
Abstract
Intracerebral hemorrhage (ICH) is a devastating clinical event without effective therapies. Increasing evidence suggests that inflammatory mechanisms are involved in the progression of ICH-induced brain injury. Inflammation is mediated by cellular components, such as leukocytes and microglia, and molecular components, including prostaglandins, chemokines, cytokines, extracellular proteases, and reactive oxygen species. Better understanding of the role of the ICH-induced inflammatory response and its potential for modulation might have profound implications for patient treatment. In this review, a summary of the available literature on the inflammatory responses after ICH is presented along with discussion of some of the emerging opportunities for potential therapeutic strategies. In the near future, additional strategies that target inflammation could offer exciting new promise in the therapeutic approach to ICH.
Introduction
Stroke is a major cause of morbidity and mortality and is the second leading cause of death worldwide (Ingall, 2004). Strokes that are caused by the breakage of a blood vessel in the brain are called hemorrhagic strokes. Spontaneous intracerebral hemorrhage (ICH) accounts for 15% to 20% of all strokes and affects approximately 120,000 people in the United States each year (Broderick et al, 1999; Ribo and Grotta, 2006). Moreover, the incidence of ICH is expected to grow, given the aging of the population. Depending on the underlying cause of bleeding, ICH is classified as either primary (unrelated to congenital or acquired lesions) or secondary (directly related to congenital or acquired lesions). Primary ICH accounts for 78% to 88% of cases and originates from the spontaneous rupture of small arteries or arterioles damaged by two major causes: hypertensive arteriolosclerosis and amyloid angiopathy (reviewed in Mayer and Rincon, 2005; Qureshi et al, 2001b; Sutherland and Auer, 2006). Secondary ICH occurs in a minority of patients in association with coagulopathy, brain tumors, aneurysms, vascular anomalies, and thrombolytic treatment of ischemic stroke (Mayer and Rincon, 2005; Qureshi et al, 2001b; Sutherland and Auer, 2006; Wang and Tsirka, 2005a). In all cases, the extravasated blood accumulates and compresses the surrounding brain tissue. Treatment for ICH is primarily supportive, and the clinical outcome is poor with potential extensive burden for the caretakers. To improve the clinical outcome of ICH, better understanding of the pathogenesis of ICH-induced brain injury is needed. Increasing evidence suggests that inflammatory mechanisms are involved in ICH-induced brain injury (Aronowski and Hall, 2005; Wang and Tsirka, 2005a; Xi et al, 2006). This review will summarize the available literature on the inflammatory responses after ICH and discuss the emerging opportunities for novel therapeutic strategies. The focus will be on cellular (leukocytes and microglia) and molecular (cytokines, proteases, and reactive oxygen species (ROS)) components of inflammation after ICH.
Animal Models of Intracerebral Hemorrhage
Current insight into the events that occur in the cerebral microvasculature after ICH mostly stems from experimental animal models of ICH. There are two main models available to mimic spontaneous ICH. One is an autologous whole-blood model, and the other uses collagenase to induce intracerebral bleeding; both require stereotactic injections. The autologous whole-blood model is one of the earliest models used for the study of ICH in rats (Bullock et al, 1984; Nath et al, 1986; Wu et al, 2003), rabbits (Kaufman et al, 1985; Koeppen et al, 2004), dogs (Qureshi et al, 2001a; Steiner et al, 1975), and pigs (Thiex et al, 2003; Wagner et al, 1996), and has recently been adapted for use in mice (Lee et al, 2006; Nakamura et al, 2004b). It consists of whole blood drawn from the animal being injected directly into the cerebrum. In the second model, collagenase, a proteolytic enzyme that dissolves the extracellular matrix around capillaries, is injected into the cerebrum, opening the blood—brain barrier and causing active intraparenchymal bleeding (Choudhri et al, 1997; Clark et al, 1998; Rosenberg et al, 1990; Tang et al, 2004; Wang et al, 2003). One feature of this model is that the hemorrhage is spontaneous in nature (Wang and Tsirka, 2005c), an attribute that allows investigation of the initial bleeding response to collagenase injection. The current animal models of ICH have been reviewed recently (Andaluz et al, 2002).
Inflammation after Intracerebral Hemorrhage
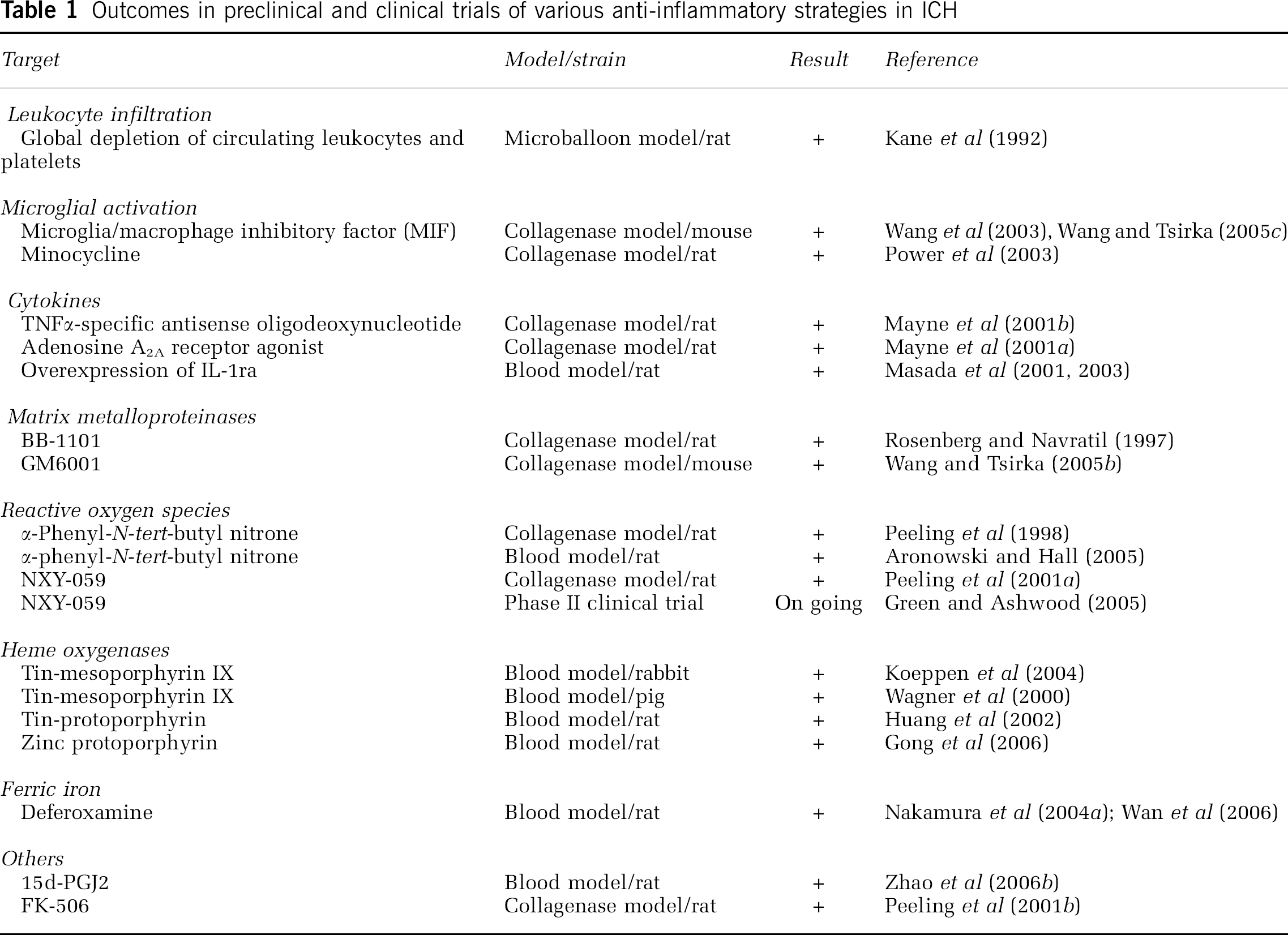
In ICH, brain tissue injury occurs after an inflammatory reaction, a common response of the cerebral parenchyma to various forms of insult. The inflammatory cascade comprises both cellular and molecular components. When ICH occurs, blood components, including erythrocytes, leukocytes, macrophages, and plasma proteins (thrombin, plasmin, etc.) immediately enter the brain. An inflammatory response follows that involves enzyme activation, mediator release, inflammatory cell migration, glial activation, brain tissue breakdown, and repair (Wang et al, 2003; Wang and Tsirka, 2005a). Understanding of the inflammation that occurs after ICH has become available from recent animal and clinical studies. Clinical studies of inflammation in ICH are usually limited to blood or cerebrospinal fluid sampling after the ictus. Relatively little histopathologic data exist concerning ICH in human post-mortem specimens. Anti-inflammatory strategies have been tested in preclinical and clinical trials (Table 1) and are discussed below.
Outcomes in preclinical and clinical trials of various anti-inflammatory strategies in ICH
Cellular Components of Inflammation
The major inflammatory cells that are activated and accumulate within the brain after ICH are blood-derived leukocytes, macrophages, and resident microglia. Leukocytes clearly perform important roles in normal host defense. Mounting evidence suggests that neutrophils in particular might be mediators of secondary brain damage after ICH. Microglia, which constitute as many as 12% of the cells in the central nervous system (Gonzalez-Scarano and Baltuch, 1999), are the first non-neuronal cells to respond to central nervous system injury. When fully activated by either neuronal cell death or other processes, they become phagocytic. Infiltrating leukocytes, macrophages, and activated glial cells are the major central nervous system sources of cytokines, chemokines, and other immunomolecules (Aronowski and Hall, 2005; Barone and Feuerstein, 1999; Emsley and Tyrrell, 2002).
Leukocytes
Animal models of ICH provide substantial evidence for the presence of white blood cell infiltration into the hematoma (Del Bigio et al, 1996; Gong et al, 2000; Mayne et al, 2001a; Peeling et al, 2001b; Wang and Tsirka, 2005b; Xue and Del Bigio, 2000a, b ; Zhao et al, 2006b). In a rat autologous blood model, neutrophil infiltration was found in and around the hematoma, which begins in less than 1 day, peaks at 2 to 3 days, and almost disappears at 3 to 7 days (Gong et al, 2000; Xue and Del Bigio, 2000a, b ). The neutrophils themselves are expected to die by apoptosis within 2 days of entering the hematoma (Savill and Haslett, 2001). An influx of neutrophils was found in a collagenase- or collagenase/heparin-induced model of ICH in rat (Del Bigio et al, 1996; Mayne et al, 2001a; Peeling et al, 2001b). Moreover, neutrophil infiltration has been shown to have similar temporal patterns in both rat ICH models (Peeling et al, 2001b; Xue and Del Bigio, 2000a). In a collagenase-induced ICH model in mice, we observed that infiltrating neutrophils were present in and around the hematoma as early as 4 h after collagenase injection and that the level peaked at 3 days (Wang and Tsirka, 2005b) (Figure 1A). Using the same model of ICH in rat, Mayne et al (2001b) found that, at 4 h after ICH, cells resembling leukocytes and bordering the hematoma were positive for tumor necrosis factor α (TNFα). It has also been shown that infiltrating neutrophils can damage brain tissue directly by generating ROS and secreting pro-inflammatory proteases (Weiss, 1989). In addition, the contents of dying leukocytes can promote inflammatory tissue injury indirectly by stimulating macrophages to release pro-inflammatory mediators (Stern et al, 1996). Taken together, these findings indicate that infiltrating neutrophils may play a role in ICH-induced early brain injury.
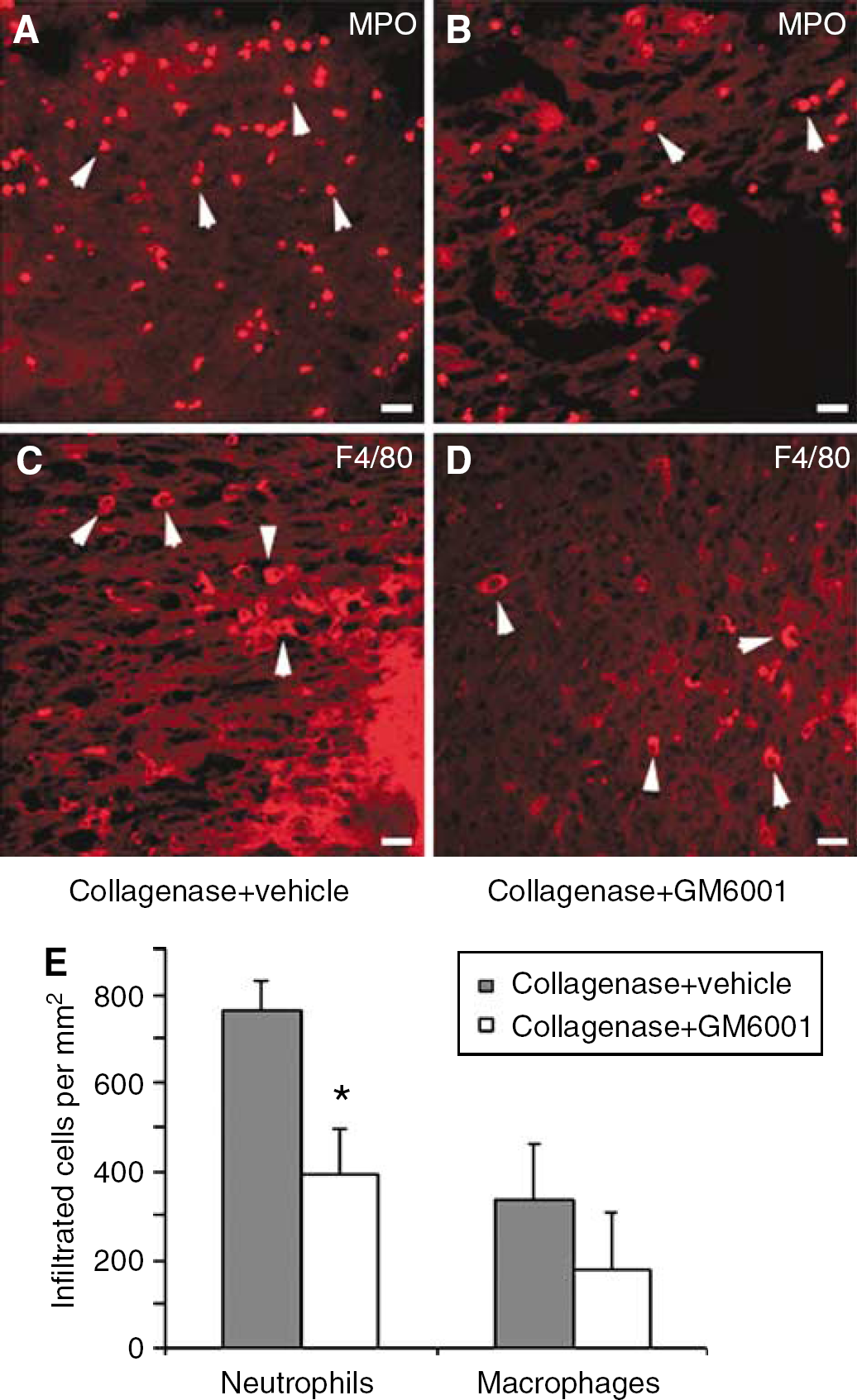
GM6001 decreased leukocyte recruitment after ICH. On day 3 after ICH induction in mice, infiltrated neutrophils (arrowheads, MPO+,
Clinical studies have provided evidence that supports the role of leukocytes in ICH. Early studies by Molle (1942) and Lee et al (1975) showed that leukocyte counts in cerebrospinal fluid, especially the polymorphonuclear neutrophilic leukocyte counts, were frequently elevated after ICH. Infants with intraventricular hemorrhage also have greater total numbers of leukocytes than infants without intraventricular hemorrhage (Paul et al, 2000). In patients with ICH, peripheral leukocyte counts were higher than normal and correlated with hematoma size (Bestue-Cardiel et al, 1999; Suzuki et al, 1995). High leukocyte count has been reported to be one of the independent predictors of early neurologic deterioration in primary ICH (Leira et al, 2004; Silva et al, 2005). An early histopathologic study by Wisniewski (1961) showed infiltration of polymorphonuclear neutrophilic leukocyte into the hemorrhagic region 2 to 4 days after ICH. This infiltration was preceded by polymorphonuclear neutrophilic leukocyte accumulation in blood vessels that bordered the hemorrhage as early as 6 to 12 h after ICH. The presence of neutrophils and/or macrophages in patients with ICH was further described by Garcia et al (1994). MacKenzie and Clayton (1999) also studied the early cellular events in the perihemorrhage region in 33 fatal cases of spontaneous ICH, and found that leukocyte infiltration appeared as early as 5 to 8 h and disappeared by 72 h after ICH. Thus, human and animal model data support a role for leukocytes in ICH pathophysiology, although the exact mechanisms remain the subject of much ongoing work.
Antileukocyte Strategies
Although antileukocyte strategies have been protective in various experimental ischemia models (Bowes et al, 1995; Hartl et al, 1996; Jiang et al, 1995; Matsuo et al, 1994), the data for such strategies in ICH are extremely limited. Mendelow's group (Kane et al, 1992) reported that in an ICH rat model produced by inflation of a microballoon specifically to mimic the mass effect of ICH, global depletion of circulating leukocytes and platelets by whole-body irradiation was protective against both ischemia and edema formation. Zhao et al (2006b) found more recently that intrastriatal injection of 15d-Prostaglandin J2 at the onset of ICH reduced neurobehavioral deficits and neuronal damage. These beneficial effects were associated with activation of peroxisome proliferator-activated receptor-gamma, elevation of catalase expression, suppression of nuclear factor-kappaB activity, and restricted neutrophil infiltration. We and others have reported that inhibition of matrix metalloproteinases (MMPs) provided neuroprotection against ICH-induced early brain injury (Power et al, 2003; Rosenberg and Navratil, 1997; Wang and Tsirka, 2005b). This beneficial effect coincided with amelioration of neutrophil infiltration (Figures 1A, 1B, and 1E) and production of oxidative stress (Wang and Tsirka, 2005b), further supporting the role of leukocytes in ICH-induced early brain injury. Therefore, antileukocyte interventions merit further evaluation as an alternative or adjunctive therapeutic approach to ICH.
Microglia
Most of the data pertaining to microglia in ICH derive from animal, rather than human studies. Under normal conditions, ‘resting,’ or ramified, microglia constitute 5% to 20% of the total central nervous system glial population (Lawson et al, 1992). After brain injury, the microglia become activated, a state that can be identified by changes in morphology. Such changes include enlarged size with stout processes, upregulation of specific genes or proteins, a migratory and proliferative response, and phagocytic behavior (reviewed in Wang and Tsirka, 2005a).
Although the primary role for microglial activation after ICH is to clear the hematoma, these activated microglia also express and release a variety of cytokines (Gregersen et al, 2000; Hanisch, 2002; Stoll et al, 2004), ROS (Banno et al, 2005; Min et al, 2006; Wang and Tsirka, 2005c), nitric oxide (Khan et al, 2005; Mander et al, 2005), and other potentially toxic factors, suggesting that activated microglia/macrophages might contribute to ICH-induced early brain injury (Power et al, 2003; Wang et al, 2003). In addition, a greater degree of microglial activation has been found in aged rats after ICH than in young rats, suggesting that activated microglia might be a contributing component to enhanced brain injury in aged rats (Gong et al, 2004). More recently, studies by Yang et al (2006a, b ) have shown that complement activation may affect inflammatory responses, including microglial activation and neutrophil infiltration, thereby contributing to ICH-induced brain injury.
The relevance of activated microglia to the onset of ICH-induced early brain injury has been suggested previously (Aronowski and Hall, 2005; Hickenbottom et al, 1999; Keep et al, 2005; Wang et al, 2003). It has been shown that systemic administration of the tirilazad-like antioxidant U101033E to counteract the generation of detrimental ROS by microglia attenuates the production of stress response proteins, including heat shock proteins 32, 47, and 70, in experimental subarachnoid hemorrhage in rats (Turner et al, 1999). Interestingly, downregulation of the expression of TNFα (a proinflammatory mediator) produced by microglia appears to reduce the hematoma volume and improve the neurobehavioral score of the animals after ICH (Mayne et al, 2001a, b ).
Investigations have been undertaken to determine the time course of hematoma clearance after ICH. In a mouse collagenase-induced ICH model, hematoma volumes remained unchanged for the first 3 days, and the majority of the hematoma was removed by day 7 (Wang et al, 2003; Wang and Tsirka, 2005c). In a porcine ICH model, the hematoma volumes remained unchanged for the first 7 days but were nearly absent by day 14 (Wagner and Broderick, 2001). Considering these findings, we suggest that the role of microglial activation after ICH might be time dependent, although Keep et al (2005) proposed that the role of microglial activation after ICH might depend on hematoma size. In a recent study of microglial activation in a mouse ICH model, we found that microglial activation appeared in the peri-ICH region 1 to 2 h after collagenase injection (unpublished data), was prominent at day 1, peaked at day 7, and returned to normal by 21 days (Wang et al, 2003; Wang and Tsirka, 2005c). In an autologous whole-blood ICH model in rat, microglial activation appeared in the peri-ICH region as early as 1 to 4 h after the insult (Hickenbottom et al, 1999; Xue and Del Bigio, 2000a), reached a maximum at 3 to 7 days, and persisted for 4 weeks (Gong et al, 2000; Hickenbottom et al, 1999; Xue and Del Bigio, 2000a). These combined findings indicate that microglial activation occurs very early after the onset of ICH and persists for 3 to 4 weeks. Therefore, the time cutoff for microglial activation between harm and protection should be clarified in ICH.
Antimicroglial Activation Strategies
Given the proposed detrimental effect of microglial activation in ICH-induced early brain injury, it is important to clarify the therapeutic potential of treatments based on the inhibition of microglial activation shortly after the onset of ICH. In a series of experiments, we infused mice centrally with MIF (macrophage/microglia inhibitory factor, tuftsin fragment 1 to 3, Thr-Lys-Pro) to inhibit microglial activation either 2 days before or 2 h after the onset of collagenase-induced ICH (Wang et al, 2003; Wang and Tsirka, 2005c). The results revealed reduced injury size and edema, decreased neuronal degeneration, and improved neurologic functions (Figure 2). These findings further support the possibility that microglial activation promotes inflammatory reactions after ICH, an effect that can cause undesirable early brain injury.
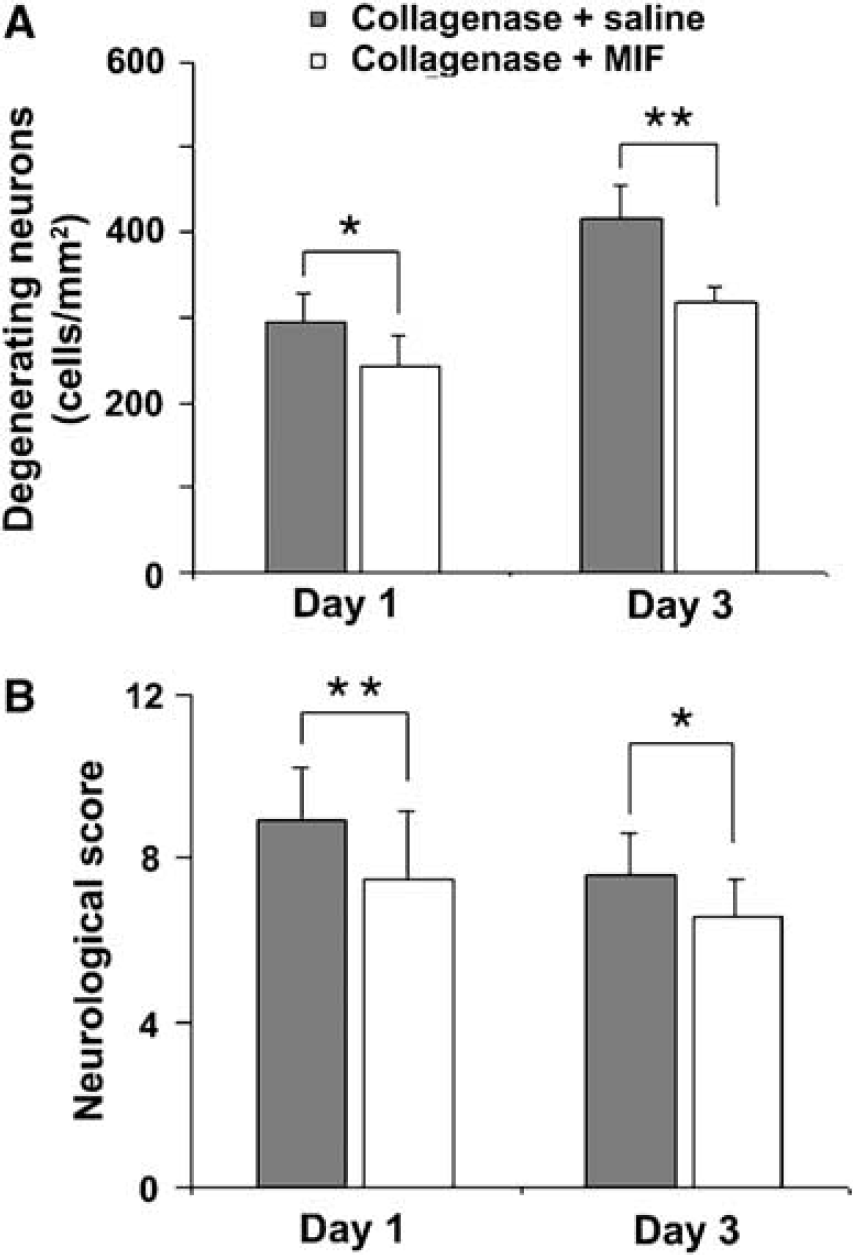
Macrophage/microglia inhibitory factor (MIF) attenuated the injury and improved the outcome of ICH. (
Other examples of treatments that target microglial activation are the tetracycline derivatives minocycline and doxycycline, broad-spectrum antibiotics that have anti-inflammatory effects independent of their antimicrobial activity (Van Den Bosch et al, 2002; Wu et al, 2002a; Yrjanheikki et al, 1998). Evidence suggests that tetracycline derivatives provide neuroprotection against global brain ischemia (Yrjanheikki et al, 1998), amyotrophic lateral sclerosis (Van Den Bosch et al, 2002), and Parkinson disease (Wu et al, 2002a), and that this neuroprotection involves the inhibition of microglial activation. Recently, a study by Power et al (2003) demonstrated that, in a rat model of ICH, 7-day treatment with minocycline initiated 1 h after collagenase injection protected the morphology of neurons and improved functional recovery. These beneficial effects coincided with amelioration of microglial activation and downregulation of MMP-12 expression (Power et al, 2003), although other mechanisms, such as inhibition of cytochrome c release (Zhu et al, 2002), could also underlie the benefits. Because of its safety record and ability to penetrate the blood—brain barrier, minocycline might be considered for human clinical trials to protect the brain against ICH-induced early brain injury. However, long-term inhibition of microglial activation and macrophage infiltration may be unwarranted because of the potential to eliminate neuroprotective benefits of microglia/macrophages as phagocytes and suppliers of neuroprotective molecules (Elkabes et al, 1996; Nakajima et al, 2001; Rapalino et al, 1998; Wang and Tsirka, 2005c).
Microglia—Astrocyte Interactions
Astrocytes, the predominant glial cell type found in the central nervous system, are known to carry out critical functions (maintenance of ionic hemostasis, metabolism of toxins, regulation of scar tissue, prevention of neovascularization, and support of synaptogenesis and neurogenesis) that are vital for normal brain functions and the outcome of stroke injury (Panickar and Norenberg, 2005). It has been shown that astrocytes have stronger antioxidative potential than neurons (Lucius and Sievers, 1996). During brain injury, for example, astrocytes can directly modulate neuronal survival by producing angiogenic and neurotrophic factors (Dhandapani et al, 2003; Swanson et al, 2004). Also they can regulate expression of the NMDA receptor subunit (Daniels and Brown, 2001) and the glutamate transporter excitatory amino acid carrier (Canolle et al, 2004), which influences neuronal sensitivity to glutamate toxicity. Thus, astrocytes likely influence neuronal survival in the post-ICH period because in the presence of astrocytes, neurons become resistant to oxidative stress (Swanson et al, 2004; Tanaka et al, 1999). In addition, astrocytes can indirectly affect neuronal injury by modulating brain inflammation via decreases in the expression of microglial inflammatory mediators (Aloisi et al, 1997; Pyo et al, 2003). New findings show that astrocytes could cooperate with microglia to prevent inflammatory responses in the brain by regulating microglial ROS production (Min et al, 2006). Therefore, modulating microglial activation through astrocytes could be a novel method to minimize the brain injury caused by ICH-induced inflammation.
Molecular Components of Inflammation
Inflammatory Gene Expression
Previous DNA microarray analysis indicated that after ICH, numerous pro-inflammatory genes are upregulated, including transcription factors, heat shock proteins, cytokines, chemokines, extracellular proteases, and adhesion molecules (Lu et al, 2006). Many such genes, including TNFα, IL-1β, nitric oxide synthase, and intercellular adhesion molecule-1, are regulated in vitro by nuclear factor-kappaB (Barnes and Karin, 1997; Emsley and Tyrrell, 2002). Nuclear factor-kappaB also is induced in blood vessels or infiltrating white blood cells of human peri-ICH cerebral cortex, which is collected during hematoma evacuation (Aronowski and Hall, 2005). The experimental work addressing nuclear factor-kappaB and inflammation after ICH has been reviewed recently (Aronowski and Hall, 2005). In the following section, the focus will be primarily on cytokines, proteases, and ROS after ICH.
Cytokines
Cytokines are mostly glycoproteins that play crucial roles in cell-to-cell signaling and are generally associated with inflammation, immune activation, and cell differentiation or death (Turrin and Plata-Salaman, 2000). Although cytokines can be released by many cell types, including microglia, astrocytes, neurons, and endothelial cells, their principal sources in the brain are activated microglia/macrophages (Emsley and Tyrrell, 2002). Nevertheless, evidence also supports the involvement of peripherally derived cytokines in brain inflammation. After ICH in humans, the blood—brain barrier permeability increases (Lampl et al, 2005). Therefore, peripherally derived mononuclear phagocytes, T-lymphocytes, natural killer cells, and polymorphonuclear neutrophilic leukocytes, which produce and secrete cytokines, can all cross the blood—brain barrier and contribute to brain inflammation (Barone and Feuerstein, 1999).
Cytokines are usually divided into pro- and anti-inflammatory mediators, based on their ability to promote or suppress immune activation. Evidence for the contribution of pro-inflammatory cytokines to brain inflammation associated with ICH is derived from animal and clinical studies. It has been demonstrated that pro-inflammatory cytokines induce or potentiate the production of other cytokines; in fact, cytokines can induce each other mutually (e.g., TNFα and IL-1β) and activate a positive feedback of cellular activation (Turrin and Plata-Salaman, 2000). The balance between pro- and anti-inflammatory mediators is probably a crucial determinant of neuronal injury and neurologic outcome after ICH.
Tumor necrosis factor α and IL-1β have been shown repeatedly to be elevated in various experimental models of brain injury. Tumor necrosis factor α is a pleotropic cytokine released in response to diverse stimulation by a number of brain cells, such as neurons, astrocytes, activated microglia/macrophages, and leukocytes (Munoz-Fernandez and Fresno, 1998). It exerts a multiple array of biologic activities, including stimulation of acute phase protein secretion and vascular permeability (Burger and Dayer, 2002). Interleukin-1 exists in two forms (IL-1α and IL-1β) that act mainly through two receptor types (types 1 and 2), which are regulated by the endogenous IL-1 receptor antagonist (IL-1ra). Just as in ischemic stroke (Allan et al, 2005; Barnes and Karin, 1997; Emsley and Tyrrell, 2002), the classic, pro-inflammatory cytokines TNFα and IL-1β have been proposed to exacerbate ICH-induced brain injury.
Many investigators have studied the effect of ICH on changes in cytokine expression. In a rat model of ICH induced by double autologous intrastriatal blood injection, a dramatic and long-lasting 8- to 40-fold increase in IL-1β was observed at 3 and 24 h after the injection (Aronowski and Hall, 2005). After intrastriatal injection of collagenase/heparin into rats, expression of TNFα was elevated in neutrophils at 4 h and in microglia/macrophages at 8 h (Mayne et al, 2001b). In another study that used rats, a single intrastriatal injection of blood increased TNFα after only 2 h (Xi et al, 2001). Such increases in TNFα might contribute to post-ICH brain edema formation (Hua et al, 2006). More recently, Lu et al (2006), using Affymetrix microarrays in the same ICH model, found that IL-1β, macrophage inflammatory protein-1α, and TNF receptor expression were upregulated at 24 h after the event. Similarly, in a porcine model of white matter ICH, induced by injection of autologous whole blood, IL-1β gene expression was increased after 16 h (Wagner et al, 2006). However, in a study that induced ICH in dogs by basal ganglia injection of autologous blood, no changes in TNFα, IL-1β, or other proinflammatory cytokines, such as IL-6, were observed in blood, cerebrospinal fluid, or brain extracts at 1 h after the ictus (Qureshi et al, 2001a). Therefore, the time course of cytokine expression may vary depending on the species used in the ICH model (Aronowski and Hall, 2005).
To date, only a few groups have published measurements of cytokines in patients with ICH. In a small study of 29 patients with ICH, serum IL-6 concentrations were increased significantly at day 1 and tended to decrease thereafter (Kim et al, 1996). However, levels remained significantly higher than those of the control group, even at day 7. In that same study, the level of TGFβ was significantly decreased at days 1 and 3 but then returned toward the control value. In a larger study of 124 patients with ICH, the elevated plasma concentrations of TNFα and IL-6 detected in patients 12 to 24 h after onset correlated with the magnitude of the subsequent perihematomal brain edema (Castillo et al, 2002), and subsequent enlargement of the hematoma (Silva et al, 2005). The latter study supports the findings of earlier animal experiments in which injections of IL-1 and TNFα into the brain induced opening of the blood—brain barrier, leading to vasogenic edema (Gordon et al, 1990; Holmin and Mathiesen, 2000; Megyeri et al, 1992). An elevated circulating IL-6 level in patients 7 days after ICH was also found to correlate with blood volume, mass effect (Dziedzic et al, 2002), and severity of brain injury, as assessed by the Glasgow Coma Scale score and mortality (Woiciechowsky et al, 2002). These clinical findings support the deleterious role of proinflammatory cytokines in human ICH-induced brain injury, suggesting that they could be potential therapeutic targets against ICH-induced early brain injury.
Cytokine targets: Animal studies have provided evidence that manipulation of cytokine pathways can be beneficial in cases of ICH. Studies by Mayne et al showed that in rats, intrastriatal infusion of TNFα-specific antisense oligodeoxynucleotide (Mayne et al, 2001b) or adenosine A2A receptor agonists (Mayne et al, 2001a) reduced TNFα mRNA and protein production in brain tissue surrounding a collagenase-induced hematoma. The result was significant reduction of both peri-ICH cell death and neurobehavioral deficits. Although inhibition of TNFα might be a useful approach to treating ICH, the situation is complicated by an apparent dual role for TNFα in which it can potentially repair or damage injured brain tissue (Hallenbeck, 2002; Shohami et al, 1999). To date, no clinical trials of anti-TNFα agents have been conducted in patients with ICH, but TNFα might offer another potential therapeutic target once its beneficial and deleterious effects in ICH have been better defined.
Interleukin-1 has gained much more attention than TNFα as a therapeutic target for stroke (Allan et al, 2005) because it has a minor role in normal brain function, and its blockade is unlikely to be associated with substantial adverse effects. Evidence has shown that inhibiting IL-1 release or activity markedly reduces ischemic cerebral damage (Rothwell, 2003). Use of IL-1ra is the most advanced approach to modifying IL-1 action. It has been reported that overexpression of IL-1ra via an adenovirus vector attenuated brain edema formation and thrombin-induced intracerebral inflammation in an autologous blood model of ICH in rat (Masada et al, 2001, 2003). Recombinant human IL-1ra (rhIL-1ra; injected subcutaneously) has been used successfully in patients with rheumatoid arthritis (Bresnihan, 2001), and no major adverse-event profiles have been reported in septic patients (Opal et al, 1997), suggesting that IL-1ra could be considered as a potential therapeutic agent for patients with ICH and could be the focus of additional preclinical and clinical research.
Proteases
Proteases have complex functions in the mammalian brain under normal and pathologic conditions. Abundant evidence indicates that extracellular proteases are involved in and mediate neuronal cell death in many neurologic diseases (Gingrich and Traynelis, 2000; Nakanishi, 2003; Yong, 2005; Yong et al, 2001). We have previously reviewed the physiologic and pathologic functions of proteases in the brain (including tissue plasminogen activator, thrombin, and cathepsins) and recent findings that link extracellular proteases with neuronal cell death in ischemic or hemorrhagic stroke (Wang and Tsirka, 2005a).
Matrix Metalloproteinases
Matrix metalloproteinases are a large family of zinc-dependent endopeptidases capable of degrading many components of the extracellular matrix. To date, approximately 24 different vertebrate MMPs have been identified, of which 23 are found in humans (Gueders et al, 2006; Nelson et al, 2000). They have been categorized into four groups of enzymes: collagenases, stromelysins, gelatinases, and membrane-type metalloproteinases (Nelson et al, 2000). Matrix metalloproteinases are important for normal physiologic brain function, but in the early stage of ICH, they can be detrimental. The activity of MMPs might be controlled by free radicals, either through activation of the latent forms or by induction of mRNA through signaling via the nuclear factor-kappaB site (Yong et al, 2001). As a result, MMPs can cause an increase in capillary permeability, thereby contributing to brain edema that is secondary to acute ischemic and hemorrhagic brain injury (Gasche et al, 1999; Liu and Rosenberg, 2005; Rosenberg and Navratil, 1997). Many investigators have reviewed the role of MMPs in mediating acute brain injury after ischemia and trauma (Cunningham et al, 2005; Gasche et al, 2006; Rosenberg, 2002; Rosenberg and Mun-Bryce, 2004; Wang and Lo, 2003), but because MMPs are essential for neurogenesis, myelin formation, and axonal growth (Cunningham et al, 2005; Kaczmarek et al, 2002; Pepper, 2001; Yong, 2005), there are concerns that inhibiting MMPs during recovery from ICH could do more harm than good. Despite a growing body of evidence regarding the detrimental properties of MMPs in acute ischemic stroke, data on their role in ICH are still limited.
Experimental studies of MMPs in ICH have focused on MMP2, 3, 9, and 12 (Lu et al, 2006; Mun-Bryce et al, 2004; Power et al, 2003; Rosenberg and Navratil, 1997; Tang et al, 2004; Wang and Tsirka, 2005b). In a collagenase-induced ICH model that produced edema, necrosis, and blood—brain barrier disruption, endogenous production of MMP9 was identified in brain tissues (Lu et al, 2006; Rosenberg and Navratil, 1997; Wang and Tsirka, 2005b). In 1997, Rosenberg and Navratil (1997) first demonstrated that MMP2 and 9 activation increased 16 to 24 h after collagenase-induced ICH in rats. Extending this work, a study by Power et al (2003) indicated that collagenase-induced hemorrhage in rat increased brain MMP2, 3, 7, and 9 mRNA levels at 24 h, but MMP12 (macrophage metalloelastase) was the most highly induced (>80-fold) at day 7. Immunohistochemistry showed MMP12 to be localized in activated microglia/macrophages surrounding the hematoma. Surprisingly, Wells et al (2005) found that collagenase-induced hemorrhage in mouse increased mRNA of MMP3 and 12, but not of MMP2 or 9. We also have observed a dramatic increase of MMP9 activity by gel and in situ zymography at 24 to 72 h in the same ICH model (Wang and Tsirka, 2005b) (Figure 3). Early increases in MMP9 mRNA and activity have been confirmed in other animal ICH models, including mouse (Tang et al, 2004), rat (Lu et al, 2006), and pig (Mun-Bryce et al, 2004). Other reports suggested that increased vascular MMP2 or MMP9 expression, stimulated by Aβ-amyloid peptides, might play a role in the pathogenesis of spontaneous ICH in patients with cerebral amyloid angiopathy (Jung et al, 2003; Lee et al, 2003). Overall, these preclinical findings support the view that some of the MMPs, especially MMP3, 9, and 12, might play an important role in the pathophysiology of ICH.
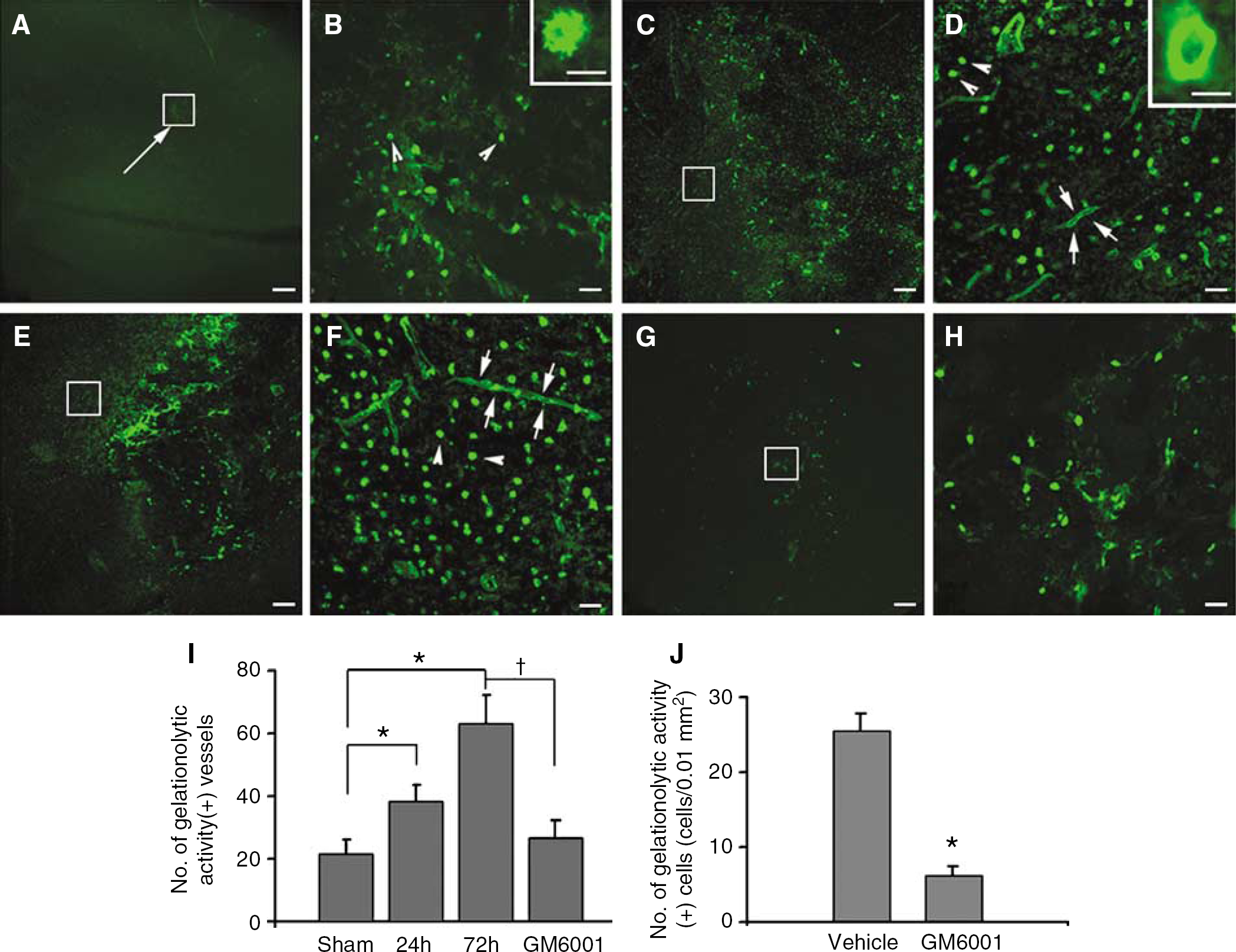
Increased in situ gelatinolytic activity after ICH. (
Several clinical studies have reported increases in blood MMP9 levels in patients with ICH (Abilleira et al, 2003; Alvarez-Sabin et al, 2004; Silva et al, 2005). Among patients with deep ICH, this increase was associated with perihematomal edema and deterioration of neurologic function within the acute stage (Alvarez-Sabin et al, 2004). Subsequently, Alvarez-Sabin et al (2004) detected the temporal profile of MMPs and their natural inhibitors in patients with acute ICH and found that increased MMP9 was associated with perihematomal edema, and that both MMP3 and 9 positively correlated with the residual scar volume at 3 months. Additionally, high levels of MMP3 were associated with mortality. Two new studies of MMPs in human brain after ICH (within the first 6 h after death) found that tissue around the intracranial hemorrhage had higher MMP9 levels than did the contralateral hemisphere (Rosell et al, 2006; Tejima et al, 2006) (Figure 4). Taken together, the animal and clinical data suggest deleterious roles for MMP3 and 9 in acute tissue damage after ICH; however, the role of MMP12 in acute ICH has yet to be established in clinical studies.
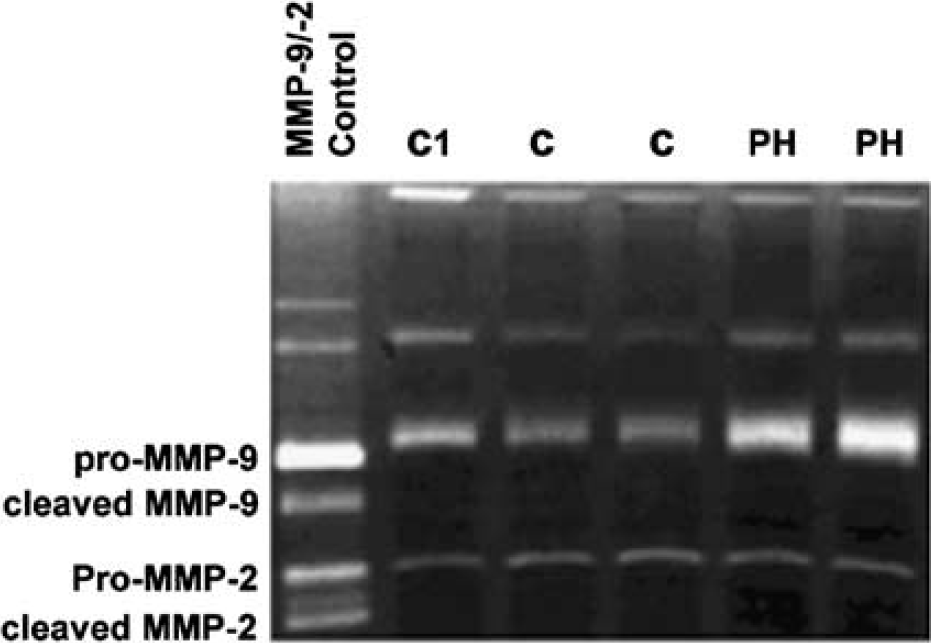
Zymograms corresponding to one representative hemorrhagic patient. Substrate-specific zymography for determination of gelatinolytic activity of MMP-2 and MMP-9 was performed on SDS-PAGE gels (10%) containing 0.1% gelatin. Human gelatinase control was also run with samples. The results indicate that gelatinolytic activity was highest in the perihematomal region. C1, control cases; C, contralateral; PH indicates perihematoma. Published with permission (Rosell et al, 2006).
Matrix metalloproteinase targets: Many investigators have used inhibitors to block MMP activity in animal studies of ICH. Rosenberg and Navratil (1997) first demonstrated that BB-1101 (a broad-spectrum hydroxamic acid-based MMP inhibitor) reduced brain edema when administrated 6 h after the onset of hemorrhage. The administration of BB-94 reduced recombinant tissue plasminogen activator-induced hemorrhage after thromboembolic stroke (Lapchak et al, 2000) and recombinant tissue plasminogen activator-mediated mortality after middle cerebral artery occlusion (Pfefferkorn and Rosenberg, 2003). Power et al (2003) showed that MMP12 was highly upregulated 7 days after ICH in rats, but that suppression of this protease with minocycline protected the morphology of neurons and improved functional recovery. In our studies that showed activation and upregulation of MMP9 after ICH, the broad-spectrum MMP inhibitor GM6001 ameliorated dysregulated gelatinase activity (Figure 3), neutrophil infiltration (Figure 1), production of oxidative stress, and brain edema, and decreased the number of degenerating neurons (Wang and Tsirka, 2005b). The animals also experienced functional improvement and a decrease in injury volume as a result of GM6001 treatment. In combination, these findings suggest that MMPs may play an important role in the pathogenesis of ICH and that interference with some of their functions might decrease ICH-induced early brain injury. However, long-term inhibition of MMPs may be inappropriate because of the potential to inhibit the beneficial roles of MMPs in regulating neurogenesis, myelin formation, and axonal growth (Cunningham et al, 2005; Kaczmarek et al, 2002; Pepper, 2001; Yong, 2005). Therefore, instead of MMP inhibitor therapies for acute ICH, strategies that focus on modulating the activity of MMPs may be considered for improving recovery and enhancing long-term functional outcome, as suggested in ischemic stroke (Zhao et al, 2006a).
Reactive Oxygen Species
After brain injury, ROS are released by neutrophils, vascular endothelium, and activated microglia/macrophages (Facchinetti et al, 1998; Weiss, 1989). It is well known that production of ROS is an inevitable consequence of normal oxidative metabolism, but high ROS levels can be lethal (Juranek and Bezek, 2005). Reactive oxygen species are considered to be an important contributor to ischemic brain injury (Crack and Taylor, 2005; Saito et al, 2005) and might also contribute to the outcome of ICH (Aronowski and Hall, 2005; Green and Ashwood, 2005; Peeling et al, 1998; Wang and Tsirka, 2005a). As a result of hemorrhage, the extracellular spaces of the brain become exposed to hemoglobin and its breakdown products. Iron and iron-related compounds, including hemoglobin, catalyze hydroxyl radical production and lipid peroxidation (Sadrzadeh et al, 1987; Sadrzadeh and Eaton, 1988), which expose the brain cells to increased levels of oxidative stress. Indeed, high levels of oxidative stress, as measured by protein carbonyl formation, have been found shortly (within minutes) after the onset of autologous blood injection in pig (Hall et al, 2000; Wagner et al, 2002). In addition, intracerebral infusion of lysed erythrocytes into the rat striatum induced marked brain edema and profound neurologic deficits (Wu et al, 2002b). In this setting, increased oxidative stress, measured by protein carbonyl formation, might be associated with reduced Mn-superoxide dismutase and CuZn-superoxide dismutase contents and increased DNA damage. Furthermore, we recently observed that, on days 1 and 3 after ICH, the increased ethidium (oxidized hydroethidine, a marker for ROS production detected in situ) persisted in the peri-ICH region, whereas, in the contralateral (vehicle control) side, only a few ethidium-positive cells were detected, and the signal was confined to the perinuclear area (Wang and Tsirka, 2005b, c ). Such animal studies provide substantial evidence that ROS may contribute to ICH-induced brain injury.
To date, only one clinical study has been conducted to ascertain whether free-radical-induced oxidative damage exists in human brain tissue after ICH. Mantle et al (2001) found that brain cortex samples from the perihematomal region of 10 patients with spontaneous ICH had proteins with evidence of oxidative damage. Surprisingly, tissue samples from six control patients (normal tissue obtained during tumor removal or aneurysmal repair) had equally high levels of oxidized proteins. In addition, no significant differences were observed between ICH and control brain tissue in the levels of the following antioxidants: glutathione, glutathione peroxidase, glutathione reductase, catalase, and superoxide dismutase. The possible reason for this unexpected similarity and the high levels of oxidative stress in the control tissues might be that the tissue used as control (peritumor and aneurysmal tissue) was pathologically compromised with potentially elevated levels of oxidative stress and, therefore, did not represent an appropriate control (Aronowski and Hall, 2005).
Reactive oxygen species target: Given the potential contribution of free radicals to ICH-induced brain injury, efforts have been made to ameliorate the damage with free radical scavengers. Using a rat collagenase model of ICH, Peeling et al (1998) demonstrated a significant behavioral improvement after treatment with dimethylthiourea and α-phenyl-N-tert-butyl nitrone, which quench free radicals. Later, Aronowski and Hall (2005) observed similar results in rats that had been subjected to ICH induced by double-blood injection. These results suggest that compounds that interrupt the free radical cascade might improve ICH outcome. Moreover, treatment with α-phenyl-N-tert-butyl nitrone reduced recombinant tissue plasminogen activator-induced hemorrhage, infarction, and neurologic deficits in a rat embolic focal ischemia model (Asahi et al, 2000). Peeling et al (2001a) additionally examined the effect of another free-radical-trapping agent, disodium 4-[(tert-butylimino)methyl]benzene-1,3-disulfonate N-oxide (NXY-059), in their rat collagenase-induced ICH model and found that it did not affect the size or resolution of the hemorrhage, but 48 h posthemorrhage, it significantly reduced the neutrophil infiltration observed in the vicinity of the hematoma and the number of TUNEL-positive cells observed at the hematoma margin. The NXY-059-treated rats also performed better on beam walking, circling, and posture-reflex tests than the control group. A Phase II study of the safety and tolerability of NXY-059 in patients with ICH (CHANT) is ongoing (Green and Ashwood, 2005).
Considering the potential sources of ROS production in ICH, other strategies that target pro-oxidant heme or iron have been tested. Because heme oxygenase metabolizes heme to release iron, regulation of this enzyme might decrease ICH-induced heme-related toxicity (Wang et al, 2006). In support of this possibility, several investigators have shown that nonselective inhibitors of heme oxygenase (tin-mesoporphyrin IX, tin-protoporphyrin, and zinc protoporphyrin) decreased ICH-induced brain injury, including brain edema and neurologic deficits (Gong et al, 2006; Huang et al, 2002; Koeppen et al, 2004; Wagner et al, 2000). Deferoxamine, a ferric iron chelator, was shown to have a similar neuroprotective effect after ICH (Nakamura et al, 2004a; Wan et al, 2006). These findings suggest that ROS could be a potential target for ICH therapy.
Summary
Several recent animal and clinical studies have provided important information about the mechanisms of ICH-induced brain injury. Current evidence indicates that various cellular and molecular components of inflammation are associated with ICH-induced brain injury. Inflammation is increasingly recognized as a key factor in the pathogenesis and outcome of ICH. Using laboratory and imaging techniques in carefully selected clinical populations, it is now possible to translate experimental approaches to human studies. Better understanding of the role of inflammation and its potential for modulation could have profound implications for treatment of patients after ICH. In the near future, additional strategies that target inflammation in the very early phase of hemostasis (Mayer et al, 2005) could offer exciting new promise in the therapeutic approach to ICH.
Literature Search Strategy and Selection Criteria
References for this review were identified by searches of Ovid, MEDLINE, and PubMed with a time frame of January 1966 to May 2006. The research terms used were ‘intracerebral hemorrhage,’ ‘inflammation,’ ‘leukocytes,’ ‘microglia,’ ‘cytokines,’ ‘proteases,’ and ‘reactive oxygen species.’ Articles also were identified through searches of the authors’ own files and from references lists of articles identified in the literature search. Only papers published in English were reviewed. Articles were selected for their conceptual importance and primacy, and the final reference list was generated based on relevance to the topics covered in this review.
Footnotes
Acknowledgements
We thank Claire Levine for her assistance in preparing this manuscript. The authors report no conflicts of interest.