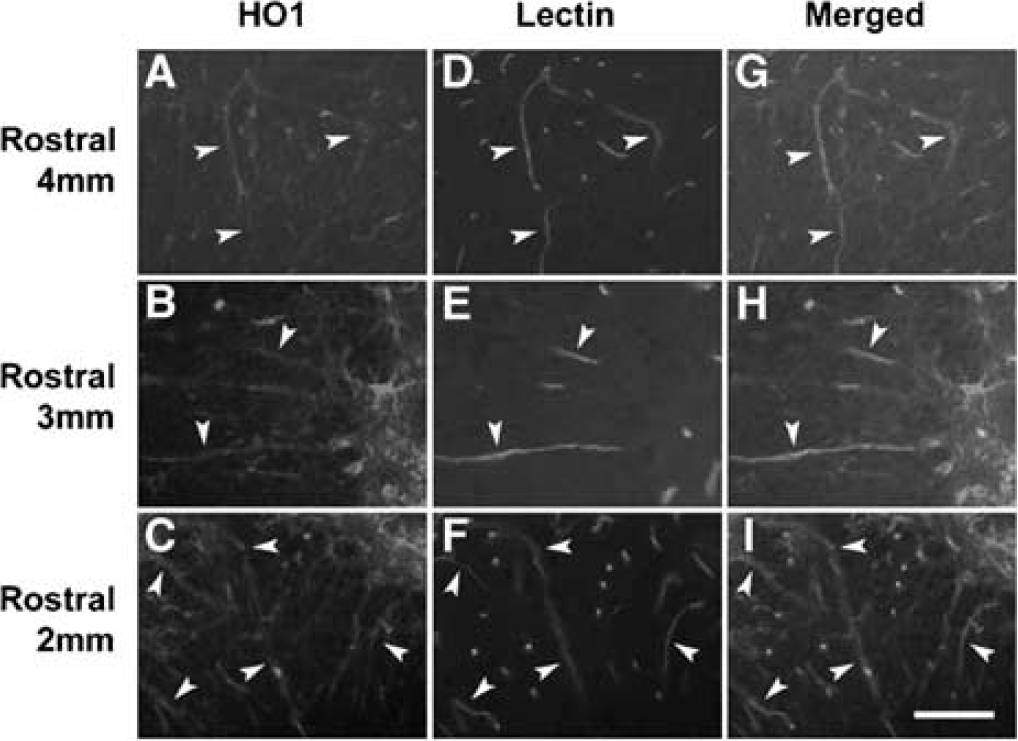
Co-localization of HO-1 in blood vessels within the lesioned epicenter at 24 h postinjury in WT mice. Heme oxygenase-1 expression (
Double-labeling experiments were next used to study the expression of HO-1 in perivascular astrocytes and neurophils. Heme oxygenase-1 was induced in astrocytic processes that were typically oriented parallel to the endothelium. Intimate contact was noted between these processes and the vessel wall (Figure 2). Infiltrated neutrophils likewise expressed HO-1 (Figure 3).
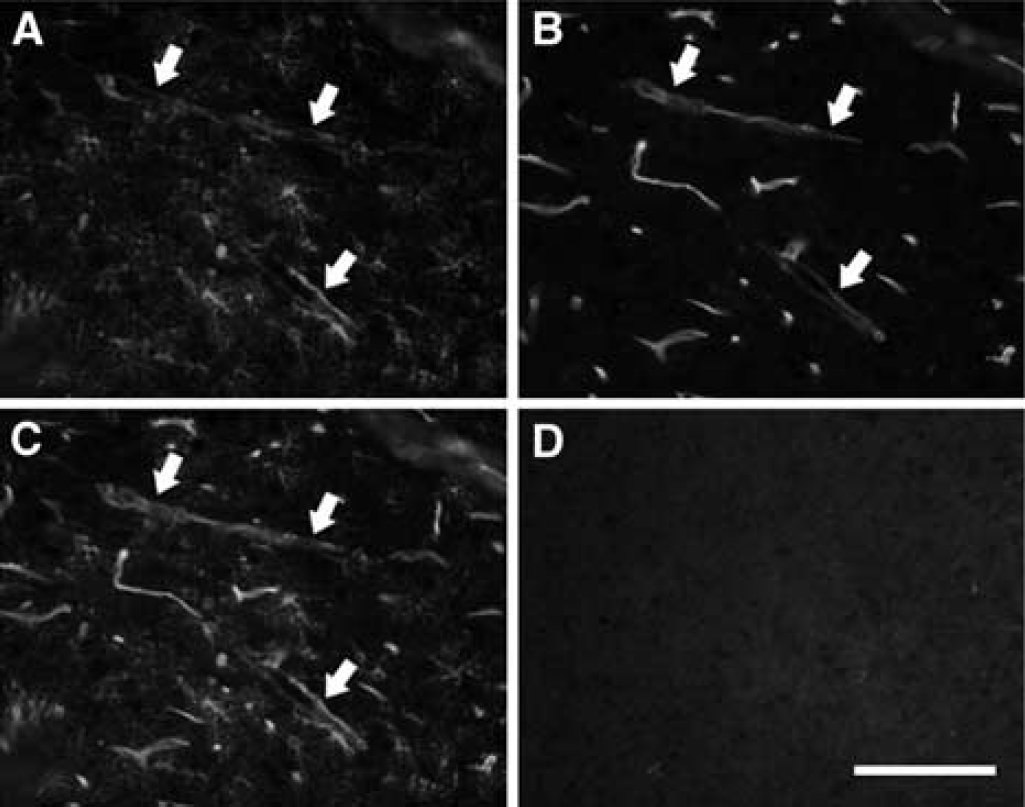
Double-labeling for HO-1 (
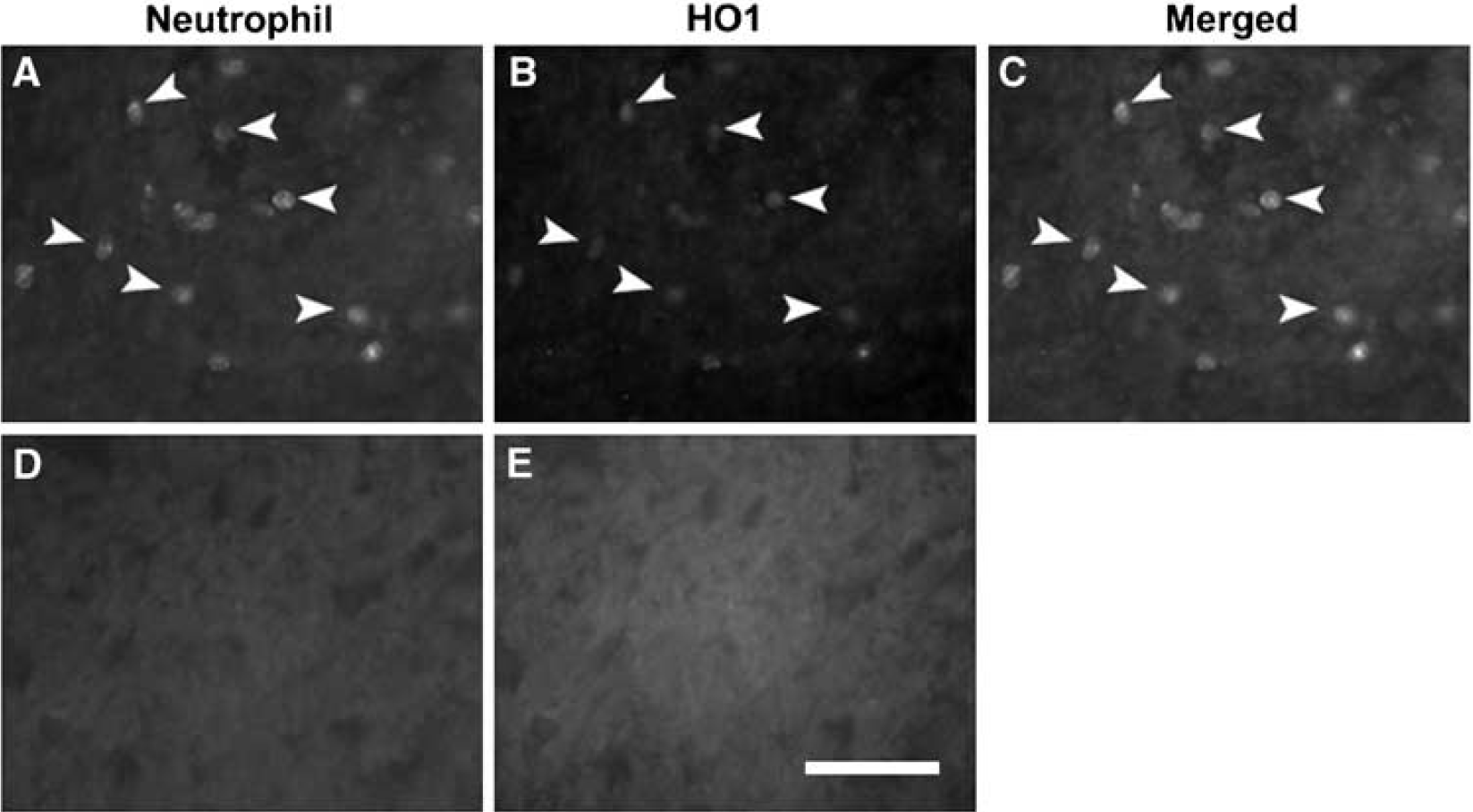
Co-localization of HO-1 in neutrophils in the lesioned epicenter at 24 h postinjury in WT mice. A subset of neutrophils (
Heme Oxygenase-1 Protein and Activity are Decreased in HO-1 +/– Mice after Injury
Heme oxygenase-1 protein was examined by Western immunoblots in uninjured (control) mice and in HO-1 +/– and WT mice 24 h after spinal cord injury (Figure 4). As expected, HO-1 protein (32 kDa) was minimally detected in the uninjured spinal cord. After spinal cord injury, however, HO-1 protein was markedly increased in the injured WT cords. In contrast, no overt change in HO-1 protein was noted in the injured cords harvested from HO-1 +/– mice.
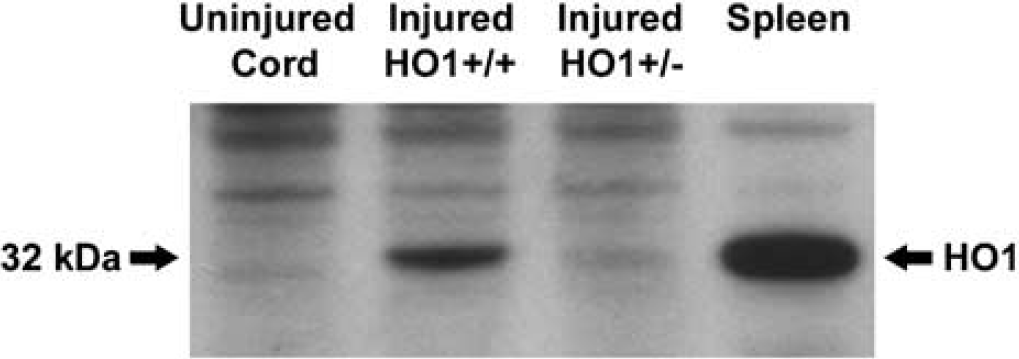
Western immunoblot of HO-1, prepared from WT (HO-1 +/+) and HO-1 +/– mice. Modest expression is noted in the uninjured spinal cord; whereas pronounced expression is shown in the spleen, a positive control. HO-1 is markedly induced within the lesioned epicenter at 24 h postinjury in WT mice. However, expression within the lesioned epicenter of HO-1 +/– mice is similar to that seen in the uninjured spinal cord.
When total HO activity in HO-1 +/– and WT mice was measured, we found no differences in HO activity between these genotypes in the uninjured spinal cord (Figure 5A). In contrast, there were significant increases within the lesioned segments in the injured WT mice relative to the uninjured WT controls (P = 0.0068, Figure 5A) and relative to the cervical cord (P = 0.0192, Figure 5B). There was a trend toward a reduction in HO activity in the injured HO +/– relative to the injured WT (Figure 5A, P = 0.0670). No difference was noted in the injured HO-1 +/– mice as compared with uninjured HO-1 +/– controls (Figure 5A, P = 0.3746) or to cervical values (Figure 5B, P = 0.2346).
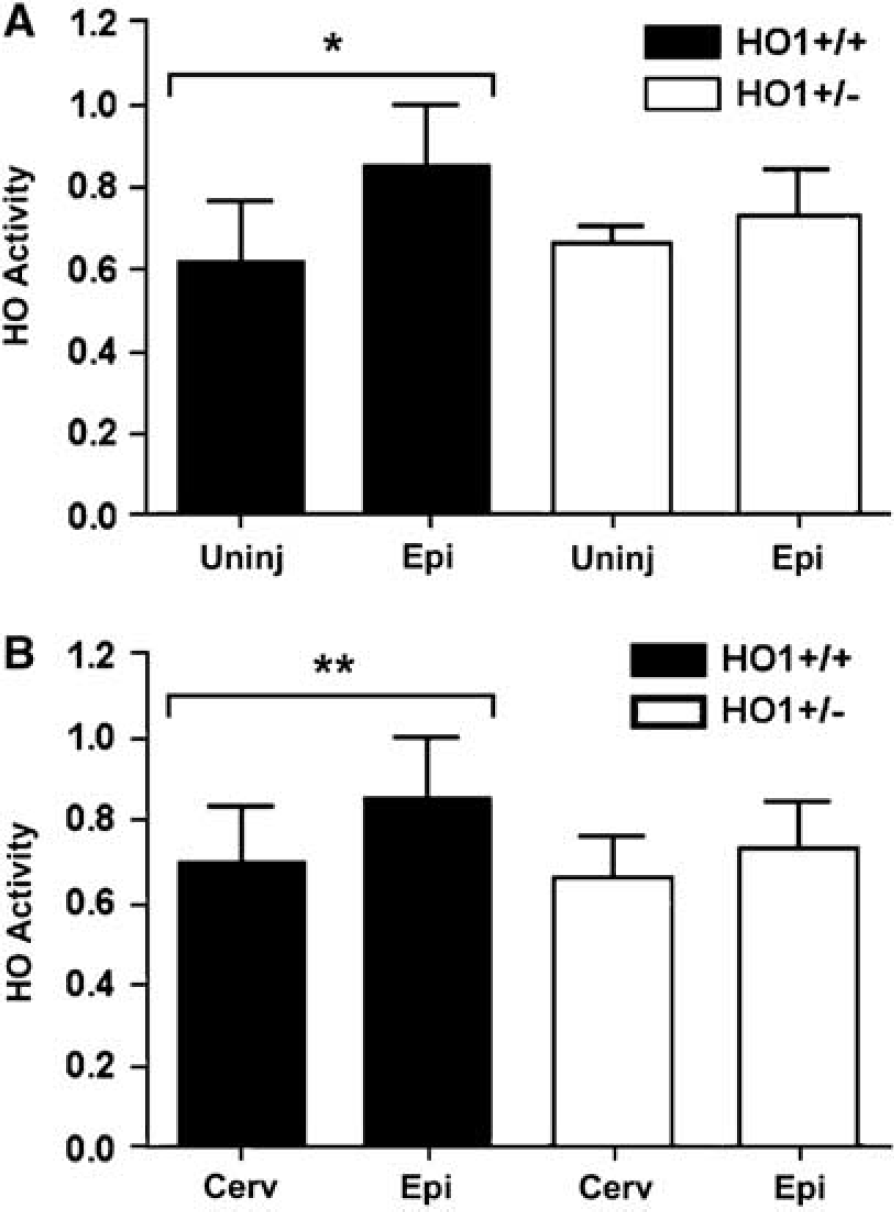
HO activity was quantified in the intact (Uninj) spinal cord and at 24 h postinjury within the lesioned epicenter (Epi) and a cervical segment (Cerv). There is a significant increase in activity within the epicenter of WT mice (HO-1 +/+), as compared with the uninjured spinal cord (
Heme Oxygenase-1 Induction Stabilizes the Blood—Spinal Cord Barrier and Attenuates Neutrophil Infiltration
The expression of HO-1 in vascular-related structures (endothelium and astrocytic processes) as well as in neutrophils suggests that HO-1 may influence vascular responses and leukocyte infiltration after spinal cord injury. We therefore compared barrier permeability and leukocyte infiltration between HO-1 +/– and WT mice. To compare barrier permeability, we asked if the trajectories along the axis of the cord differed between the two genotypes. Based on two-way analysis of variance, we found no significant interaction (P = 0.587). Main effects show on average that the values for the HO-1 +/– animals are significantly higher than that of the WT (P = 0.042) (Figure 6).
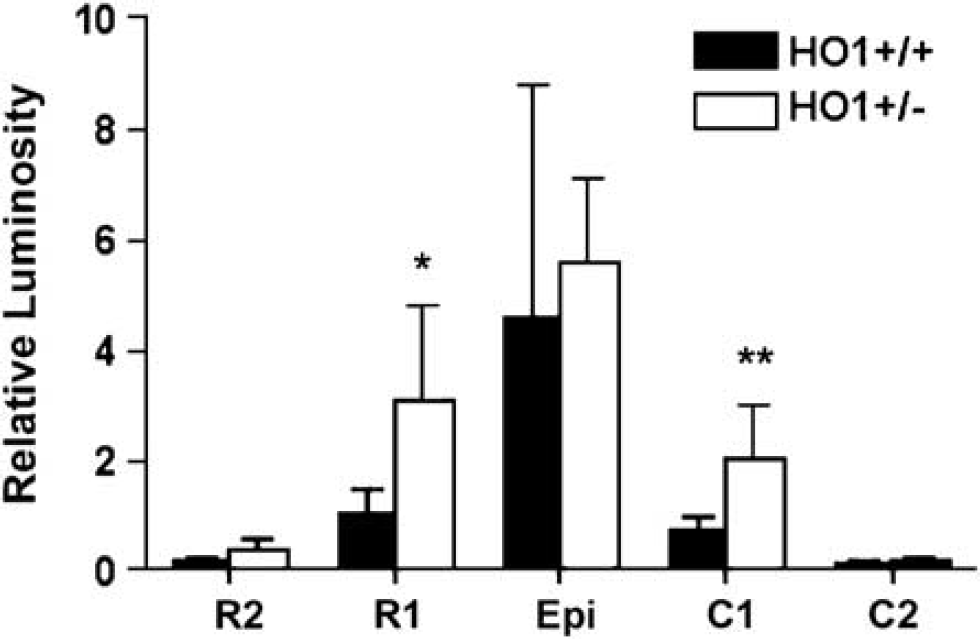
Comparison of blood—spinal cord barrier breakdown to luciferase in the spinal cord at 24 h postinjury in WT (HO-1 +/+) and HO-1 +/– mice. Based on two-way analysis of variance, the trajectories along the axis of the cord are similar between the two genotypes (P = 0.587). Main effects show on average that the values for the HO-1 +/– animals are significantly higher than that of the WT (P = 0.042). Values are shown as mean ± s.d.
The early inflammatory response was next compared in spinal cord-injured HO-1 +/– and WT mice. Neutrophil infiltration in the injured cords was significantly less in the WT as compared with HO-1 +/– mice (Figure 7).
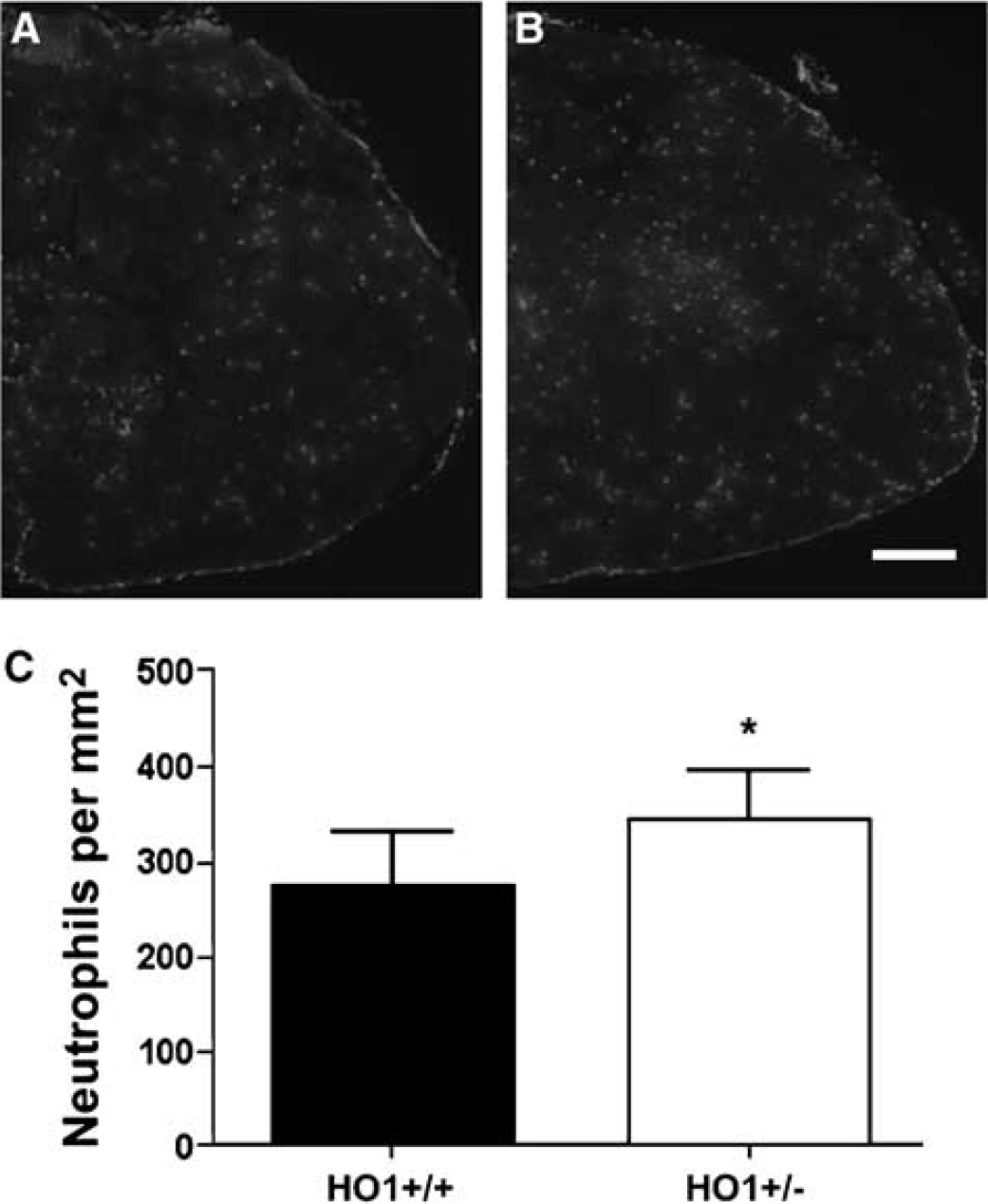
Comparison of neutrophil infiltration in the spinal cord at 24 h postinjury in WT (HO-1+/+) (
Heme Oxygenase-1 Reduces Oxidative Stress after Spinal Cord Injury and Limits the Degradation of Myelin Basic Protein
There is evidence that HO-1 confers neuroprotection in part by degrading the pro-oxidant heme and -generating bilirubin, which has been shown to be a potent antioxidant (Dore et al, 1999; Vreman et al, 1998). We therefore examined the expression of HNE-modified proteins in spinal cord-injured HO-1 +/– and WT mice by Western immunoblots and immunocytochemistry. Of the four bands analyzed, only one, a 10 kDa HNE-modified protein, was significantly higher in HO-1 +/– compared with WT mice (Figures 8A and 8B, P < 0.05)). 4-Hydroxynoneal was primarily immunolocalized in cells with a rounded phenotype in the white matter (Figure 8C); whereas no staining was observed in the control tissue (Figure 8D).
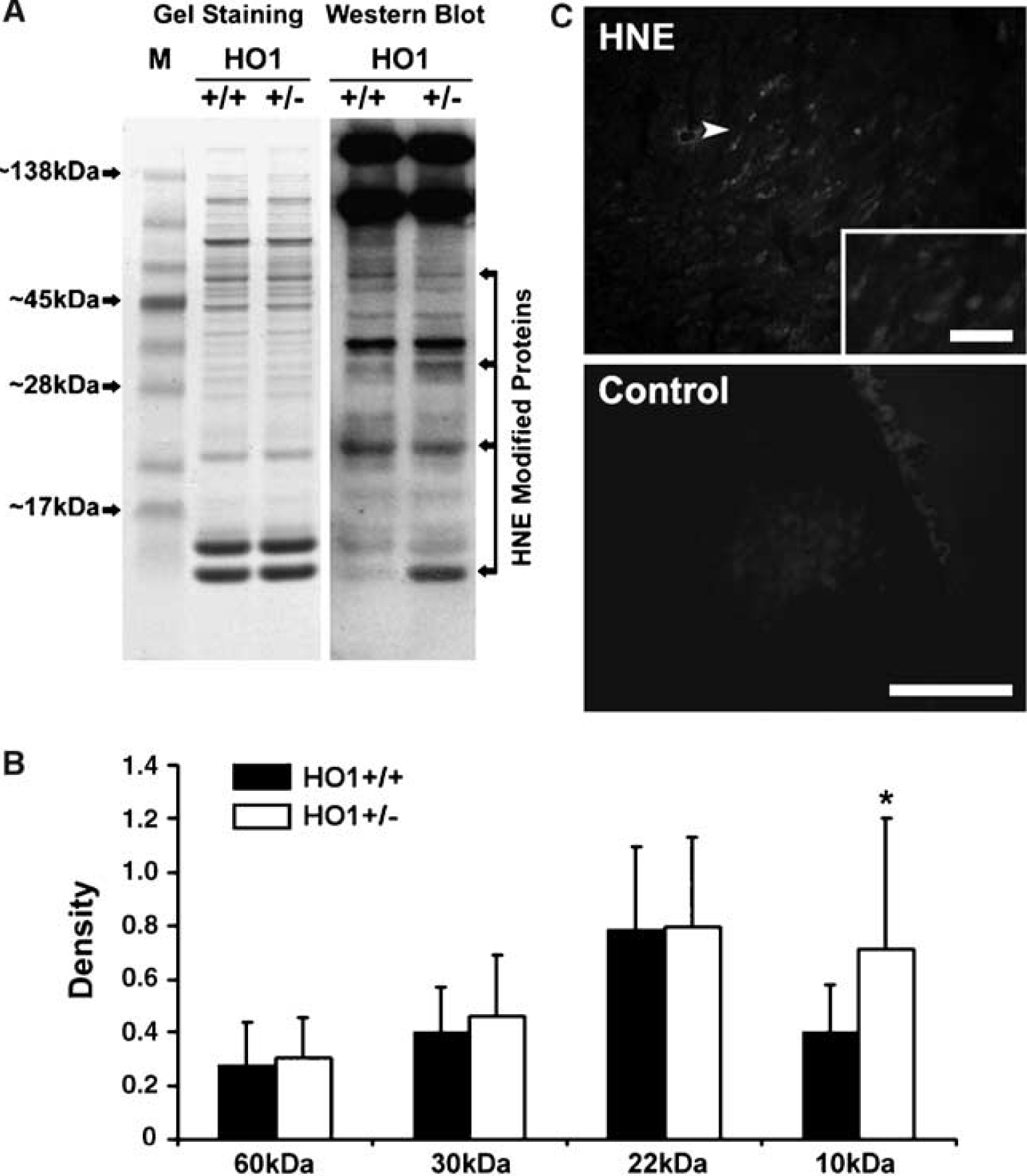
Immunoexpression of HNE within the lesioned epicenter at 24 h postinjury. Western immunoblot for HNE-modified proteins from WT (HO-1 +/+) and HO-1 +/– mice (
Of the six bands analyzed for MDA-modified proteins, only one, a 10 kDa modified protein, was significantly higher in the HO-1 +/– as compared with WT mice (Figures 9A and 9B, P = 0.043). Malondialdehyde was likewise immunolocalized in white matter structures that had a rounded phenotype (Figure 9C) and no staining was observed in the control tissue (Figure 9D).
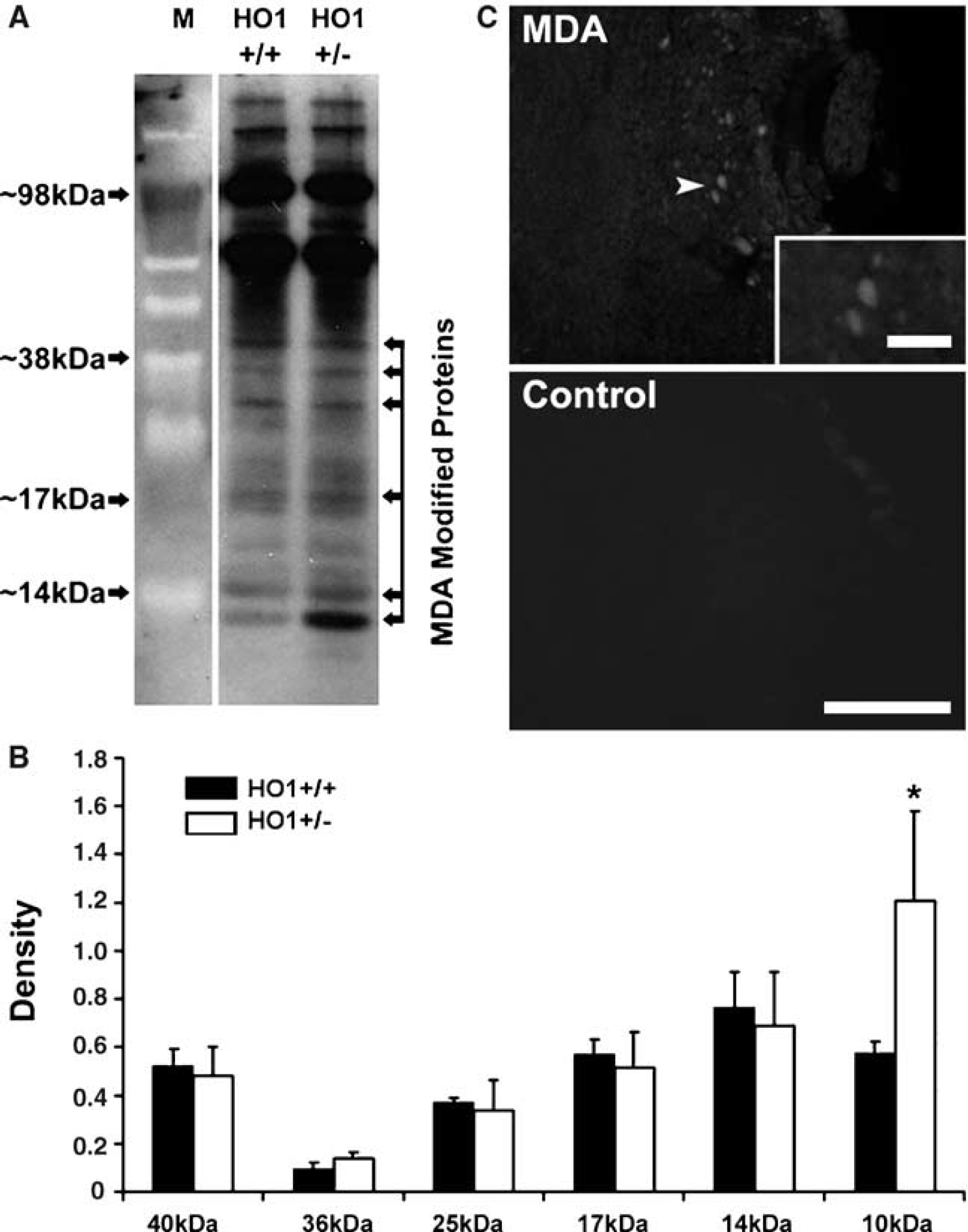
Western immunoblot for MDA, prepared from WT (HO-1 +/+) and HO-1 deficient (+/–) mice (
We next determined if the limited expression of HNE- and MDA-modified proteins in the white matter of acutely injured, HO-1 +/– mice correlated with greater protection to white matter structures. To test this possibility, we examined the expression of MBP and its degradation products by Western immunoblots (Figure 10). Myelin basic protein appeared as four isoforms of 21.5, 18.5, 17, and 14 kDa by Western blots (Figure 10A). An approximate 10-kDa protein degradation band was detected in both spinal cord-injured HO-1 +/– and WT mice. Densitometric analysis revealed significantly greater expression of this degradation product in HO-1 +/– as compared with WT mice (P = 0.049; Figure 10B).
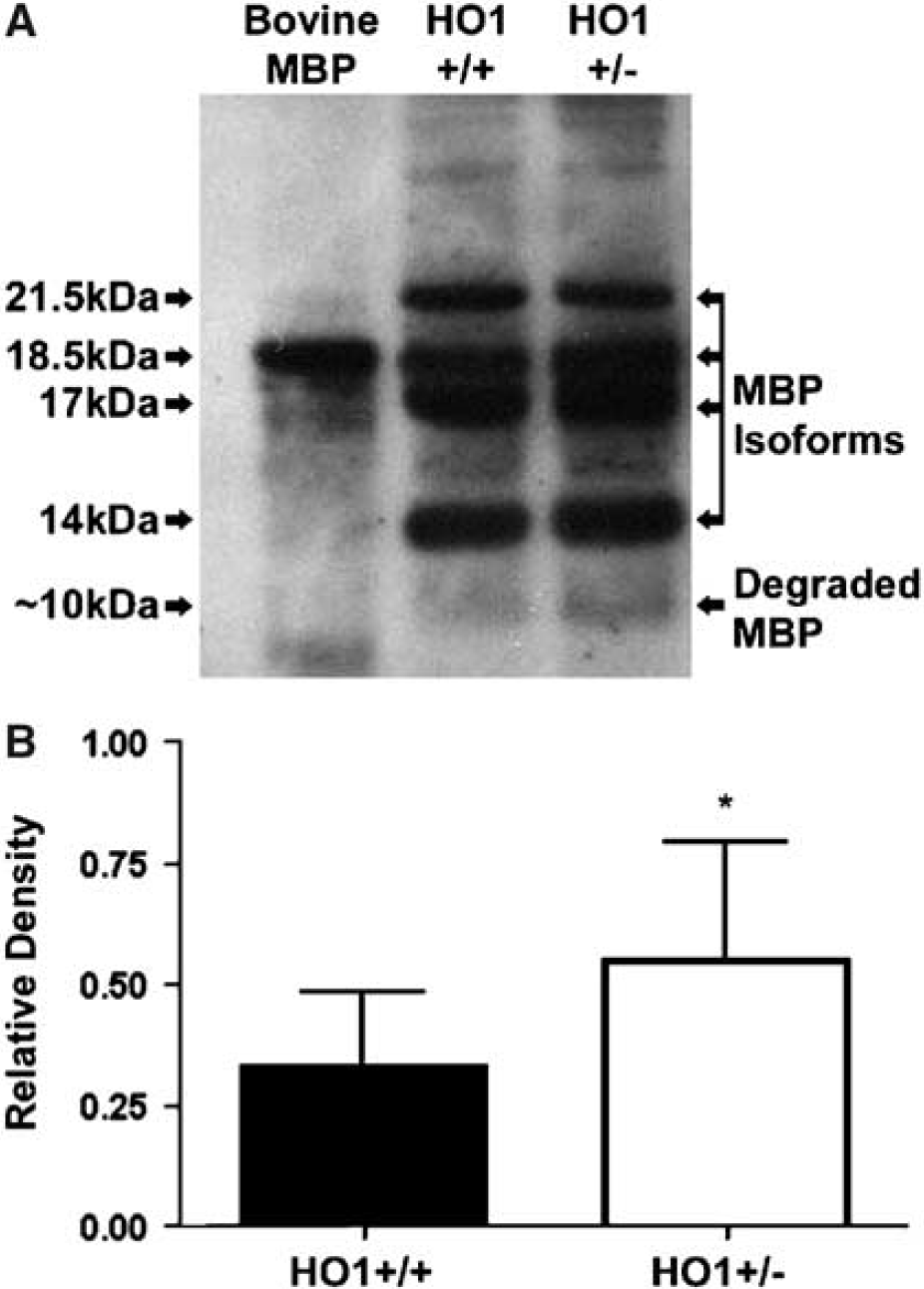
Western immunoblot for MBP, prepared from WT (HO-1 +/+) and HO-1 deficient (+/–) mice (
Discussion
Here, we show that HO-1 modulates barrier function, neutrophil infiltration, and oxidative stress in the acutely injured spinal cord. Heme oxygenase-1 is expressed in the vascular compartment including perivascular astrocytes and in recruited neutrophils after this injury. Abnormal barrier permeability is significantly greater in spinal cord-injured, HO-1 +/– mice. 4-Hydroxynoneal and MDA, indicators of oxidative stress, are also increased in spinal cord-injured HO-1 +/– mice. Together, these observations suggest that HO-1 protects the acutely injured cord by stabilizing the barrier, limiting neutrophil infiltration, and modulating oxidative stress. Consistent with this hypothesis, we show that degradation of MBP, a surrogate marker for white matter injury, is significantly greater in spinal cord-injured, HO-1 +/– mice.
Heme Oxygenase-1 and the Injured Spinal Cord
Heme oxygenase-1 is minimally detected in the uninjured spinal cord by Western immunoblots and immunocytochemistry and is markedly induced in spinal cord-injured WT mice. These findings are consistent with previous studies (Mautes et al, 2000). A similarly overt pattern of induction is not seen in spinal cord-injured HO-1 +/– mice.
Both HO-1 and HO-2 catalyze the same biochemical reaction and thus contribute to total HO activity. We found that expression of HO-1 protein correlates with activity of the enzyme in vitro, as measured through the quantitation of CO by gas chromatography (Vreman and Stevenson, 1999).
The total HO activity was unchanged in the HO-1 +/– relative to the WT. There are several scenarios that might explain this observation. There may have been a compensatory increase in HO-2 in the HO-1 +/–. Since we did not evaluate the protein levels of HO-2 in this study, we cannot rule out this possibility. In vitro studies would suggest otherwise. Chen-Roetling et al (2005) have shown that HO-2 protein in astrocytes is not altered by HO-1 gene deletion. Similarly, Kapturczak et al found no difference in HO-2 in the spleens of HO-1 homozygote knockout mice relative to WT controls. Alternatively, the absence of a difference between the groups may reflect residual HO-1 activity that persists in the heterozygote knockout. Finally, the absence of any difference may be because of the much higher level of HO-2 activity in the brain relative to HO-1. A majority of the HO activity in the brain is attributed to HO-2 (Maines, 2000). As such, partial loss of HO-1 may have had minimal impact on the measurable levels of HO activity in the brain.
Heme Oxygenase-1 and Neutrophil Infiltration
We found that neutrophil infiltration is markedly reduced in spinal cord-injured WT mice compared with HO-1 +/– mice. Heme oxygenase-1 is an important determinant in the inflammatory cascade. Elevated levels of HO-1 result in marked suppression of the inflammatory response; whereas inhibition of HO-1 potentiates this response (Willis et al, 1996). Recent studies have begun to address the mechanisms whereby HO-1 may influence inflammatory events. Heme oxygenase-1 inhibits transmigration of inflammatory cells (Ishikawa et al, 1997). This inhibition is attributed to the altered expression of specific adhesion molecules expressed on the vasculature. For example, HO decreases heme-induced expression of intracellular adhesion molecule 1 (Wagener et al, 1999), a molecule that mediates neutrophil adhesion to the endothelial surface and is necessary for the transmigration of neutrophils into the parenchyma (Justicia et al, 2003).
Heme oxygenase-1 may also modulate neutrophil transmigration through downregulation of MMPs. Matrix metalloproteinase-9 is actively expressed within the acutely injured spinal cord (Noble et al, 2002). A likely source for this early increased activity is the pool of neutrophils that have infiltrated into the injured tissue. Neutrophils are thought to use this MMP to transmigrate across vessels into the injured spinal cord (Noble et al, 2002). Consistent with this hypothesis is the finding that blockade of MMP activity attenuates neutrophil infiltration (Noble et al, 2002). In a recent study, Lee and Chau (2002) have shown that HO-1 mediates the anti-inflammatory effects of IL-10 via suppression of MMP-9 and reinforces the hypothesis that HO-1 functions as an immunomodulator through its ability to limit transmigration of leukocytes across the blood—brain/spinal cord barrier.
Heme Oxygenase-1 and Barrier Function
The blood—brain/spinal cord barrier consists of specialized endothelial cells and a surrounding basal lamina that is contacted by perivascular endfeet (Abbott, 2002). Each of these structures contributes to the barrier's ability to regulate permeability of proteins across its interface. Here, we show induction of HO-1 in both endothelial cells and the adjacent astrocytic processes in the injured spinal cord. We further show that barrier disruption to the protein luciferase is significantly increased in HO-1 +/– mice. These observations implicate HO-1 in modulation of barrier function after spinal cord injury.
Insights can be gained from studies where systemic administration of agents induces HO-1 in the vasculature (Lu et al, 2002; Yamauchi et al, 2004). Lu et al (2002) evaluated the expression of heat shock proteins in both normal and ischemic brains in animals pretreated with the neuroprotective steroid, estradiol. A surprising finding was that estradiol upregulated heat shock proteins including HO-1 in blood vessels. In a complementary study, systemic administration of hemin, 24 h before spinal cord injury, resulted in the specific induction of HO-1 in spinal cord blood vessels and a significant attenuation of barrier disruption and neutrophil infiltration (Yamauchi et al, 2004). Collectively, these studies support the hypothesis that vascular induction of HO-1 stabilizes this interface against ischemia and trauma and the ensuing early inflammatory response.
We can only speculate on the mechanisms by which HO-1 modulates barrier function. One scenario is that HO-1 modulates barrier function through its interactions with proteases. This is depicted in the role of HO-1 as an upstream regulator of MMP-9 (Lee and Chau, 2002), a protease that degrades critical components of the barrier (Mun-Bryce and Rosenberg, 1998) and that has been shown to mediate barrier disruption after spinal cord injury (Noble et al, 2002). Heme oxygenase-1 may also stabilize barrier function by modulating the expression of pro-inflammatory cytokines such as tumor necrosis factor-α (Lee and Chau, 2002), which is produced by activated leukocytes, astrocytes, microglia, and neurons (Bethea et al, 1999), is increased in the injured spinal cord (Yune et al, 2003), and has been shown to disrupt the blood—brain barrier (Mayhan, 2002).
Heme Oxygenase-1: Oxidative Stress and White Matter Injury
Heme oxygenase-1 deficiency resulted in a significant increase in the generation of HNE- and MDA-modified proteins. 4-Hydroxynoneal covalently modifies proteins on cysteine, lysine, and histidine residues (Camandola et al, 2000) and binds to cytoskeletal proteins, such as neurofilaments, microtubule-associated proteins, and glial fibrillary acidic protein (Mattson et al, 1997; Neely et al, 1999). Modification of these proteins leads to neuronal, axonal, and glial cell damage (McCracken et al, 2000). In the present study, we detected modified proteins with molecular weights of approximately 13 kDa. The question arises as to the identity of these proteins. Microtubule-associated proteins are one possibility. Low molecular weight light chains (28 and 30 kDa) of microtubule-associated proteins have been detected by Western blot in bovine brain (Vallee and Davis, 1983) and a 16.4 kDa light chain 3 of the neuronal microtubule-associated proteins has been reported in rodent brain (Mann and Hammarback, 1994).
Our findings suggest that HO-1 modulates early oxidative stress. Beneficial or detrimental consequences of this modulation likely depend on how the end products of heme metabolism, including bilirubin and ferrous iron, interact in the injured cord. Whereas bilirubin has been shown to be neuroprotective (Dore et al, 1999), ferrous iron can be damaging. Cellular iron is a detrimental factor because it elicits the formation of free radicals, with consequent damage to DNA, proteins, and lipids (Ferris et al, 1999; Vreman et al, 1998). Whether this occurs in the setting of spinal cord injury depends in part on how iron is handled. For example, the toxic effects of free iron can be countered by upregulation of intracellular ferritin (Hoekstra et al, 2004) and HO-1-mediated efflux of cellular iron (Ferris et al, 1999). In this study, we show that HO-1 activity increases in the acutely injured cords of WT animals and is associated with reduced oxidative stress, relative to that found in the spinal cord injured, HO-1 deficient animals. These findings suggest that the over-all net effect of HO-1 activity is to protect the injured cord from early oxidative damage.
Heme oxygenase-1 mediated protection is further validated in measures of white matter injury. We found that degradation of MBP is significantly increased in HO-1 +/– mice after spinal cord injury. Increased oxidative stress may augment ongoing demyelination. It is also possible that HO-1 operates upstream from proteases that selectively degrade myelin. Myelin basic protein, a substrate for MMP-9 (Hartung and Kieseier, 2000), is elevated in the acutely injured spinal cord (Noble et al, 2002) at a time when HO activity is increased. Degradation of MBP is attenuated in spinal cord injured MMP +/– animals (Trivedi et al, 2005). Importantly, in an in vivo model of inflammation HO-1 downregulates MMP-9 (Lee and Chau, 2002).
Several intermediate molecules, including tumor necrosis factor-α, link HO-1 to MMP-9. Tumor necrosis factor-α induces MMP-9 in a variety of cell types including astrocytes (Arai et al, 2003) and endothelial cells (Harkness et al, 2000). Heme oxygenase-1 and its downstream end product CO reduce the level of tumor necrosis factor-α (Lee and Chau, 2002), and as such, establish a mechanistic link between HO-1 and MMP-9.
In summary, our findings suggest that HO-1 plays an important protective role in the acutely injured spinal cord. This suggests that HO-1 may be a suitable target for therapeutic intervention. As proof of concept we have shown that heme-mediated induction of HO-1, before spinal cord injury, is neuroprotective (Yamauchi et al, 2004). Whether a post-treatment paradigm would yield similar results has yet to be evaluated. Several issues should be considered in this context. First, it is possible that a post-treatment paradigm will target different inflammatory events. For example, we have shown that induction of HO-1 in blood vessels before injury, results in reduced infiltration of neutrophils into the injured cord (Yamauchi et al, 2004). Delayed induction of HO-1 in blood vessels may not effectively address the transmigration of neutrophils that typically peaks within the first day postinjury. However, it may alter the transmigration of other leukocytes, including monocytes and lymphocytes that exhibit a more delayed temporal course of infiltration. Second, the beneficial effects of HO must be tempered by pharmacologic studies, relying on HO inhibitors such as tin-protoporphyrin that have shown neuroprotection (Huang et al, 2002; Kadoya et al, 1995; Panizzon et al, 1996). Whether this benefit is achieved through blockade of HO remains unclear. Tin-protoporphyrin as well as other protoporphyrins acts as nonspecific inhibitors of heme-binding proteins (Imai et al, 1990; Wong et al, 2000). Importantly, they can directly inhibit lipid peroxidation and as such could confer neuroprotection.
Footnotes
Acknowledgements
We also thank Jung-Euk Lee and Nino Maida for their superb technical contributions to this study.