
Abstract
A brief period of bilateral carotid occlusion (BCO)-induced forebrain ischemia in gerbils triggers neuronal degeneration and the subsequent expression of amyloid precursor protein (APP), b-amyloid protein (b-AP), and apolipoprotein E (APO-E) in the selectively vulnerable CA, region of the hippocampus. The increase in immunoreactivity is secondary to the postischemic degeneration of the CA1 neurons and is largely astrocyte-derived as evidenced by a simultaneous increase in glial fibrillary acidic protein (GFAP) staining. Oxygen radical-induced lipid peroxidation has been strongly suggested to play a role in postischemic neuronal damage and Alzheimer's disease. Recent literature suggests a possible link between early oxidative stress and APP overexpression. Therefore, the present investigation examined the effect of two novel brain-penetrating pyrrolopyrimidine lipid peroxidation inhibitors (PNU-101033E and PNU-104067F) on CA1 neurodegeneration and the subsequent increase in APP, b-AP, APO-E, and GFAP immunostaining at 4 days after a 5-minute episode of forebrain ischemia. Using an antibody for lipid peroxidation–derived malondialdehyde (MDA)-modified proteins, the authors also examined the effects of PNU-104067F on MDA immunostaining 2 days after ischemia, before completion of the neuronal loss. At 2 days, the authors also evaluated microglial activation using an antibody to surface major histocompatibility complex class II antigen expressed by activated microglia. Gerbils were treated at 30 mg/kg orally 30 minutes before the BCO and 2 hours after ischemia, followed by daily dosing for the next day (microglia and MDA) and the successive 3 days for APP, b-AP, APO-E, and GFAP immunostaining. APP and APO-E staining was significantly suppressed by 50% and 66%, respectively, with either compound. b-AP immunoreactivity was decreased 56% with both compounds, and GFAP expression was significantly decreased 53% (PNU-101033E) and 60.5% (PNU-104067F). There was a concomitant partial sparing of the CA1 hippocampal neurons by both PNU-101033E and PNU-104067F (P<.01) as determined by cresyl violet histochemistry. PNU-104067F significantly inhibited lipid peroxidation-derived MDA immunostaining and microglia activation (P<.05) at 48 hours after ischemia. Brain-penetrable lipid peroxidation inhibitors may provide attenuation of various glial response proteins after ischemic injury, probably secondary to neuronal protection.
Keywords
Amyloid precursor protein (APP) is a large transmembrane glycoprotein which is the source of the b-amyloid (b-AP), a 40–42 amino acid peptide, known to accumulate in the senile plaques and blood vessels in individuals with Alzheimer's disease (AD) (Selkoe, 1993). Deposition of b-AP after proteolytic cleavage of APP may play a key role in the pathogenesis of this disease (Hardy and Allsop, 1991; Younkin, 1991; Selkoe, 1994). Regulation of APP expression is also of interest in the context of the secondary pathophysiology of traumatic and ischemic brain injury since recent studies have revealed abnormal expression of APP in neurons and glial cells after various brain insults, including excitotoxic injury (Nakamura et al., 1992; Topper et al., 1993), mechanical trauma (Otsuka et al., 1991; Wallace et al., 1991; Sherriff et al., 1994;), and global or focal ischemia (Abe et al., 1991; Nukina et al., 1992; Stephenson et al., 1992; Wakita et al., 1992; Hall et al., 1995b). In addition, b-AP deposits have been shown to be present in 30% of fatally head-injured patients, and occur in association with increased expression of APP (Gentleman, 1993; Roberts et al., 1994). This seems consistent with the fact that prior head injury is a risk factor for development of AD (Mortimer et al., 1985; Henderson, 1988; Merz, 1989; Graves et al., 1990).
Apolipoprotein E (APO-E), is a 34-kd protein that mediates the clearance of several plasma lipoproteins (Mahley, 1988) and appears in three isoforms (E2, E3, E4). There is an increased frequency of the E4 allele in sporadic and both early and late-onset familial AD patients (Corder et al., 1993; Houlden et al., 1993; Poirier, 1993; Saunders et al, 1993). In vitro experiments indicate that the APO-E4 form, when oxidized, binds to b-AP causing aggregation into amyloid filaments much more rapidly than the more common APO-E3 form (Strittmatter et al., 1993a; Sanan et al., 1994). These in vitro data suggest that APO-E may play a direct role in b-AP deposition and partially explain the association of APO-E4 and AD. Co-localization of APO-E4 and b-AP in the plaques of AD patients support this concept (Namba et al., 1991). Using brief forebrain ischemia in gerbils, we have previously shown an increase in APP, b-AP, and APO-E immunoreactivity in the selectively vulnerable CA1 region of the hippocampus at 4 days after the ischemic injury (Hall et al., 1995b, Ali et al., 1996). This follows the prior 84% loss of the CA1 neurons.
In rodent forebrain ischemia models, there is a significant increase in membrane lipid peroxidation within minutes after reperfusion which continues for several days, and is believed to play a role in the delayed neuronal damage observed by 4 days after injury (Floyd and Carney, 1991, Hall et al., 1993a, Sakamoto et al., 1991). Therefore, the present investigation examined the ability of two novel brain-penetrating pyrrolopyrimidine lipid peroxidation inhibitors, PNU-101033E and PNU-104067F (Hall et al., 1995a; 1997a; Bundy et al., 1995) (Fig. 1) to decrease delayed postischemic CA1 damage and the expression of APP, APO-E, and b-AP. We also evaluated the effects of PNU-104067F on lipid peroxidation-derived malondialdehyde (MDA), and microglial activation using immunocytochemical methods.
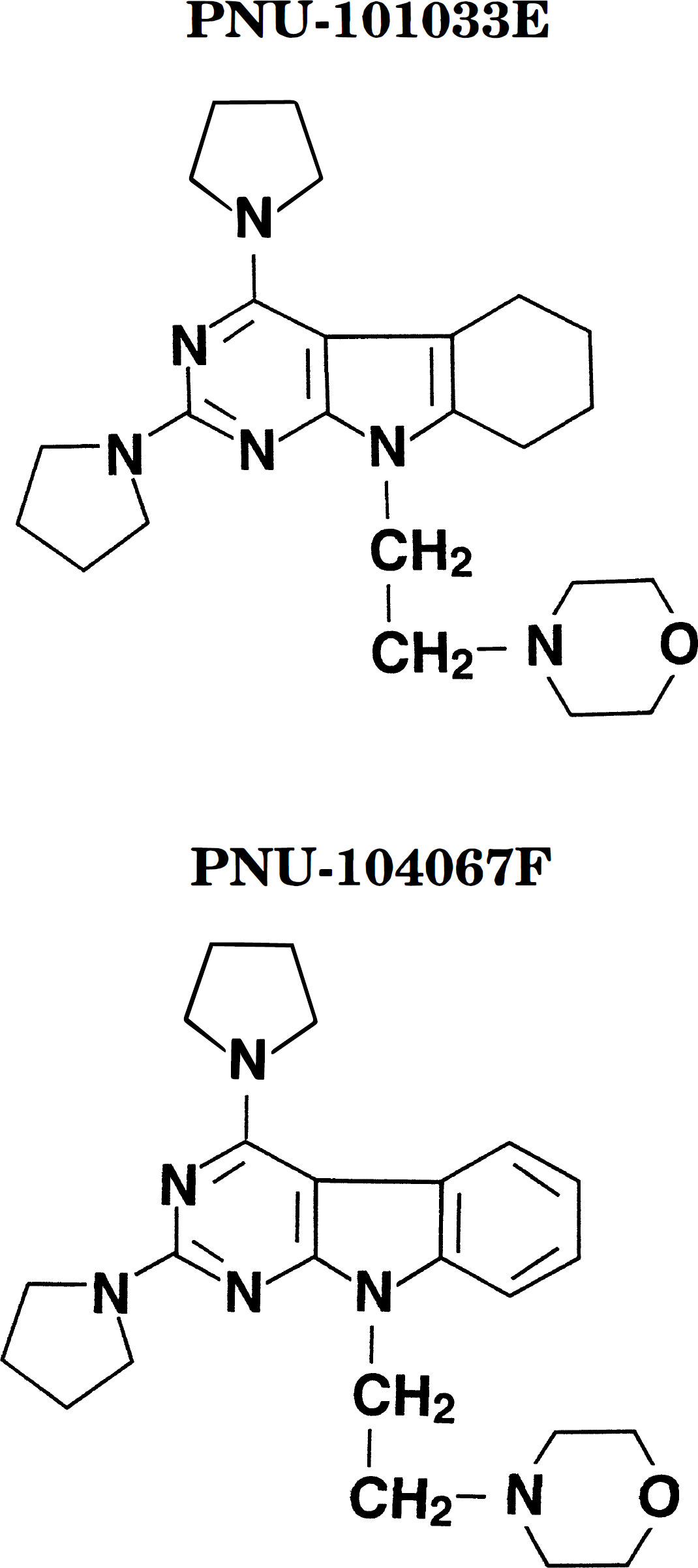
Chemical structures of the pyrrolopyrimidines PNU-104067F and PNU-101033E. The suffixes represent the salts of each compound: E=di-HCI; F = mono-HCI.
MATERIALS AND METHODS
Bilateral carotid occlusion global forebrain ischemia model
All experiments were performed in strict compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Pharmacia & Upjohn, Inc.
Male Mongolian gerbils (Meriones unguiculatus), obtained from Tumblebrook Farms (West Brookfield, MA) weighing 50 to 60 g were anesthetized with methoxyflurane. A 1- to 2-cm midline throat incision provided access to both carotid arteries, which were clamped with microaneurysm clips. Gerbils were placed in a warming box with an ambient temperature maintained at 37°C via a heating chamber that holds brain temperature at 35.5°C for the duration of the bilateral carotid occlusion (BCO) (Hall et al., 1993b). After 5 minutes of BCO, the clips were removed and reperfusion was allowed for either 48 or 96 hours. The animals remained in the heating chamber until they recovered from anesthesia and could regulate their own body and brain temperatures.
Tissue preparation
After either 48 or 96 hours of reperfusion (see explanations below), the animals were deeply anesthetized and perfused intracardially. Animals were perfused with cold oxygenated Krebs-Ringer bicarbonate, pH 7.2 (Sigma, St. Louis, MO) until the effluent was cleared of blood (2 minutes), followed by perfusion with cold 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4) for 8 minutes (approximately 200 mL). Brains were removed, blocked, and post-fixed for 4 hours at 4°C. Tissues were equilibrated in increasing concentrations of sucrose in phosphate-buffered saline (PBS), pH 7.4 (10,20,30%) over a 3-day period. After complete equilibration in 30% sucrose, the brains were frozen in liquid nitrogen vapor for 10 minutes and stored at -70°C until processing. Brains were sectioned at 25 μm on a Leitz sledge microtome with a freezing stage into 0.01 mol/L PBS at 4°C (pH 7.4). The sections were mounted on gelatin-coated slides and air dried.
Immunocytochemical procedures
After rinsing in 0.01 mol/L PBS pH 7.4 (buffer), endogenous peroxidase activity was quenched by incubating the slides in 0.3% H2O2 in methanol for 30 minutes. After buffer rinse, the sections were blocked in normal goat serum for 20 minutes, then incubated overnight at room temperature in the primary antibody diluted in buffer. The primary antibodies were anti-APP (clone 22C11; Boehringer Mannheim), anti-b-AP (Boehringer Mannheim), anti–glial fibrillary acidic protein (GFAP) (clone G-A-5; Boehringer Mannheim), and anti-APO-E (clone 1-H4; Organon Teknika), all used at a 1:2000 dilution. The class II surface major histocompatibility complex antibody used for assessing microglial activation was MRC OX3 (code# MCA 45G; Harlan Bioproducts for Science) at 1:100. The polyclonal anti-MDA, which has been recently described (Hall et al., 1997b), was also used at a 1:100 dilution. The slides were rinsed in buffer and then incubated in secondary antibody [affinity-purified biotinylated antimouse or antirabbit immunoglobulin G (goat); ABC Elite Kit, Vector Laboratories, Inc, Burlingame, CA] according to the recommended dilutions for 2 hours. After a 10-minute buffer rinse, slides were incubated for 1 hour in the Vectastain Elite ABC Reagent according to recommended procedures. Immunocytochemical controls were also run for sham and each of the postischemic time points that were only exposed to the secondary antibody. After a 10-minute buffer rinse, the immunoreactivity was visualized by incubation for 4.5 minutes in the ABC peroxidase substrate solution with nickel chloride added for the computer-assisted densitometric analysis. Slides were rinsed in ddH2O, dehydrated in increasing concentrations of ethanol, cleared in xylene, and mounted in DPX.
Neuronal cell counts
For the investigation of the effects of PNU-101033E and PNU-104067F on hippocampal CA1 neuronal preservation, a 96-hour time point was selected. This was based on our previous demonstration that postischemic CA1 degeneration with the gerbil forebrain ischemia model in our hands is complete by 72 hours (i.e., it is not greater at either 48 or 168 hours) (Hall et al., 1995b). The animals were treated with either PNU-101033E (30 mg/kg orally, n=18), PNU-104067F (30 mg/kg orally, n=20), or vehicle (40% aqueous hydroxypropyl-b-cyclodextrin; Encapsin™HPB, American Maize-Products Company, Hammond, IN, U.S.A.) (n = 20) 30 minutes before the BCO and 2 hours after the BCO, and daily thereafter for 3 days. The doses were selected based on the previous demonstration of their protective efficacy (Hall et al., 1995 a; 1997a). A group of untreated, nonischemic (sham) animals (n = 9) was also included as a control. Numbers of normal appearing medial CA1 neurons were blindly counted in sham, vehicle, and drug-treated animals in 25-μm sections stained with cresyl violet. The number of neurons/315 mm was counted in the medial CA1 region and averaged for the two hemispheres.
MDA and microglial immunocytochemical analysis
For the investigation of effects of PNU-104067F on MDA immunostaining and microglial activation, animals were treated with vehicle (n=8), PNU-104067F (n=8), or sham (n = 7) using the same protocol except that sacrifice occurred at 48 hours after injury. This time point was selected for two reasons. The first is that both microglial activation (Morioka et al., 1991; Gehrmann et al., 1992) and MDA immunostaining (Hall et al., 1997b) are significantly increased by 48 hours after brief forebrain ischemia, thus constituting an adequately developed end point for drug therapy. The second reason is that we wanted to investigate drug effects on these parameters at a time that preceded completion of CA1 degeneration so that possible mechanistic conclusions could be drawn concerning the effects of pyrrolopyrimidine administration and its possible association with CA1 preservation. The number of microglia were manually counted across the hippocampus at 200X final magnification in a 620 mm x 1240 mm area. The raw numbers were converted to the number of microglia/mm2. All evaluations were blinded. Semiquantitative analysis of MDA immunostaining was made in the right medial CA1 region of the hippocampus. MDA slides were blindly evaluated and scored from 0 to 4, based on the intensity of immunoreactivity as shown in Fig. 2 and previously described (Hall et al., 1997b).
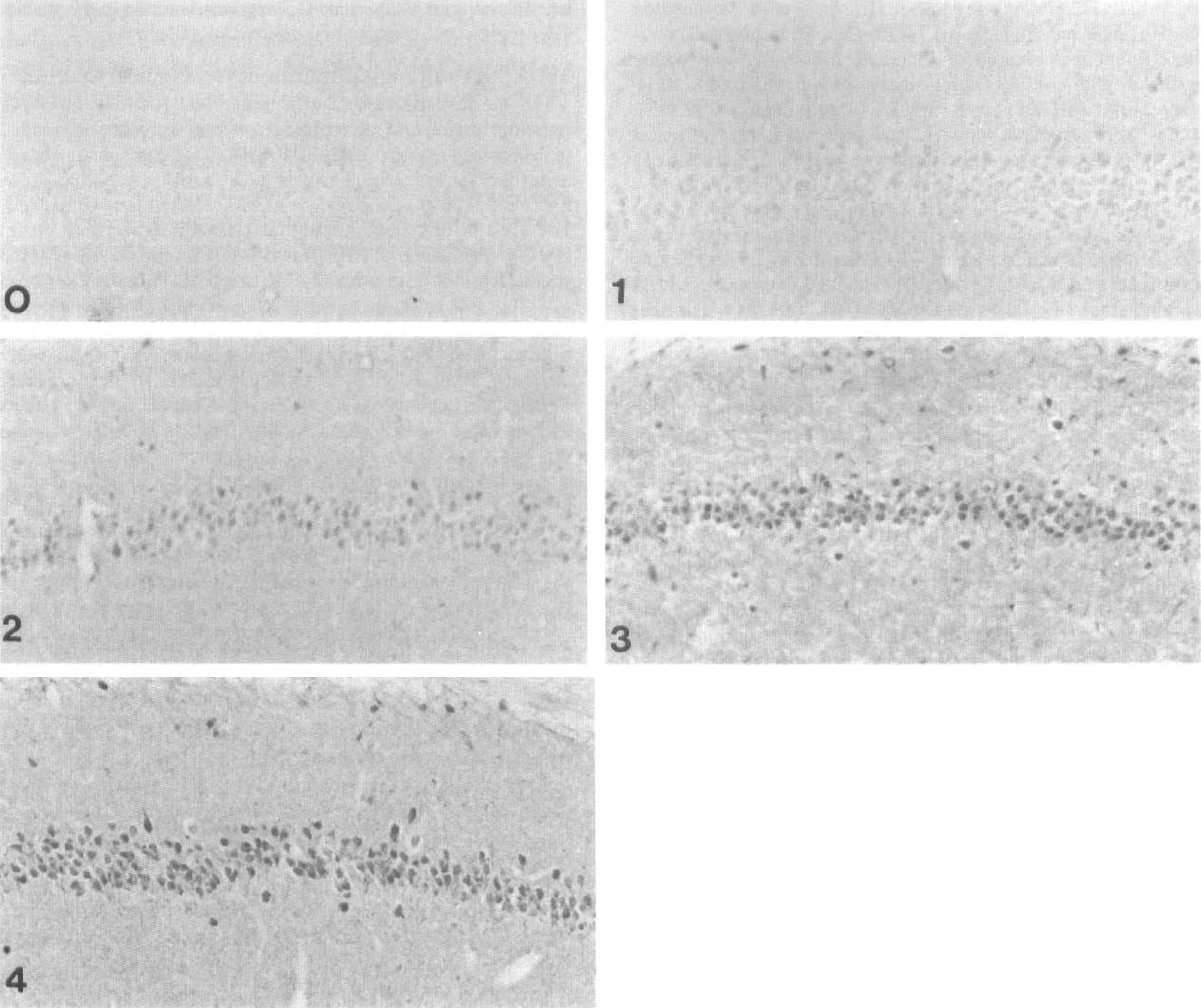
Photomicrographic examples of the semiquantitative grading scale of the CA1 region of the hippocampus for malondialdehyde immunostaining from 0 (no staining) to 4 (most intense staining).
Quantitative computer-assisted densitometric immunocytochemical analysis of APP, APO-E, b-AP, and astrocyte activation
Immunostaining intensity for APP, APO-E, b-AP, and astrocyte activation (GFAP) in the CA1 region of the hippocampus was measured using an Image One analysis system (Universal Imaging Corporation, Image One, West Chester, PA, U.S.A.) attached to an Olympus microscope. A 96-hour postischemic time was selected for these studies based on the prior demonstration that these parameters are significantly and maximally increased by that point (Hall et al., 1995b). The immunostained areas of the digitized CA1 region of the hippocampus were outlined. The adjacent nonimmunostained area representing the background was outlined separately. The optical density (OD) of each of these areas was calculated by subtracting the OD of the adjacent background from the OD for the CA1 area. For each animal, the background-corrected OD was taken for the right and left CA1 sector and the mean used as the value for that animal. The output of the video camera was determined to be linear over the range of the OD measured in this study. A normalizing function was applied to correct for variation in intensity of illumination between sections.
Statistical analysis
All data are expressed as mean ± SD. Differences in CA1 neuronal counts and each of the immunocytochemical end points between sham-operated (nonischemic) and postischemic animals were statistically evaluated using a one-way analysis of variance with a P<.05 required for significance.
RESULTS
Effects on 96-hour postischemic CA1 neuronal damage
Figure 3 shows that by 96 hours after ischemia, there is an 84% loss of medial CA1 neurons in the vehicle-treated animals (sham=82.6 ± 11.8, vehicle-treated ischemic = 13.2 ± 15.5 neurons/315 μm). This is consistent with previously published reports concerning the loss of neurons in the CA1 region after a 5-minute ischemic gerbil injury (Kirino, 1982; Hall et al., 1995a, b ). Both PNU-101033E and PNU-104067F provided significant protection against neuronal loss. The former attenuated the CA1 damage to 44% and the latter decreased it to 60%. Figure 4 is a cresyl violet–stained photomicrograph depicting the effects of PNU-104067F on CA1 neuronal cell loss 96 hours days after 5 minutes of forebrain ischemia.
Effects on 48-hour postischemic MDA immunoreactivity
At 48 hours after a 5-minute period of forebrain ischemia, the CA1 neuronal loss is approximately 50% (data not shown). Figure 5 shows that by 48 hours, the mean intensity score for lipid peroxidation-related MDA immunoreactivity in vehicle-treated animals was increased more than five-fold over sham-treated animals (P<.01 versus sham). PNU-104067F significantly decreased the postischemic immunostaining by 48% (P<.05 versus vehicle) roughly proportional to the degree of CA1 neuroprotection assessed at 96 hours.
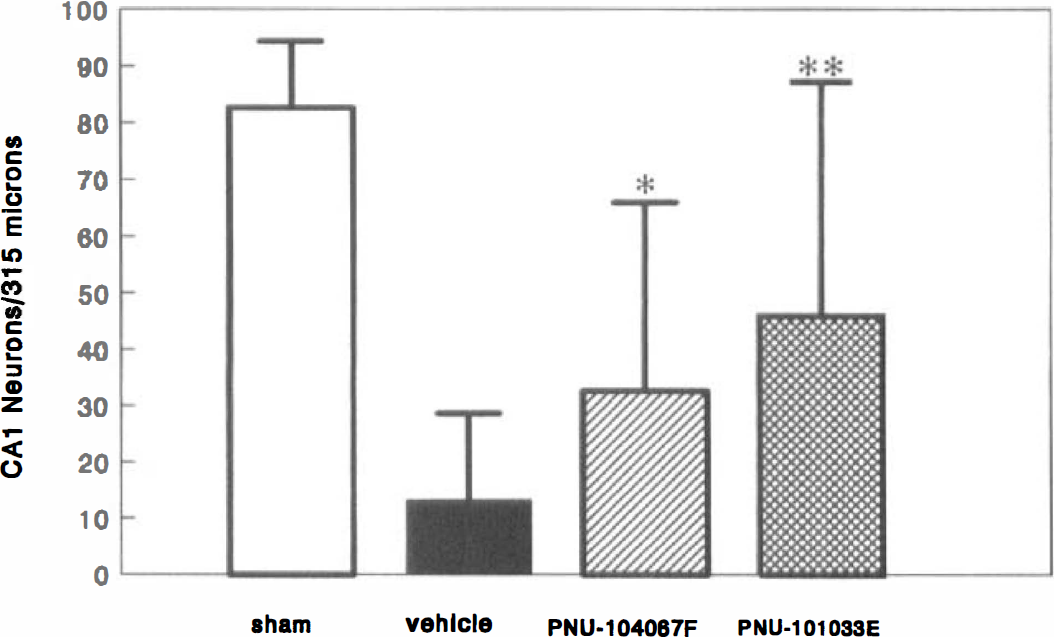
Effects of PNU-101033E and PNU-104067F on neuronal damage in the medial hippocampal CA1 region at 96 hours after a 5-minute period of forebrain ischemia in the gerbil. Values = mean ± SD for 9 to 20 animals.
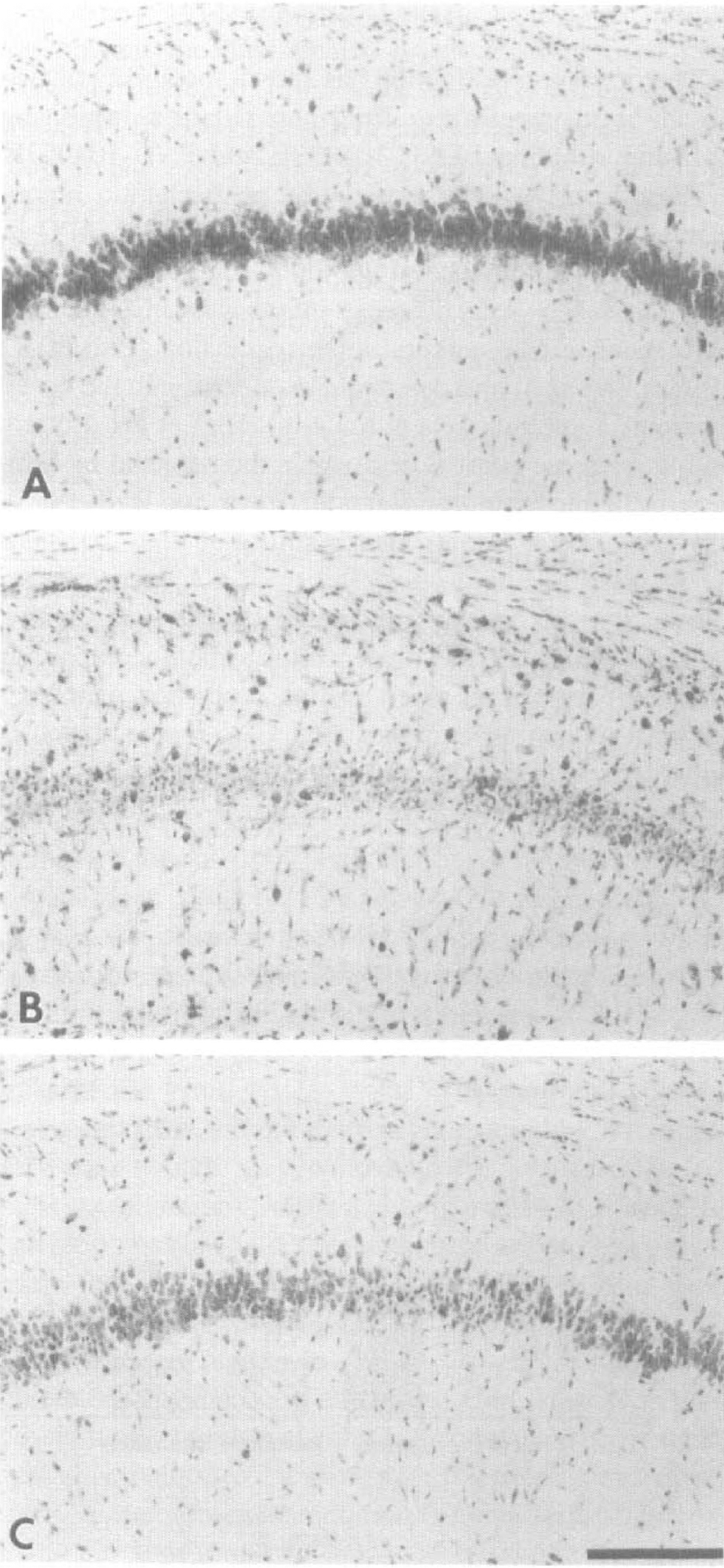
Photomicrographic examples of 96-hour postischemic necrosis in the medial hippocampal CA1 region in (A) sham-, (B) vehicle-, and (C) PNU-104067F-treated animals. Calibration bar = 250μm.
Effects on 48-hour postischemic microglial activation
Characteristic microglial activation (major histocompatibility complex II immunostaining) in the medial CA1 region of a sham and a vehicle-treated animal is shown in Fig. 6. No activated microglia were observed in animals 48 hours after a sham-BCO. In contrast, vehicle-treated animals had 94±21 activated microglia/mm2 throughout the medial CA1 region. Treatment with PNU-104067F significantly decreased this activation by 31%, as quantified in Fig. 7.
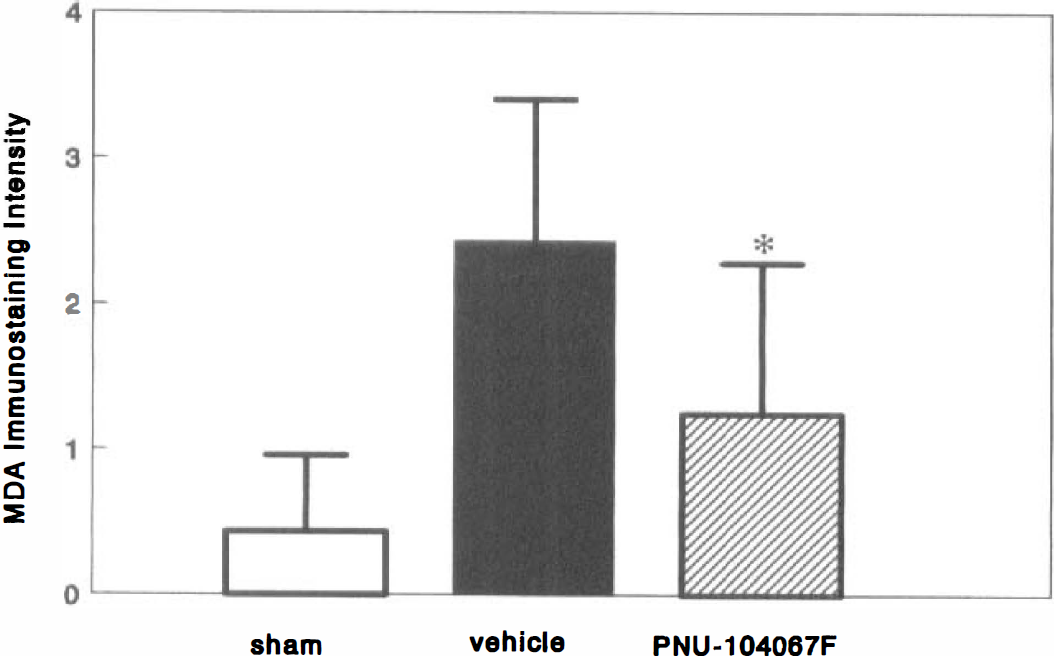
Effect of PNU-104067F (30 mg/kg orally) on the expression of malondialdehyde immunoreactivity at 48 hours after ischemia. Values = mean ± SD for 7 to 8 animals.
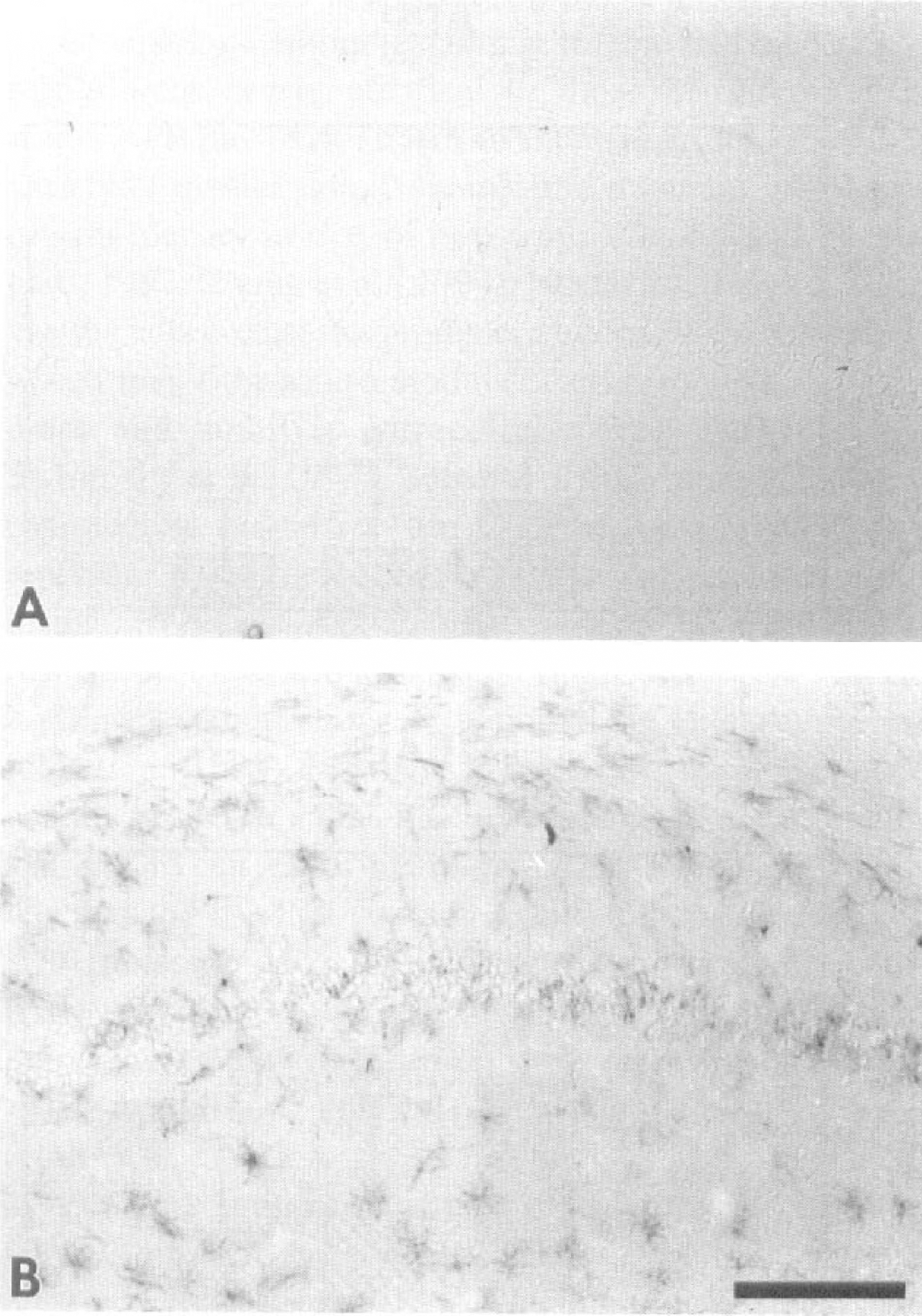
Photomicrographic example of microglial activation in the medial hippocampal CA1 region of gerbils 48 hrs after a 5-minute period of forebrain ischemia in (A) sham-operated and (B) vehicle-treated animals. Calibration bar = 500 μm.
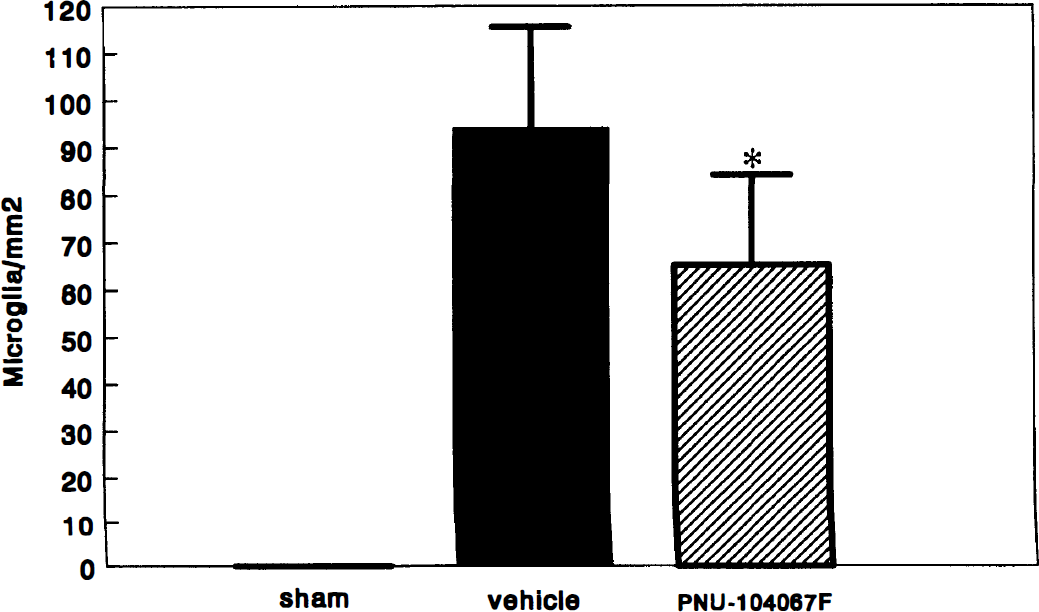
Effects of PNU-104067F (30 mg/kg orally) on microglia activation in the medial hippocampal CA1 region at 48 hours after ischemia. Values = mean ± SD for 7 to 8 animals.
Effects on postischemic APP, b-AP, APO-E, and GFAP immunoreactivity
In Fig. 8, the effects of PNU-101033E and PNU-104067F on the intensity of delayed (96-hour) postischemic CA1 APP, b-AP, APO-E, and GFAP immunoreactivity are presented. The immunostaining for all four parameters was significantly (P<.01 versus sham) increased in vehicle-treated gerbils. However, both PNU-101033E and PNU-104067F significantly suppressed the mean increase in APP, b-AP, APO-E, and GFAP immunostaining intensity.
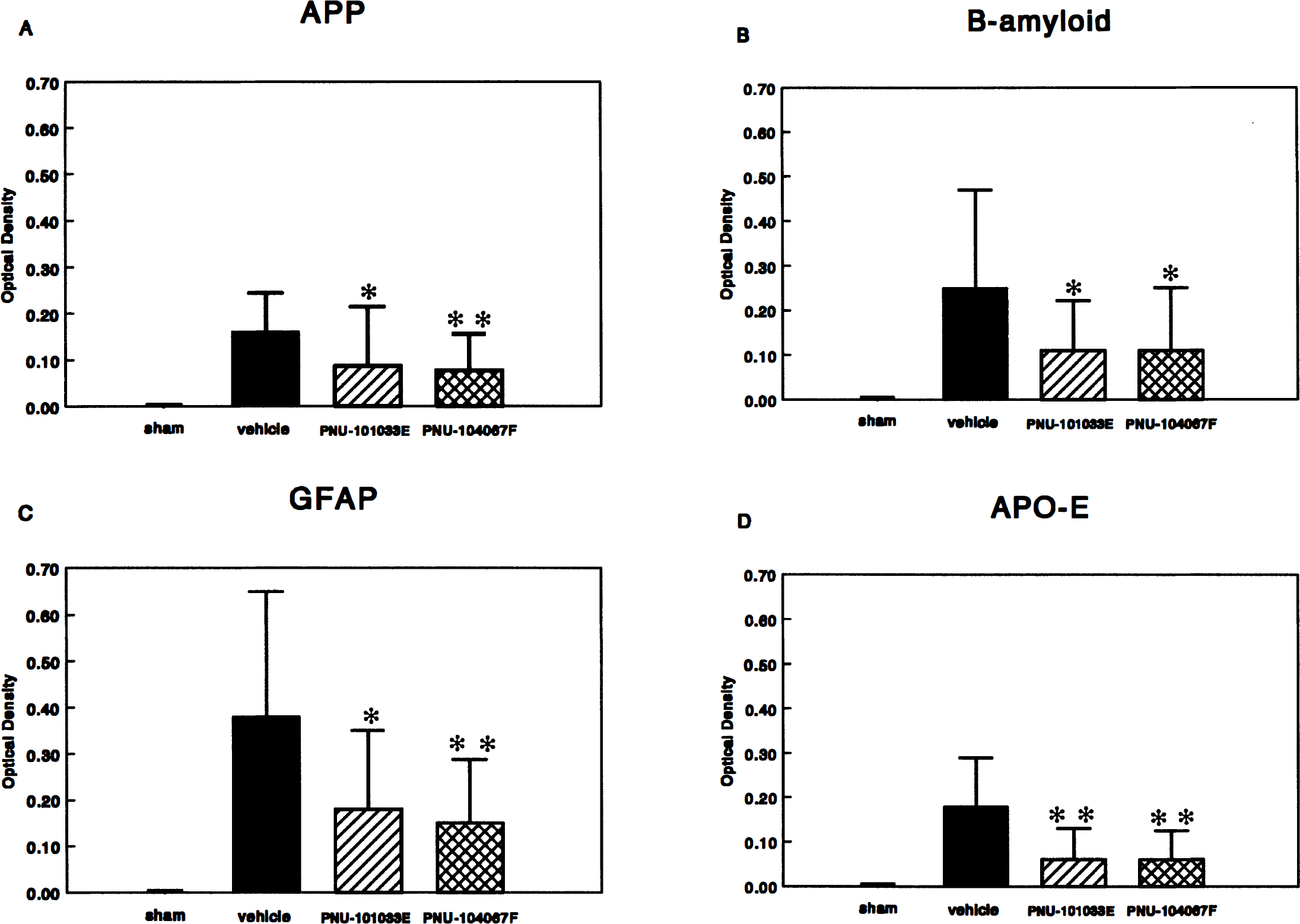
Effects of PNU-101033E and PNU-104067F on (A) APP, (B) b-amyloid, (C) APO-E, and (D) GFAP immunostaining in the medial hippocampal CA1 region at 96 hours after a 5-minute period of forebrain ischemia. Values = mean ± SD for 9 to 20 animals.
DISCUSSION
The present investigation has shown the ability of two novel brain-penetrating lipid-peroxidation–inhibiting pyrrolopyrimidines, PNU-101033E and PNU-104067F, to significantly attenuate delayed postischemic hippocampal CA1 neuronal loss as well as APP, b-AP, APO-E, and GFAP immunostaining. PNU-101033E has been previously shown to decrease infarct size in mice after permanent middle cerebral artery occlusion (Hall et al., 1995a), to lessen hippocampal CA1 necrosis in the gerbil 5-minute forebrain ischemia model (Hall et al., 1995a), and to decrease cortical necrosis in the neonatal rat hypoxic-ischemic brain injury model (Fleck and Hall, 1995). The present study confirms the ability of PNU-101033E to decrease selective vulnerability of CA1 neurons and shows that this property is shared by the close analog PNU-104067F. In the present study, both compounds provided significant, albeit partial, protection of the selectively vulnerable hippocampal CA1 region which is consistent with a role of oxygen radical-induced lipid peroxidation in postischemic neuronal damage (Floyd and Carney, 1991; Hall et al., 1993a; Hall, 1996). While the present mechanistically oriented study used preischemic treatment, prior work has shown that, at least for PNU-101033E, postischemic administration beginning as late as 4 hours after reperfusion still results in significant neuroprotection in the gerbil 5-minute forebrain ischemia model (Hall et al., 1995a).
In the forebrain ischemia model, there are increases in oxygen radical formation and membrane lipid peroxidation, as well as disturbances in membrane function within the first minutes after reperfusion which precede delayed CA1 neuronal death (Floyd and Carney, 1991; Hall et al., 1993a; Kirino et al., 1993). The increased level of lipid peroxidation persists for several days after brief forebrain ischemia (Floyd and Carney, 1991). The present results show immunocytochemical evidence of CA1 neuronal lipid peroxidative injury by 48 hours after reperfusion as judged by the significant increase in MDA-related immunostaining. The immunostaining is increased significantly when only 50% of the CA1 neurons are lost. Because this increase occurs during the active period of degeneration, it further indicates a participatory role of lipid peroxidation in the postischemic degenerative process. PNU-104067F significantly decreased the postischemic MDA immunostaining, which is consistent with an antioxidant neuroprotective mechanism of action of the pyrrolopyrimidines.
There are several potential sources of oxygen radicals in ischemic brain (Hall, 1996). The neuronal changes that result from ischemia are accompanied by microglial activation which may contribute to postischemic free radical production (Colton and Gilbert, 1987). Microglial activation has been documented previously to be an early event in the CA1 region of rats subjected to brief forebrain ischemia (Morioka et al., 1991; Gehrmann et al., 1992), and microglia are perhaps the initial responding glial element after neuronal injury. Consistent with their possible role in postischemic CA1 damage is their widespread activation coincident with the time course of active CA1 neuronal degeneration. The present study confirms the appearance of activated microglia throughout the hippocampus 48 hours after the injury which corresponds to the midpoint of neuronal cell loss. PNU-104067F was shown to suppress microglial activation, possibly contributing to the neuroprotection observed. Moreover, the fact that a lipid peroxidation inhibitor can suppress microglial activation suggests that lipid peroxidation products may play a role in triggering microglial activation although this possibility requires further study.
In addition to the potential significance of the present study to selective neuronal vulnerability in the context of brief global ischemia, the current results may also have relevance to AD. First of all, within the hippocampus, selective CA1 degeneration, as observed after brief forebrain ischemia, is also seen in AD brains (West et al., 1994). Thus, CA1 protective efficacy in the gerbil model may be relevant to the possibility of CA1 neuronal preservation in AD. Second, oxygen radicals and lipid peroxidation have recently been suggested to play a role in the development of AD. Support for this view includes the findings that (1) brain reactive oxygen levels increase with age (Blass, 1993); (2) antioxidant enzymes such as superoxide dismutase are altered in AD brains (Pappolla et al., 1992); (3) there is an increase in lipid peroxidation products in the brains of AD patients (Subbarao et al., 1990; Götz et al., 1992); (4) the AD brain is more susceptible to lipid peroxidative processes (Subbarao et al., 1990; Götz et al., 1992); and (5) there is an increased level of total iron in various regions of AD brains which can drive lipid peroxidation reactions (Richardson, 1993).
Another feature of the gerbil forebrain ischemia paradigm and the present results which may be of relevance to AD concerns the demonstrated coincident increases in immunostaining for APP, b-AP, and APO-E in the selectively vulnerable CA1 region of the hippocampus. Our previous studies have shown an increase in APP, b-AP, and APO-E immunoreactivity which is delayed until 72 hours after ischemia by which time CA1 pyramidal neuronal degeneration is complete (Hall et al., 1995b). The timecourses of immunostaining in the current experiments were nearly identical to those in our previous work. APP has been shown to increase in response to a variety of insults, both ischemic and traumatic. APP processing occurs in one of two ways (Gandy and Greengard, 1992; Zhong et al., 1994). Normally, it is processed via the a-secretase to produce a secreted form of APP which may function to modulate cell responses to glutamate and stabilize intracellular free Ca2+ (Smith-Swintosky et al., 1994). Because of these potentially neuroprotective properties, the secreted form of APP may represent a trophic response after a variety of cerebral insults. However, APP can alternatively be processed to form neurotoxic amyloidogenic fragments, most notable b-AP, under conditions of neuronal degeneration (Iverfeldt et al., 1993). However, it must be cautioned that we cannot be certain that the increases in b-AP immunostaining in our experiments represent free b peptide or whether the b-AP is contained within the context of the APP molecule. Most likely some of the increase in b-AP observed from 3 to 7 days after brief forebrain ischemia represents free b-AP based on some reported differences in the overall time courses of APP versus b-AP increase (Hall et al., 1995b).
Astrocyte proliferation, as evidenced by increases in GFAP, has also been documented in the hippocampus after ischemia (Petito et al., 1990; Schmidt-Kastner et al., 1990, Hall et al., 1995b), as confirmed in the present experiments. The coincidence of astrocytic activation and the APO-E increase suggest that astrocytes are the main source of APO-E (Boyles et al., 1985; Pitas et al., 1987). Astrocytes are the main source of APO-E in the brain, and studies have shown a relationship between increased astrocytic activation and the levels of hippocampal APO-E mRNA after injury-induced degeneration (Ignatius et al., 1986; Poirier et al., 1991; Poirier et al., 1993; Ali et al., 1996). It has been suggested that APO-E may play a role in the post-traumatic neuronal repair by increasing lipid transport. Thus, the postischemic expression of APO-E, similar to that of APP discussed earlier, could represent a repair or a trophic response. Alternatively, the increased production of certain APO-E isoforms coincident with increased b-AP could conceivably be detrimental (Strittmatter et al., 1993a; 1993b; 1994). This latter possibility seems more relevant to the current results because the increase in APO-E does not occur until after CA1 degeneration is complete.
In summary, the pyrrolopyrimidine lipid peroxidation inhibitors PNU-101033E and PNU-104067F significantly decreased both microglial activation and lipid peroxidation–related immunostaining in the hippocampal CA1 region 48 hours before completion of CA1 neuronal degeneration. At 96 hours, a decrease in CA1 neuronal loss was effected by both compounds. Also at 96 hours after the ischemic insult, both PNU-101033E and PNU-104067F significantly attenuated APP, b-AP, APO-E, and GFAP immunostaining which normally increases secondarily to the degeneration of the hippocampal CA1 neurons. The results suggest that a decrease of oxidative damage and subsequent neuronal preservation may proportionately decrease the potentially deleterious glial expression of APP, b-AP, and APO-E. Such an action may not only have an impact on delayed postischemic functional recovery but also help to retard the amyloidogenic progression of AD.
Footnotes
Acknowledgment
The authors thank Dr. Craig Thomas from Hoechst-Marion-Roussel for the supply of polyclonal antibody to malondialdehyde-modified proteins.