
Abstract
The present study describes, for the first time, a temporal and spatial cellular expression of erythropoietin (Epo) and Epo receptor (Epo-R) with the evolution of a cerebral infarct after focal permanent ischemia in mice. In addition to a basal expression of Epo in neurons and astrocytes, a postischemic Epo expression has been localized specifically to endothelial cells (1 day), microglia/macrophage-like cells (3 days), and reactive astrocytes (7 days after occlusion). Under these conditions, the Epo-R expression always precedes that of Epo for each cell type. These results support the hypothesis that there is a continuous formation of Epo, with its corresponding receptor, during the active evolution of a focal cerebral infarct and that the Epo/Epo-R system might be implicated in the processes of neuroprotection and restructuring (such as angiogenesis and gliosis) after ischemia. To support this hypothesis, a significant reduction in infarct volume (47%; P < 0.0002) was found in mice treated with recombinant Epo 24 hours before induction of cerebral ischemia. Based on the above, we propose that the Epo/Epo-R system is an endogenous mechanism that protects the brain against damages consequent to a reduction in blood flow, a mechanism that can be amplified by the intracerebroventricular application of exogenous recombinant Epo.
Cerebrovascular accidents, defined as ischemic and hemorrhagic stroke, represent the third leading cause of death after coronary heart disease and cancer. Various potential treatment approaches have been developed to reduce the extent of tissue injury, approaches that have been derived essentially from experimental models of focal cerebral ischemia. Although in stroke, severe tissue hypoxia and hypoglycemia lead to rapid and irreversible cell death, part of the neuronal damage is secondary to, and outside of, the primary focus of damage. Accordingly, a penumbral region has been identified in which neuronal loss can be reduced by therapeutic intervention. Neurodegeneration has been ascribed to the excessive release of endogenous excitatory amino acids (e.g., glutamate); however, it is apparent that the mechanisms of neuronal damage depend also on complex interactions between neurotransmitters, neuropeptides, and inflammatory molecules that modify survival and repair. Indeed, it is now clear that cytokines participate in the inflammatory response associated with cerebral ischemia (Rothwell et al., 1994; Arvin et al., 1996). Besides the proinflammatory cytokines such as interleukin-1 (IL-1), IL-6, tumor necrosis factor-α (TNF-α), and transforming growth factor-β (TGF-β), recent studies underline the possible implication of hematopoietic cytokines in the processes of development and repair in the brain. For example, neurotrophic effects have been reported for IL-3, granulocyte-macrophage colony-stimulating factor, and erythropoietin (Epo) (Konishi et al., 1993). In parallel among the novel approaches to stroke therapy, considerable attention has been brought to the identification of genes specifically expressed by hypoxia. For example, the transcription factor hypoxia-inducible factor-1 activates a number of genes including several glycolytic enzymes, inducible nitric oxide synthase, vascular endothelial growth factor (VEGF), and Epo (Wenger and Gassmann, 1997), all of which have been postulated to serve as defense mechanisms against local and systemic hypoxia.
Erythropoietin has been recognized as the main regulator of erythropoiesis and was thought to be exclusively produced in fetal liver and adult kidney. However, there is growing evidence that Epo, like other hematopoietic factors, is also implicated in the CNS. Erythropoietin mRNA is expressed in both rodent and primate brain, and its expression is increased by hypoxia (Tan et al., 1992; Digicaylioglu et al., 1995; Marti et al., 1996). It has been detected in cerebrospinal fluid collected from patients with traumatic brain injury (Marti et al., 1997). In vitro, astrocytes have been shown to express Epo and this can be upregulated by decreased oxygen levels (Masuda et al., 1994; Marti et al., 1996). Moreover, Epo receptors (Epo-R) have been detected, not only in neural cell lines (Masuda et al., 1993; Juul et al., 1998), but also in hippocampal and cortical neurons (Morishita et al., 1997; Juul et al., 1998). Specific Epo binding sites have been found in defined areas of the adult mouse brain including the hippocampus and neocortex (Digicaylioglu et al., 1995), two structures known to be sensitive to global ischemia (Pulsinelli et al., 1982). Overall, these findings suggest that Epo could act on neurons in a paracrine fashion. This notion has been supported by the in vitro and in vivo neuroprotective effects of Epo, based on studies in global cerebral ischemia, not thought to be a model of stroke (Morishita et al., 1997; Sakanaka et al., 1998). Given these different findings, we hypothesized that the Epo/Epo-R system could also play a role in focal cerebral ischemia, which is a model of stroke. Accordingly, we have investigated the temporal, spatial, and cellular profiles of Epo/Epo-R expression at both mRNA and protein levels, after permanent focal middle cerebral artery occlusion (MCAO) in mice. We have, furthermore, analyzed the physiologic role of Epo in cerebral ischemia by examining whether the histologic damage after an ischemic insult is reduced by pretreatment with intracerebroventricularly injected Epo. Finally, and to further investigate the mode of action by which Epo exerts its effects in vivo, we studied the effect of Epo on neuronal cell death in vitro.
MATERIALS AND METHODS
Focal cerebral ischemia
Surgical protocols were approved by the local ethics committee and governed by the pertinent national legislation. Focal ischemia was induced in OF1 mice (Iffa Credo, L'Arbresle, France) by the permanent occlusion of the left middle cerebral artery under chloral hydrate anesthesia as reported previously (Welsh et al., 1987; Nawashiro et al., 1997). Briefly, a skin incision was made between the orbit and ear. Under an operating microscope, an incision was made dividing the temporal muscle, and the left lateral aspect of the skull was exposed by reflecting the temporal muscle and surrounding soft tissue. The distal course of the middle cerebral artery was then visible through the translucent skull. A small burr-hole craniectomy was performed with a dental drill. The left middle cerebral artery was coagulated by bipolar diathermy. The muscle and soft tissue were replaced and the incision was sutured. In this model, ischemia is restricted to the neocortex. At different times after occlusion, mice were anesthetized and the brains were removed.
Semiquantitative reverse transcription-polymerase chain reaction
Total RNA was prepared from cerebral cortices and cultured cells by phenol-chloroform extraction using the RNAxel extraction kit (Promega, Charbonnieres, France). Reverse transcription and amplification by polymerase chain reaction (PCR) were performed as described previously (Digicaylioglu et al., 1995). Primers for Epo, Epo-R, and β-actin have been used as published (Digicaylioglu et al., 1995; Ringheim et al., 1995; Yamaji et al., 1996), All samples were normalized to the PCR signal obtained for the housekeeping gene, β-actin.
In situ hybridization
The technique used for in situ hybridization was essentially that described by Breier et al. (1992). RNA probes were generated by in vitro transcription of the plasmid pBSmEPOR.17, a partial mouse Epo-R cDNA, spanning from exon 3 to exon 6 (bp 388 through 856, numbered according to D'Andrea et al., 1989), of the mouse Epo-R cDNA clone pXM190 (kindly provided by A. D'Andrea, Boston, MA, U.S.A.). Hybridization was performed on cryostat-cut coronal brain sections (10 µm) with 2.5 × 104 cpm/mL 35S-labeled RNA probe overnight at 48°C. Sections were washed, dehydrated, coated with Kodak NTB-2 emulsion (Eastman Kodak, Germany), and developed after a 3-week exposure.
Western blot analysis
Cortical tissue lysates were prepared at different times after occlusion, and 30 µg of protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis as described previously (Hayashi et al., 1997). Western blot analysis was performed using rabbit polyclonal antibodies for Epo (1:500; R&D system Europe Ltd., United Kingdom) and Epo-R (1:150; Santa Cruz, Le Perray en Yvelines, France), respectively. Peroxidase-coupled goat anti-rabbit IgG (1:80,000; Sigma, L'Isle d'Abeau Chesnes, France) was used in secondary incubations followed by the detection of reactive bands by chemiluminescence (NEN, Paris, France).
Immunochemistry and isolectin binding
After MCAO, mice were perfusion-fixed with 4% paraformaldehyde and coronal free-floating brain sections (20 µm) were cut on a vibratome. For examination of cultured cells, cells were fixed to the wells by incubation with 4% paraformaldehyde and thereafter washed with phosphate-buffered saline (PBS). The immunochemical staining was performed with the avidin-biotin-peroxidase technique as described previously (Bernaudin et al., 1998). The following primary antibodies were used: rabbit polyclonal antibodies to glial fibrillary acidic protein (GFAP; 1:5,000 dilution; Dako, France), von Willebrand Factor (1:200; Dako, Tnappes, France), Epo-R (1:100; Santa Cruz, France), and Epo (1:100; R&D system, Europe Ltd., United Kingdom) and a mouse monoclonal antibody to microtubule-associated protein-2 (MAP2; clone HM-2, 1:250; Sigma, France). Negative controls were performed by omitting the primary antibody and by preabsorbed the Epo and Epo-R antibodies with the homologous antigens. Microglia were visualized using peroxidase-linked isolectin B4 (isolated from Bandeiraea simplicifolia GS-B4, 1:100; Sigma, France).
Intracerebroventricular injections
Anesthetized mice were treated by intracerebroventricular injection of 3 µL of vehicle (PBS with 1 mg/mL of bovine serum albumin; Sigma, France) or recombinant mouse Epo (rMoEpo; 0.4 µg/kg; Boehringer, Meylan, France) 24 hours before MCAO or at the same time as the occlusion. The infarct volumes and physiologic parameters were measured 24 hours after MCAO. For the evaluation of cortical infarct volume, coronal brain sections (15 µm) were cut on a cryostat and stained with thionin. The infarcted areas of each section were measured with a Rag 200 image analyzer (BIOCOM). Total infarct volumes (mm3) were calculated after integration of infarcted areas with the distance between each section level analyzed (400 µm). Statistical significance was determined using Student's t test.
Primary cultures of neocortical neurons and experimental treatments
Dissociated neocortical cell cultures from OF1 mouse embryos at 14 to 15 days of embryonic development were prepared according to Deloulme et al. (1991). After 10 days in culture, cells were immunocytochemically identified as 85% neurons and 15% astrocytes. At this time, the cells were pretreated with different concentrations of recombinant human Epo (rHuEpo; Boehringer, France) for 24 hours before the administration of 15 µmol/L NMDA (N-methyl-
Primary cultures of astrocytes
Cortical astrocyte cell cultures were prepared from 1 to 3 postnatal 1 to 3 postnatal-day-old OF1 mice as described previously (Vivien et al., 1998). Experiments were performed on cortical astrocyte cultures after 21 days in vitro.
Endothelial cell culture
The rat brain endothelial cell line (RBE4), provided by Neurotech (Paris, France), was maintained in DMEM/Ham's F-12 (1:1) supplemented by 10% fetal calf serum, 300 µg/mL G418 geneticin, 1 ng/mL basic fibroblast growth factor, 50 U/mL of penicillin, and 50 µg/mL of streptomycin (Gibco, Cergy Pontoise, France).
RESULTS
Constitutive expression of Epo and Epo-R in brain
The brain cellular expression of Epo and Epo-R was first studied in vitro. Although astrocytes have been identified as the primary source of brain Epo in vitro (Masuda et al., 1994; Marti et al., 1996), our reverse transcription-PCR and immunocytochemistry analyses performed in neuronal, astroglial, and endothelial cell cultures revealed that in addition to astrocytes, neurons expressed Epo. In contrast, no Epo expression was detected in endothelial cells. However, Epo-R were detected on neurons, astrocytes, and endothelial cells (Fig. 1).
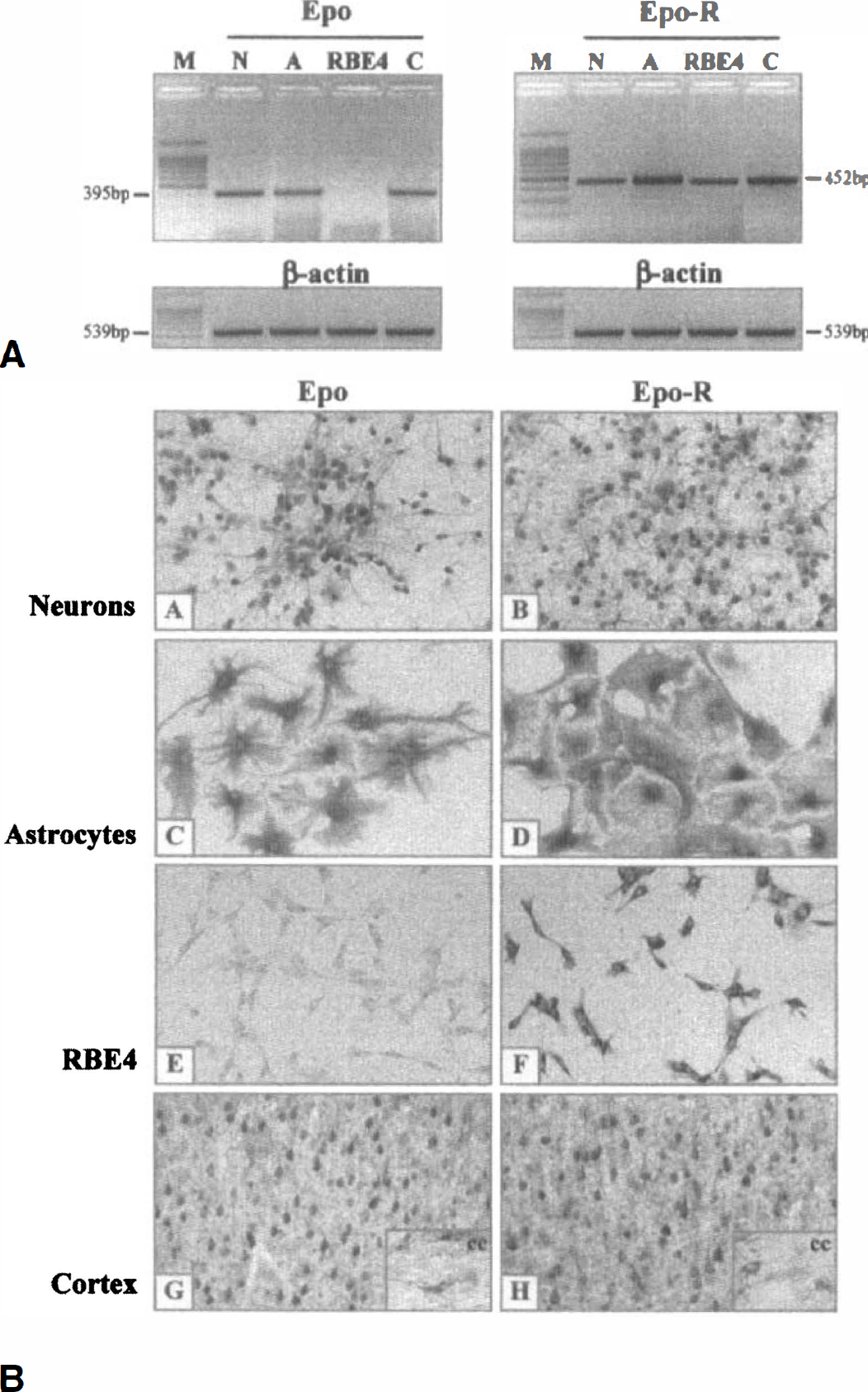
Constitutive expression of Epo and Epo-R in brain.
Under normoxic conditions, Epo and Epo-R were constitutively expressed in the cerebral cortex obtained from adult mice: both mRNA (Fig. 1A) and corresponding proteins were expressed (Figs. 1B and 2). Western blot analysis showed that brain Epo migrated as two bands with an apparent molecular size of 33 kDa and 18 kDa, corresponding to the secretable mature Epo and the nonglycosylated protein moiety of 165 residues, respectively (Fig. 2A). The difference in size between brain and rMoEpo (40 kDa) was mainly because of poor sialylation of brain Epo as described previously (Masuda et al., 1994). Our immunohistochemical study, in accordance with our immunocytochemistry results, shows a prominent staining for Epo in the normal adult mouse brain in neurons, whereas astrocytes were weakly immunostained (Fig. 1B). The protein bands corresponding to brain Epo-R were of the expected size (68, 62, 46, 43, and 29 kDa) for the different Epo-R forms described in neural or erythroid cells (Kuramochi et al., 1990; Migliaccio et al., 1991; Nagao et al., 1992; Masuda et al., 1993) (Fig. 2B). The 68-kDa protein has been described as the glycosylated mature receptor form that is translocated to the cell surface, whereas the 62-kDa protein corresponds to the native intracellular form of Epo-R. The 46-kDa to 43-kDa bands have been identified as the major proteolytic forms of Epo-R and the Mr 29,000 to the soluble receptor. A band of 37 kDa was also detected, but we do not know yet to which protein it corresponds. All these Epo-R forms are undetectable when the antibody is preabsorbed with the homologous antigen. Immunohistochemistry revealed that both neurons and astrocytes constitutively expressed Epo-R (Fig. 1B).
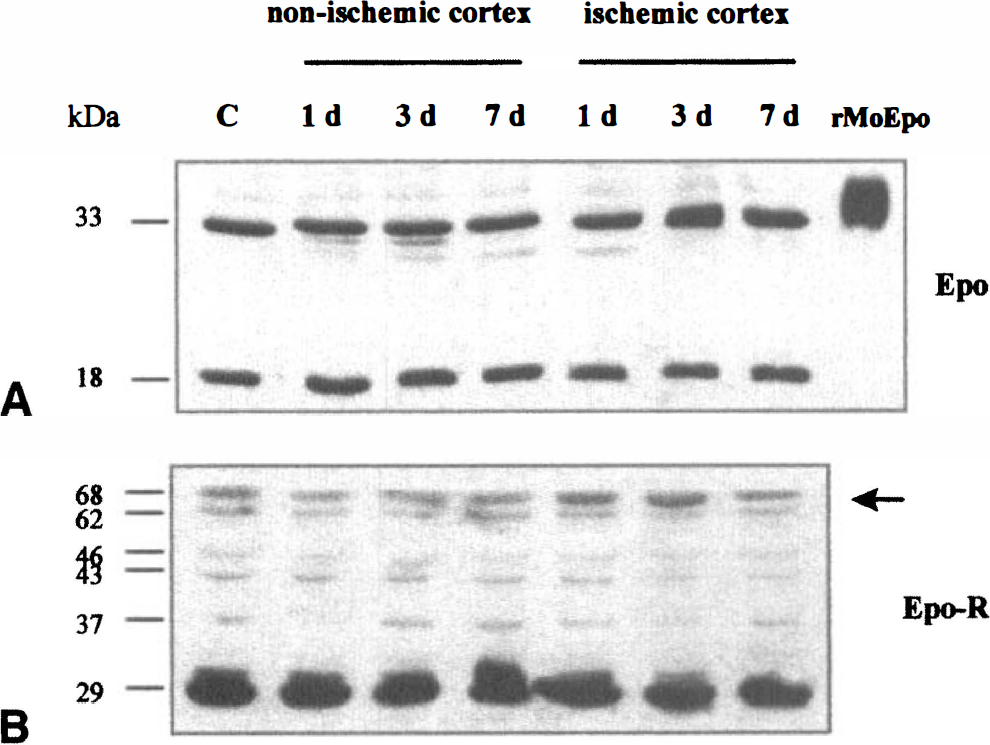
Western blot analysis of Epo and Epo-R in Iysates from nonischemic and ischemic cortex prepared after different periods of occlusion revealed two bands of 33 kDa and 18 kDa for Epo (A), corresponding to the mature and the nonglycosylated form of Epo, respectively. Recombinant mouse Epo (40 kDa) was used as positive control. Analysis of Epo-R (B) revealed six bands of 68, 62, 46, 43, 37, and 29 kDa.
Cerebral ischemia induces a modulation of Epo and Epo-R expression
We analyzed the chronologic sequence of Epo/Epo-R expression in infarcted brain after different occlusion durations, at both mRNA and protein levels. Using reverse transcription-PCR, no changes in Epo and Epo-R mRNA were detected in whole lysates of ischemic or nonischemic cortex, at any time after occlusion (data not shown). As the response to the focal ischemic insult is restricted to a small volume within the cortex, one might not expect major changes in the expression level of brain Epo and Epo-R mRNA in total cortical preparations. Accordingly, we analyzed the expression pattern of Epo and Epo-R by in situ hybridization. This approach revealed an induction of Epo-R mRNA at 12 and 24 hours after occlusion, in cells of the ischemic border zone as well as those in the core (Fig. 3). In situ hybridization for the ligand Epo identified single positive cells within the ischemic core after 24 hours of occlusion (data not shown). Unfortunately, a high background level, caused by the low sensitivity of the probes, did not allow us to further analyze the expression of Epo at the mRNA level.
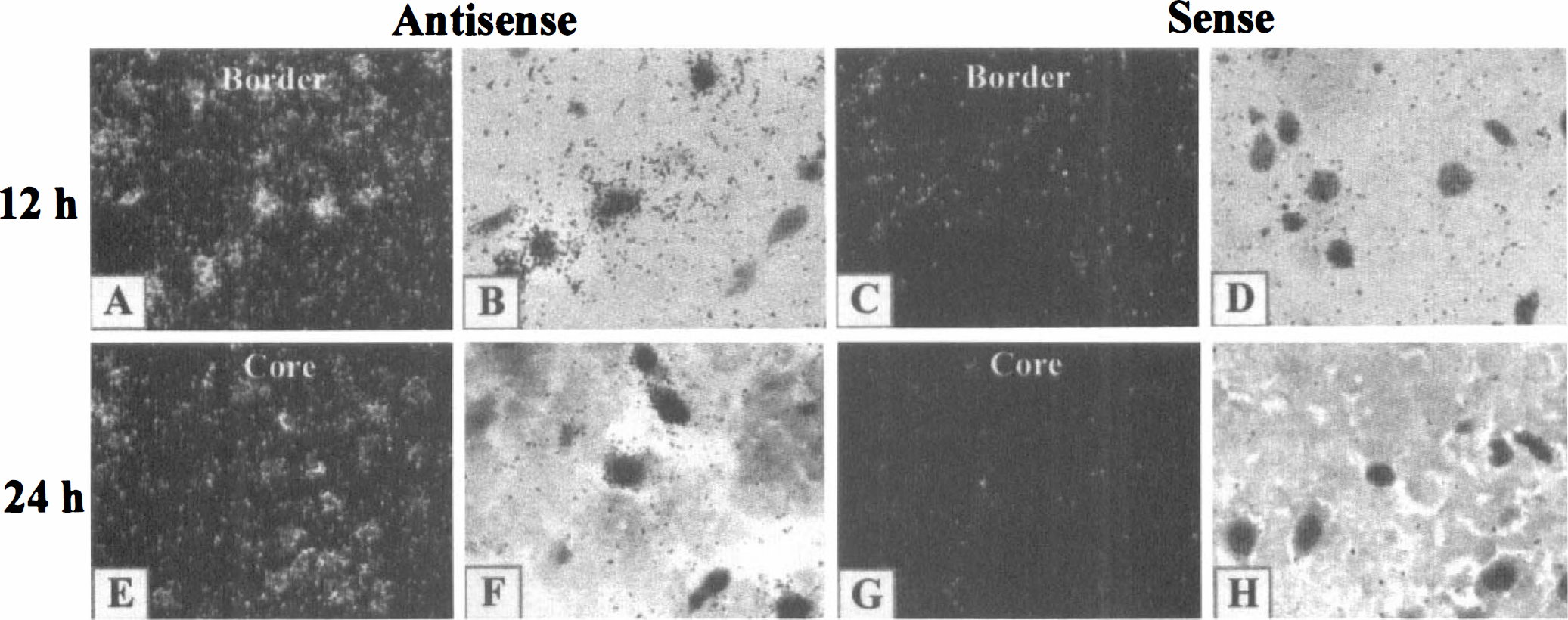
In situ hybridization of Epo-R mRNA on coronal brain section after MCAO (n = 3): positive cells were detected at 12 (
Western blot analysis showed no significant changes in the immunostaining intensity of the Epo bands after ischemia (Fig. 2A). With respect to Epo-R, the fully glycosylated form (68 kDa) showed an increase after 1 day that peaked after 3 days in the lysates taken from the ischemic cortex (Fig. 2B). No 68-kDa Epo-R form changes were detected at any time in the nonischemic, contralateral cortex.
The immunohistochemical study revealed a specific temporal cellular expression of Epo and Epo-R as a function of the duration of ischemia. Immunopositive cells for Epo and Epo-R were identified by comparing the cellular morphology, obtained along with the progression of the infarct, with specific immunomarkers for neurons (MAP2), astrocytes (GFAP), and endothelial cells (von Willebrand factor), and with isolectin for microglia/macrophage-like cells. The neuronal expression of Epo and Epo-R progressively disappeared in the infarcted zone, reflecting the neuronal death. Indeed, 12 hours after occlusion, immunoreactivity was not found in neurons within the ischemic core, whereas neurons located outside the infarct remained immunoreactive for Epo and Epo-R (Fig. 4C and 4D). However, 1 day after occlusion, endothelial cells located in the ischemic core became immunoreactive for Epo (Figs. 4E and 5E). An increasing signal and a vascular hypertrophy were observed from 3 to 7 days (Figs. 4G, 4I, 5G, and 5J). Three days after occlusion, a few microglia/macrophage-like cells expressed Epo (Fig. 5G), whereas the reactive astrocytes became positive 7 days after occlusion (Fig. 5L). The kinetic pattern of cellular expression for Epo-R was slightly different. Endothelial cells located in the ischemic core became immunoreactive for Epo-R as soon as 12 hours after occlusion (Fig. 4D) with an increasing signal from 1 to 7 days (Figs. 4F, 4H, 5F, 5H, and 5K). Positive microglia/macrophage-like cells were also detected from 1 (Fig. 5F) to 7 days (data not shown). Astrocytes surrounding the infarct were intensively immunostained for Epo-R 3 and 7 days after occlusion (Figs. 4H, 4J, 5I and5M). Astrocytes located in the thalamus became immunoreactive for Epo-R 7 days after occlusion in accordance with the late astroglial reaction caused by the retrograde degeneration of thalamocortical projections (data not shown). The differential cellular expression of Epo and Epo-R suggests a selective role for Epo after MCAO, in particular in the processes of neuroprotection and restructuring such as angiogenesis and the glial reaction.
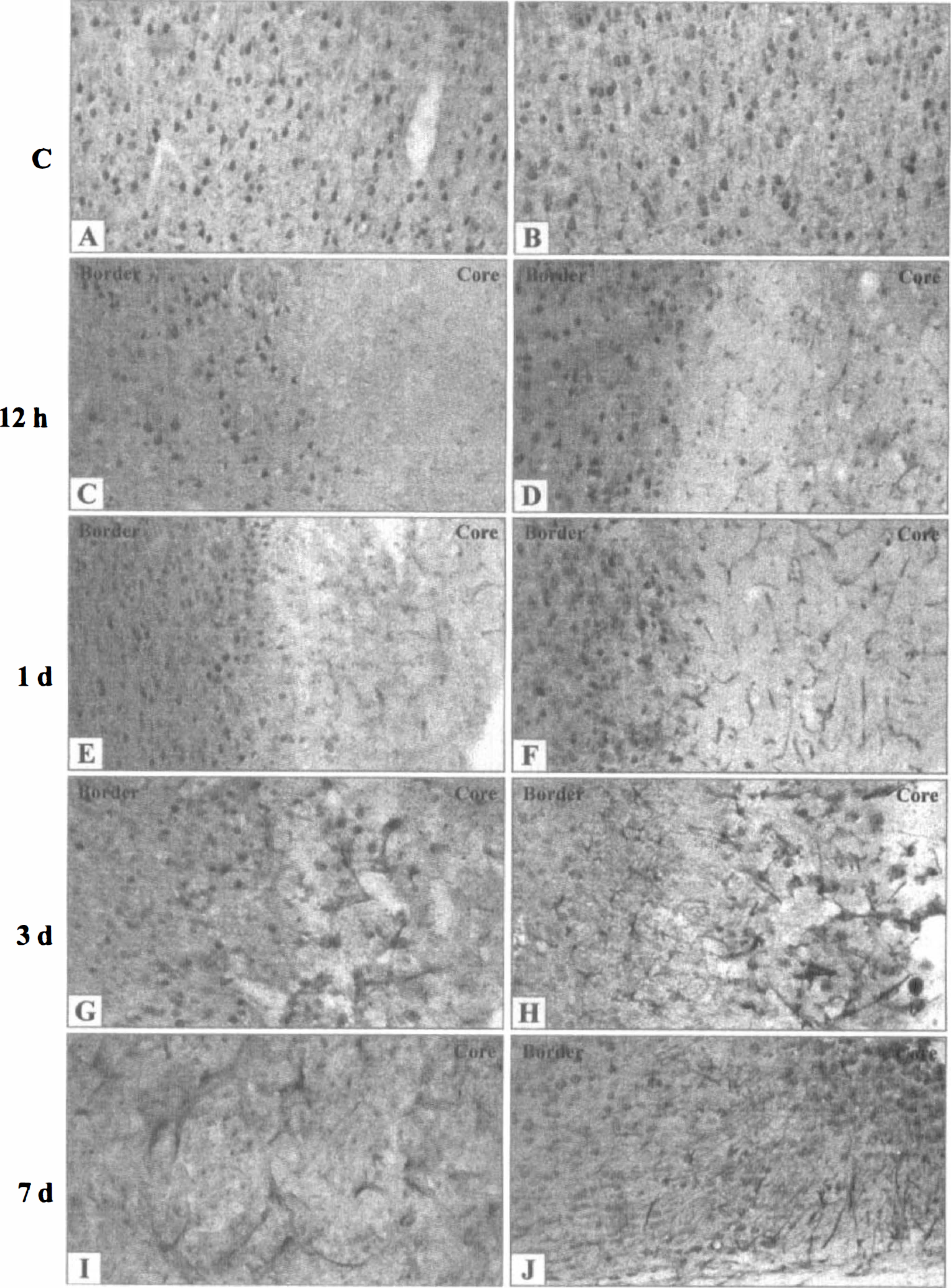
Representative photographs of immunohistochemistry of Epo and Epo-R showing the temporal and spatial evolution of immunoreactive Epo (
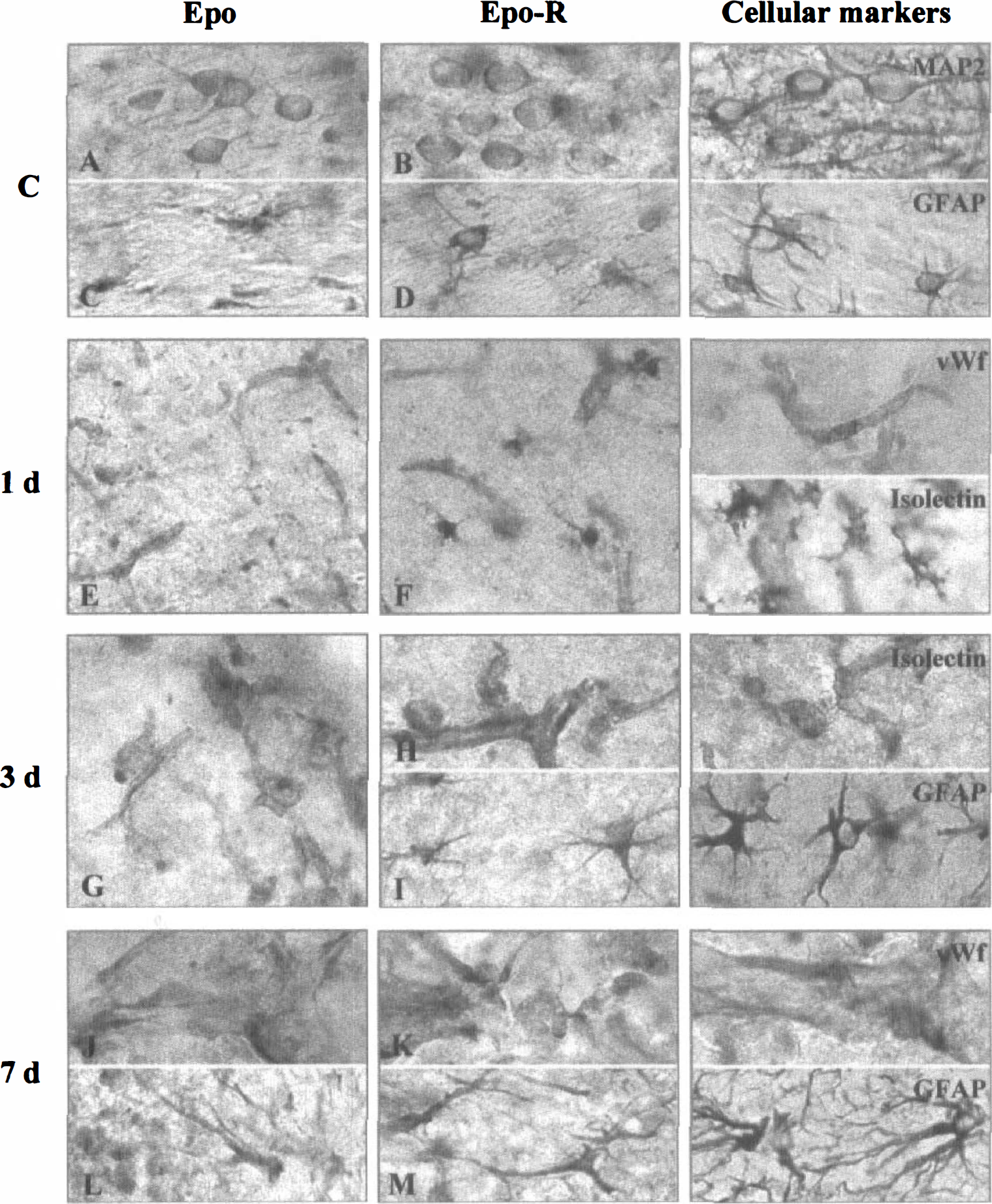
Representative photographs of immunohistochemistry of Epo and Epo-R showing the constitutive cellular expression of Epo and Epo-R in neurons (
Protective effect of Epo in cerebral ischemia
To further evaluate the role of Epo during stroke, mice were treated with rMoEpo (0.4 µg/kg intracerebroventricularly) 24 hours before the induction of focal cerebral ischemia. There were no significant differences in the physiologic parameters (mean ± SD) between Epo-treated animals and controls (pH, vehicle 7.29 ± 0.08, Epo 7.28 ± 0.07; body temperature, vehicle 37.06°C ± 0.15°C, Epo 37.07°C ± 0.12°C; Paco2, vehicle 40.2 ± 4.3, Epo 41.4 ± 4.7 mm Hg; PaO2, vehicle 130.1 ± 5.1, Epo 132.9 ± 3.2 mm Hg; hematocrit, vehicle 41.5% ± 4.7%, Epo 40.9% ± 5.9%). The volume (mean ± SD) of the cortical infarct in vehicle-treated mice was 40 ± 9 mm3 (n = 9). Pretreatment with Epo led to a significant reduction of the infarct volume by 47% (21 ± 4 mm3, n = 8, P < 0.0002). The reduction of the infarcted area by Epo is shown at different rostrocaudal coronal planes in Fig. 6. The same dose administered at the same time as occlusion was without effect.
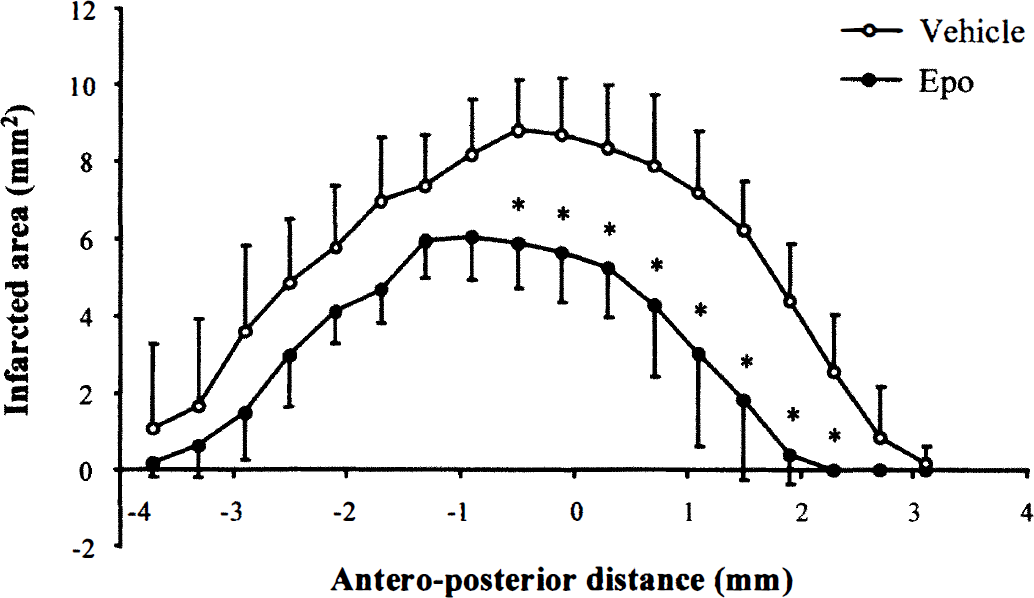
Neuroprotective effects of Epo in a mouse model of permanent focal cerebral ischemia at different rostrocaudal coronal planes (in mm from Bregma). The results are means ± SD of data obtained for nine (vehicle-treated) and eight (Epo-treated) mice. * P < 0.00277 (Bonferroni corrected critical P value = 0.5/18); significant difference between the two groups (Student's t-test with Bonferroni correction after a significant two-way repeated measures analysis of variance at P < 0.0002).
To provide further insight into the mechanisms through which Epo could exert its protective effects in cerebral ischemia, we studied in vitro the effect of Epo on neuronal death. Neuronal death was induced by exposing neocortical neurons to 15 µmol/L NMDA for 24 hours. This treatment led to the death of about 50% of all cells. The effect of NMDA was completely inhibited by the addition of 10 µmol/L of the noncompetitive NMDA antagonist, MK-801 (Fig. 7). When neocortical neurons were pretreated for 24 hours with rHuEpo at a concentration of 30 and 300 pmol/L before exposure to NMDA, a significant reduction of neuronal damage by 65% and 79%, respectively, (P < 0.001), was observed (Fig. 7).
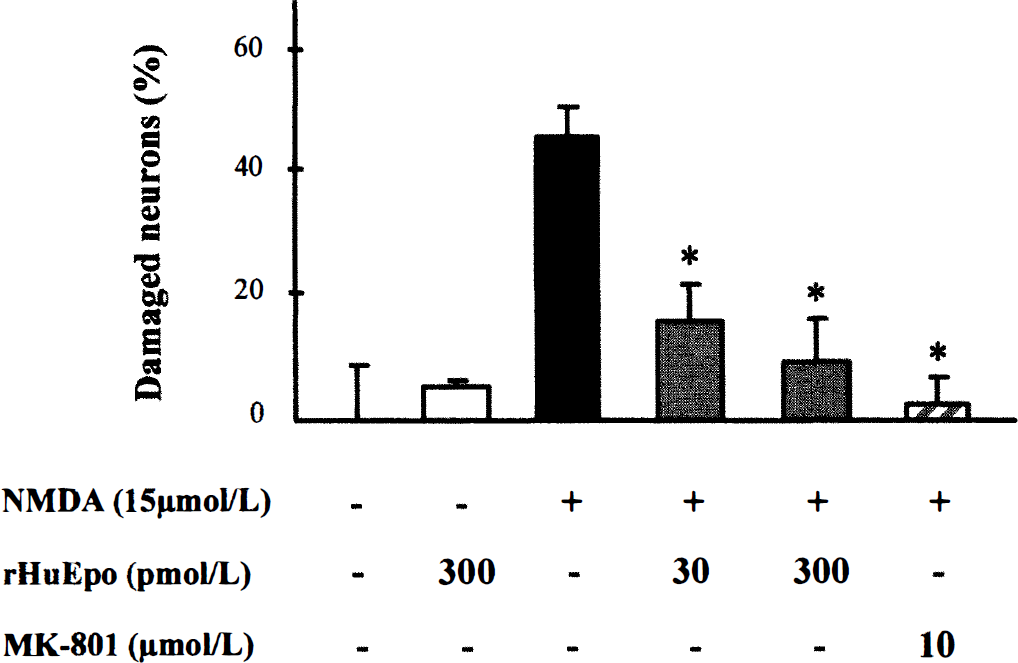
Erythropoietin protects mouse neocortical neurons from NMDA-induced injury. At 10 days in vitro, neocortical neurons were pretreated with rHuEpo 24 hours before exposure to NMDA (15 µmol/L). Neuronal death was evaluated, 24 hours later, by measuring the activity of lactate dehydrogenase released into the medium. MK-801, a noncompetitive NMDA antagonist, was used as a neuroprotective positive control. Values are indicated as mean ± SD of three experiments performed in quadruplicate. * P < 0.001 compared with the values for neurons exposed to NMDA alone (Student's t test).
DISCUSSION
Epo protects the brain from cerebral ischemia. We and others have previously shown that Epo and its receptor are expressed in the brain of man, monkey, mouse, and rat and that Epo is induced by hypoxia in this organ (Tan et al., 1992; Digicaylioglu et al., 1995; Marti et al., 1996, 1997; Juul et al., 1998). We therefore postulated that Epo might have neuroprotective effects during focal ischemia. Indeed, we demonstrate that intracerebroventricular administration of rMoEpo 24 hours before occlusion reduced the infarct volume by 47% in mice subjected to permanent MCAO. There are several possible mechanisms through which Epo could achieve this protective effect. A direct neuroprotective effect could be evoked as we have demonstrated that pretreatment of cultured mouse neocortical neurons with rHuEpo prevents NMDA-induced neuronal death. This finding is in agreement with previous reports showing a neuroprotective effect of Epo on glutamate-mediated (Morishita et al., 1997) and ischemia-induced neuronal cell death (Sakanaka et al., 1998). These authors proposed that Epo rescues neurons from nitric oxide-induced death (Sakanaka et al., 1998) and that Epo pretreatment induces a rapid and transient increase in intracellular calcium (Morishita et al., 1997), which can be compared with the in vivo neuroprotective phenomenon known as ischemic preconditioning (Kitagawa et al., 1990). One might also speculate that Epo could modulate the inflammatory response that accompanies cerebral ischemia. However, there is no evidence to suppose that Epo has an effect on the production of IL-1, IL-6, TNF-α, or TGF-β, cytokines involved in the inflammatory response in the CNS (Rothwell et al., 1994; Arvin et al., 1996). We cannot rule out the possibility that Epo influences the glial reaction, as astrocytes not only produce Epo but also express the Epo-R on their surface (see below). Finally, it is feasible that Epo could modulate angiogenesis in the ischemic brain, leading to increased blood flow and tissue oxygenation in the border zone of the ischemic area, and thus counteract the expansion of the damaged core region. To support this hypothesis, Epo has been described as stimulating in vitro both endothelial cell migration and proliferation, which are key steps in angiogenesis (Anagnostou et al., 1990; Yamaji et al., 1996). Furthermore, Krupinski et al. (1994) demonstrated that the areas in which neurons tend to survive longer are the same as those demonstrated to be highly angiogenic. In addition, the induction of VEGF, the principal regulator of angiogenesis, during cerebral ischemia (Kovacs et al., 1996; Hayashi et al., 1997) and the increased capillary density in the cerebrovascular bed induced by chronic hypoxia (Harik et al., 1995) suggest that angiogenesis is an adaptive response to hypoxic and ischemic stimuli. However, proliferation of endothelial cells and new vessel formation usually begin several days after ischemia (Chen et al., 1994; Krupinski et al., 1994). Accordingly, rather than a direct action of Epo on angiogenesis, one possible mechanism that could be evoked is that Epo, like VEGF (Hayashi et al., 1998), reduced infarct volume by protecting endothelial cells from apoptotic cell death.
Temporal and spatial expression pattern of Epo and Epo-R
Given the neuroprotective role of Epo in cerebral ischemia, we then asked whether the cell types involved in these processes (neurons, endothelial cells, microglia, and astrocytes) could express Epo and the Epo-R to be able to respond to the endogenous ligand. To clarify this issue, we studied the modulation of Epo and Epo-R expression, at both mRNA and protein levels, after permanent MCAO in mice. In situ hybridization experiments revealed some Epo-expressing cells at 24 hours after occlusion within the ischemic core and an Epo-R mRNA induction at 12 and 24 hours after occlusion in cells found in the ischemic core as well as in the border zone. In addition to the basal expression of Epo in neurons and astrocytes, we have been able to describe three chronologically distinct patterns of Epo expression, each related to a single cell type corresponding to endothelial cells (1 day), microglia/macrophage-like cells (3 days), and reactive astrocytes (7 days after occlusion). With focal cerebral ischemia, the Epo-R expression always precedes the Epo expression in the same cell type. Our results are consistent with the hypothesis that there is a continuous requirement for Epo during the active evolution of the cerebral infarct. Because astrocytes have been identified in primary cultures as the main cellular source of Epo (Masuda et al., 1994; Marti et al., 1996), it could be that, in vivo, Epo is secreted by astrocytes and detected by immunochemistry only when bound to receptors on neurons, endothelial cells, and microglia. However, our in vitro data, in accordance with those of Juul et al. (1998), are in favor of a neuronal Epo expression. The precise identification of the cellular source of Epo requires a combination of in situ hybridization with immunochemistry in the same brain tissue section, which was not feasible in our present investigation.
Potential therapeutic use of Epo
The temporal and spatial expression of Epo and its receptor in neuronal, endothelial, and glial cells parallels the occurrence of cellular responses associated with infarct progression and suggests different paracrine roles for this cytokine in these cell types after focal permanent MCAO, specifically in the neurodegenerative, angiogenetic, and glial reactions. We propose that Epo acts in multiple ways involving, at least, both neuroprotection and angiogenesis to protect the brain from cerebral ischemia. One might think of the Epo/Epo-R system as an endogenous defense system to protect the brain against damage consequent to a reduction in blood flow or chronic hypoxia. After an upregulation of the Epo/Epo-R system, the brain could counteract the detrimental effects of these events, such as occurs in the ischemic heart (Schaper and Ito, 1996). A severe ischemic insult will, of course, exhaust the self-protective capacity of the brain. Even so, the Epo/Epo-R system is activated, and by adding exogenous additional recombinant Epo, we were able to show a reduced infarct volume. Our findings might encourage a novel therapeutic approach (which might, however, require intracerebroventricular administration) in which Epo or the recently described Epo-mimicking peptides (Wrighton et al., 1996, 1997) could be tested for the treatment of cerebral ischemia and other neurodegenerative diseases.
Footnotes
Acknowledgments
The authors thank Dr. R.H. Wenger (Zürich) for plasmid pBSmEPOR.17 and Dr. A. D'Andrea (Boston) for providing mouse Epo-R cDNA. The authors also thank Dr. C. Bauer for critically reading the manuscript.